Abstract
Exposure of proliferating cells to genotoxic stresses activates a cascade of signaling events termed the DNA damage response (DDR). The DDR preserves genetic stability by detecting DNA lesions, activating cell cycle checkpoints and promoting DNA damage repair. The phosphoinositide 3-kinase-related kinases (PIKKs) ataxia telangiectasia-mutated (ATM), ATM and Rad 3-related kinase (ATR) and DNA-dependent protein kinase (DNA-PK) are crucial for sensing lesions and signal transduction. The checkpoint kinase 1 (CHK1) is a traditional ATR target involved in DDR and normal cell cycle progression and represents a pharmacological target for anticancer regimens. This study employed cell lines stably depleted for CHK1, ATM or both for dissecting cross-talk and compensatory effects on G₂/M checkpoint in response to ionizing radiation (IR). We show that a 90% depletion of CHK1 renders cells radiosensitive without abrogating their IR-mediated G₂/M checkpoint arrest. ATM phosphorylation is enhanced in CHK1-deficient cells compared with their wild-type counterparts. This correlates with lower nuclear abundance of the PP2A catalytic subunit in CHK1-depleted cells. Stable depletion of CHK1 in an ATM-deficient background showed only a 50% reduction from wild-type CHK1 protein expression levels and resulted in an additive attenuation of the G₂/M checkpoint response compared with the individual knockdowns. ATM inhibition and 90% CHK1 depletion abrogated the early G₂/M checkpoint and precluded the cells from mounting an efficient compensatory response to IR at later time points. Our data indicates that dual targeting of ATM and CHK1 functionalities disrupts the compensatory response to DNA damage and could be exploited for developing efficient anti-neoplastic treatments.
Introduction
Exposure of proliferating cells to genotoxic stresses activates a cascade of signaling events termed the DNA damage response (DDR). The DDR preserves genetic stability by detecting DNA lesions, activating cell cycle checkpoints and promoting DNA damage repair. The phosphoinositide 3-kinase-related kinases (PIKKs) ataxia telangiectasia mutated (ATM), ATM and Rad 3-related kinase (ATR) and DNA-dependent protein kinase (DNA-PK) are crucial for lesion sensing and proximal signal transduction.Citation1,Citation2 ATM and DNA-PK recognize and promote the repair of DNA double-strand breaks (DSBs). ATR is the primary responder to DNA single-strand lesions and replication stress. A variety of studies have shown evidence for significant overlap and cross-talk between these pathways.Citation3,Citation4 The checkpoint kinase 1 (CHK1) was initially characterized as part of the canonical ATR-CHK1 pathway and is activated by single-strand stretches of DNA such as those at replicative blocks generated through exposures to UV, hydroxyurea or nucleoside analogs.Citation2 CHK1 also coordinates Rad51-mediated homologous recombination (HR) involved with DSB repair.Citation5 CHK1 functionality is required for cell cycle progression, even in the absence of genotoxic assaults.Citation6 Coordinated recruitment of CHK1 and DNA methyltransferase 1 (DNMT1) to DSBs suggests a potential contribution to epigenome integrity.Citation7,Citation8 CHK1 also functions as a histone H3 kinase and, in this way, contributes to the regulation of transcriptional expression of a subset of cell cycle-dependent genes.Citation9 As part of the DDR, CHK1 plays an important role in virtually all aspects of DNA damage checkpoint signaling. The critical roles of CHK1 in cellular responses to genotoxic assaults make it an attractive pharmacological target for improving efficacy of chemotherapeutic regimens.Citation10-Citation12
Previously, a report from our laboratory documented that ATM-depleted cells (90% reduction in protein expression) display an attenuated G2/M checkpoint following irradiation, due to a compensatory DNA-PKcs-mediated mechanism.Citation13 In that study, ATM-deficient hTERT-184 cells exhibited increased levels of CHK1 pSer345 phosphorylation after IR compared with their ATM-proficient counterparts. This hyperactivation was significantly reduced and correlated with an abrogated G2/M checkpoint when IR was combined with DNA-PKcs inhibition, suggesting that increased CHK1 signaling could be contributing to checkpoint enforcement in ATM-deficient cells. Leung-Pineda and coauthors showed that CHK1 phosphorylation is kept in balance through a feedback regulatory relationship with protein phosphatase 2 (PP2A), wherein inhibition of kinase activity of CHK1 leads to reduced PP2A activity and subsequently to increased CHK1 phosphorylation by ATR.Citation14 Both ATM and CHK1 are among known PP2A substrates,Citation15 and ATM dephosphorylation by PP2A after DNA damage is required for resolution of DNA repair foci.Citation16 Moreover, PP2A is important for initiating the IR-induced G2/M checkpoint signaling response.Citation17 Therefore, disruption of CHK1 function could be expected to affect not only the G2/M checkpoint, but also the phosphorylation status of PP2A substrates, including ATM. Here we examine how disruption of either ATM or CHK1 function affects cellular responses to irradiation and investigate the role of PP2A. We show that ATM and CHK1 cooperate in enforcing the G2/M checkpoint following DSB induction, and propose that this redundancy may be exploited for developing clinically relevant anti-neoplastic treatments.
Results
Generation of stable CHK1-depleted human cell lines
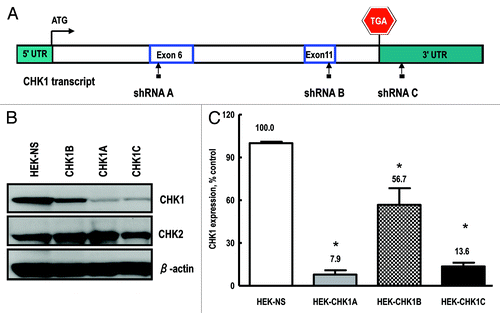
Human embryonic kidney (HEK-293T) and human mammary epithelial cell culture (HME-CC) cell lines were stably transduced with lentiviral-based constructs expressing either a non-silencing (NS) or individual CHK1-specific shRNA sequences. Three different CHK1-targeting constructs were tested, two of which mapped to exons 6 and 11 (shRNA CHK1A and shRNA CHK1B, respectively) and a third one, shRNA CHK1C, mapped to the 3′-UTR (). Whole-cell extracts from the control and CHK1-depleted cell lines were analyzed by western blot to verify the degree of target gene depletion () and normalized to β-actin to correct for loading differences. While all three tested shRNAs significantly reduced CHK1 expression, construct shRNA CHK1A was the most efficient in doing so, resulting in ~92% depletion of the target gene, with shRNA CHK1C following at ~86% depletion (). Transduction with the third construct, shRNA CHK1B, led to a reduction of about 43% in CHK1 protein level (). The CHK1A cell line was chosen for use in the experiments described in this report.
Figure 1. Generation and validation of stable cell lines. (A) Checkpoint kinase-1 (CHK1) transcript structure, depicting the regions targeted by the shRNA constructs used in the current study: exon 6 (shRNA A); exon 11 (shRNA B) and the 3′-UTR (shRNA C). Also shown are the start and stop codons. (B) Representative western blot showing CHK1 expression in cell lines transduced with either the non-silencing shRNA or the CHK1 targeting constructs (CHK1A, CHK1B and CHK1C). (C) Quantification of CHK1 expression in stable HEK293 cell lines. The experiment was performed with three independent replicates and analyzed with Image Quant software (GE). Statistically significant differences are marked by asterisks (p < 0.05).
Proliferation characteristics and sensitivity to genotoxic treatments of the CHK1-depleted stable cell line
Cell proliferation characteristics for the HEK-293T cells expressing the non-silencing shRNA (HEK-NS) and the HEK-293 cells expressing the CHK1A shRNA (HEK-CHK1A) were examined under either control conditions or following exposure to 3 Gy γ-radiation. Initially, counts of cells excluding Trypan blue were determined as an indication of numbers of viable cells in logarithmically growing cell cultures over time, as well as in cultures exposed to ionizing radiation (IR) at various times after treatment (data not shown). The resulting growth curves of the untreated cells (Fig. S2) revealed that the HEK-CHK1A cells have a faster doubling time than the HEK-NS cells, reflecting a faster progression through the cell cycle. Indeed, the calculated doubling times were 16.2 ± 3.9 h for the HEK-CHK1A line and 27.8 ± 2.1 h for HEK-NS cells, a difference that proved statistically significant (p < 0.01). Comparing the fraction of mitotic cells in both HEK-NS and HEK-CHK1A cell lines under control conditions revealed that the HEK-CHK1A cells consistently showed a statistically significant higher proportion of cells in mitosis (). IR treatment slowed growth significantly in both cell lines, with an inhibition of proliferation as reflected in an increase in the cell doubling time of 178% in the control HEK-NS cell line (48.5 ± 5.0 h) and 162% in the CHK1-deficient HEK-CHK1A cell line (27.0 ± 3.9 h) (p < 0.01 for both).
Figure 2. CHK1-depleted cells display decreased survival following exposure to ionizing radiation. (A) The proportions of mitotic cells in HEK-NS and HEK-CHK1A cell lines under control conditions. Graphed data are an average of six biological replicates. All samples were treated and processed for cycle profiles as described in “Materials and Methods.” Briefly, at the indicated time points, the cells were trypsinized, washed with PBS, fixed in 4% paraformaldehyde and stored in PBS in preparation to mitotic cell determination by flow cytometry. (B) Cells were seeded in 96-well plates and allowed to recover overnight. For each time point, two sets of plates were prepared (untreated and irradiated). Cell viability measurements were performed at the indicated time points with the CellTiter Blue assay as described in “Materials and Methods.” The bars represent ratios of irradiated/untreated readings. The measurements were performed with 12 replicates per condition and cell line and tested for reproducibility in at least three independent experiments. Statistically significant differences are marked by asterisks (p < 0.05). (C) HEK-NS and HEK-CHK1A were seeded at a density of 100 cells per 100-mm plate and allowed to attach overnight. For each cell line, one group of plates was left untreated; four other groups were irradiated with 0.5, 1, 2 or 3 Gy γ-IR. After treatment, the plates were returned to the incubator without a media change and allowed to grow undisturbed for 10–14 d. Colonies were stained with methylene blue and counted. Cellular survival was calculated relative to control, which was set to 100%. The dashed horizontal line represents the 50% survival (LD50 = lethal dose 50, used here for referring to the dose that kills 50% of the tested cellular population).
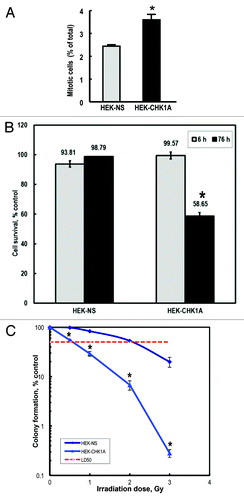
Further evaluation of the consequences of CHK1 depletion in the HEK-CHK1A cell line was performed with a CellTiter Blue assay (resazurin to rezorufin conversion) to quantify the metabolically active cells and evaluate cellular survival following exposure to 3 Gy γ-IR. The average value for untreated controls was set to 100%, and the other values being compared with control were determined relative to untreated controls. For both the control HEK-NS cell line and the CHK1-depleted HEK-CHK1A cell line, no significant change in proliferation was observed at 6 h post-irradiation relative to their respective controls. However, at 76 h post-IR, the CHK1-depleted cell line showed a statistically significant drop (41%) in the viability of metabolically active cells ().
Cell lines were tested for radiosensitivity as reflected in the ability to form colonies following treatment with IR at different doses. Clonogenic assay results showed that the HEK-CHK1A cell line displayed a markedly increased sensitivity to γ-radiation exposure as compared with the control HEK-NS cells (), with an LD50 = 0.5 Gy γ-IR, in contrast to the control cell line which had an LD50 of 2 Gy γ-IR.
G2/M checkpoint responses to irradiation following CHK1 depletion
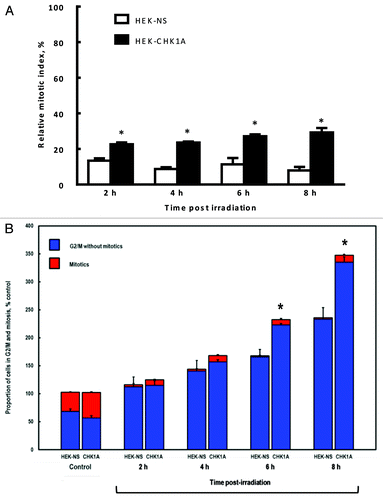
To investigate the G2/M transition regulation in CHK1-depleted cells, we examined the G2 DNA damage checkpoint in response to γ-IR in a time course (). Briefly, cells were exposed to 3 Gy γ-IR, collected at the indicated time points (2, 4, 6 and 8 h), fixed and processed for flow cytometric analysis of cell cycle profiles as described in “Materials and Methods.” The relative mitotic index (RMI) is the ratio of the number of cells in mitosis in the irradiated sample divided by the number of cells in mitosis in the untreated sample, expressed as a % of the control. A low RMI is therefore suggestive of a strong G2/M checkpoint arrest (i.e., cells are prevented from entering mitosis), whereas a higher RMI indicates checkpoint attenuation or even abrogation. CHK1 depletion resulted in an attenuated G2/M checkpoint response to radiation-induced damage, with the RMI values that were consistently 2.0–2.5-fold higher than those for the control cell line (). Also of note is that in addition to a weakened G2/M checkpoint (), CHK1-depletion (HEK-CHK1A line) led to a higher increase in the accumulation of cells in the G2 phase with time following exposure (), up to 8 h post-IR, than what was observed in the HEK-NS cells (). This suggests that CHK1-depleted cells may resume cell cycle faster, following a less efficient S phase checkpoint and a shorter S phase delay.
Figure 3. Mitotic indices of CHK1-depleted cells under control conditions and following genotoxic treatment. HEK-NS and HEK-CHK1A cells were plated, allowed to recover for 18–24 h and where appropriate, subjected to ionizing radiation (3 Gy γ-IR). All samples were treated and processed for cell cycle profiling as described in “Materials and Methods.” Briefly, at the indicated time points, the cells were trypsinized, washed with PBS, fixed in 4% paraformaldehyde and stored in PBS in preparation to mitotic cell determination by flow cytometry. (A) Relative mitotic indices of HEK-NS and HEK-CHK1A cell lines following irradiation were calculated by dividing the percentage of mitotic cells in the treated sample by the percentage of mitotic cells in its respective untreated sample and expressed as % control. Mean RMIs were then determined from at least three independent experiments and biological replicates. See supplementary files for flow cytometry profiles and Table S1 for the absolute values of mitotic cell measurements. (B) Proportions of cells in G2/M of the cell cycle following irradiation in HEK-NS and HEK-CHK1A cell lines (based on DNA content, less % mitotics based on phospho-H3). Statistically significant differences are marked by asterisks (p < 0.05).
Taken together, the above data suggest that CHK1 depletion results in enhanced radiosensitivity, lower survival rates following exposure to IR, accelerated cellular proliferation rates and altered G2/M checkpoint responses to IR-induced damage.
Cooperation between CHK1 and ATM in the G2/M checkpoint response
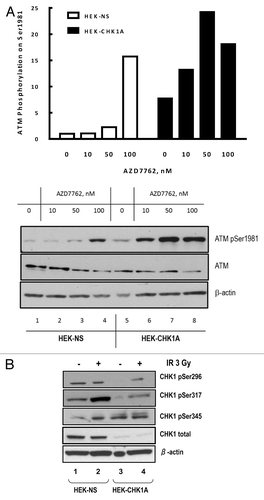
Analysis of ATM activation in response to irradiation showed that both cell lines, HEK-NS and HEK-CHK1A displayed IR-dependent increases in ATM phosphorylation (). The magnitude of ATM phosphorylation following IR treatment was higher in the CHK1-depleted cells at the early time points, showing a rapid elevation at 2 h with continued increase in the phosphorylation of ATM at 6 h post treatment (). For both cell lines, ATM phosphorylation reverted to the basal level at 24 h post-irradiation. Notably, CHK1 depletion by itself resulted in a reproducible increase in the basal level of ATM phosphorylation on Ser1981 (, time 0; , compare lanes 1 and 3; and , compare lanes 1 and 5).The partial attenuation of the G2/M checkpoint under conditions of CHK1 depletion in the HEK-CHK1A cells (), rather than a complete abrogation of the G2/M checkpoint, could thus be due to a compensatory ATM hyperactivation as reflected in this increased ATM phosphorylation on Ser1981 ().
Figure 4. ATM autophosphorylation in CHK1-depleted cells. (A) Time course of ATM activation in response to ionizing radiation as measured by phosphorylation of Ser1981 in HEK-NS and HEK-CHK1A cell lines. Graphed data are the average of three independent measurements ± SEM (B) Representative western blot showing increased ATM phosphorylation in HEK293 stable cell lines HEK-NS and HEK-CHK1A at 2 h post-irradiation. (C) Exponentially growing HEK293 cells were seeded at 50% confluency and allowed to attach overnight. Transfections were performed in 6-well dishes with 300 pmoles of non-targeting siRNA or CHK1 siRNA per well and Lipofectamine 2000 according to the manufacturer's instructions. At 48 h post-transfection the cells were pre-treated for 1 h with either vehicle (DMSO) or 10 µM Ku55933, followed by exposure to ionizing radiation (3 Gy γ-IR). The cells were harvested at 2 h post-IR and analyzed by western blot. (D) HEK-NS and HEK-CHK1A cell lines were seeded and allowed to recover for 18 h. Following irradiation at 3 Gy γ-IR, the cells received either vehicle (DMSO) or 10 µM Ku55933 after 30 min post-IR. The samples were harvested at 2 h post exposure and analyzed by western blot.
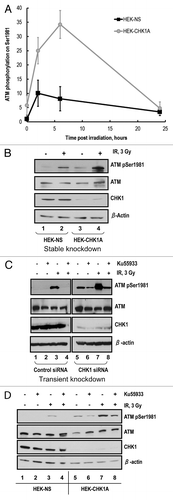
Increased ATM phosphorylation on Ser1981 following IR was also observed when CHK1 was depleted transiently (, compare lanes 3 and 7). ATM inhibition with 10 µM Ku55933 for 1 h prior to irradiation resulted in a complete block of its phosphorylation on Ser1981 in the control siRNA-transfected cells (, lane 4, left panel). In marked contrast, the CHK1-depleted cell line exhibited an elevated basal level of ATM phosphorylation on Ser1981 that was only slightly reduced by pretreatment with the ATM inhibitor (, compare lanes 1 and 2 on the left with lanes 5 and 6 on the right). There was significantly higher ATM phosphorylation on Ser1981 in response to IR that was reduced by the ATM kinase inhibitor only to the unirradiated basal levels (, compare lanes 3, 7 and 8). As Ser1981 is an ATM auto-phosphorylation site, this observation suggested an altered ATM dephosphorylation dynamics in CHK1-deficient cells.
Interestingly, Leung-Pineda et al. reported that CHK1 inhibition with Gö6976 disrupted a regulatory circuit between CHK1 and the phosphatase PP2A.Citation14 Additionally, PP2A is an ATM Ser1981 phosphatase (reviewed in ref. Citation15). We hypothesized that residual ATM auto-phosphorylation observed in CHK1-depleted cells could be due to a disruption of PP2A function and thus result in lower than normal rates of ATM dephosphorylation. To test this hypothesis, we treated control HEK-NS and CHK1-deficient HEK-CHK1A cells with the ATM inhibitor Ku55933 at 30 min post-irradiation (to allow the IR-mediated activation first) and then analyzed the ATM status at 2 h post-irradiation, to evaluate effects of CHK1 depletion on the stability of the ATM activating phosphorylation. The HEK-NS cell line showed no detectable ATM phosphorylation before irradiation, with or without treatment with Ku55933. In contrast, and as previously shown in , HEK-CHK1A cells showed detectable ATM phosphorylation in the absence of irradiation, and this phosphorylation was only partially reduced by treatment with the ATM inhibitor (). Furthermore, while addition of Ku55933 at 30 min post-irradiation resulted in a completely eliminated IR-induced ATM phosphorylation in HEK-NS cells at 2 h post-IR, treatment with the ATM inhibitor caused only a partial reduction of ATM phosphorylation in the CHK1-depleted HEK-CHK1A cells (), in agreement with the results obtained for transient transfection experiments ().
To further interrogate the steady-state phosphorylation dynamics of ATM on Ser1981, we exposed control and CHK1-depleted cells with the CHK1 inhibitor AZD7762 without additional treatment. Inhibition of CHK1 kinase function led to a dose-dependent increase in ATM phosphorylation on Ser1981 in the control HEK-NS cell line (, open bars) and this effect was further enhanced in the CHK1-depleted HEK-CHK1A cells (, black bars). It was striking to us that treatment of the HEK-CHK1A cells with the CHK1 inhibitor AZD7762 had such a dramatic effect on ATM phosphorylation, because these cells had virtually no CHK1 protein. However, it should be noted that despite the near complete depletion of CHK1, HEK-CHK1A cells still exhibit significant CHK1 phosphorylation levels under both control and irradiated conditions (). In fact, when normalized to the levels of total CHK1 protein, the phosphorylated form of CHK1 is up to 200-fold more abundant in the depleted cells than in the control HEK-NS cells (Table S2). Therefore, the residual CHK1 protein in the HEK-CHK1A cell line appears hyper-phosphorylated compared with the fraction of phosphorylated CHK1 seen in the non-silencing cell line HEK-NS. This provides a plausible explanation for the observed further effects of CHK1 inhibition following treatment with AZD7762 as reflected in increasing levels of ATM phosphorylation (). Although one possible explanation for increased ATM phosphorylation is a higher accumulation of damage due to lack of CHK1 functionality, the ability of the CHK1 inhibitor to elicit elevated ATM phosphorylation even after short exposure times and without other genotoxic treatments is indicative of effects on a feedback regulatory loop, in which impaired CHK1 function may preemptively lead to an accumulation of ATM phosphorylation.
Figure 5. CHK1 inhibition results in enhanced ATM phosphorylation. (A) HEK-NS and HEK-CHK1A cell lines were seeded and allowed to recover for 18 h. CHK1 inhibitor AZD7762 in the indicated concentrations or the vehicle control were added to the culture medium. The cells were harvested 6 h later. ATM phosphorylation on Ser1981 was analyzed by western blot and graphed as fold-change relative to the untreated control of the HEK-NS cell line. (B) CHK1 protein abundance and phosphorylation status in HEK293 stable cell lines. HEK-NS and HEK-CHK1A cell lines were seeded and allowed to recover for 18 h. The samples were harvested at 2 h post-irradiation with 3 Gy γ-IR and analyzed by western blot.
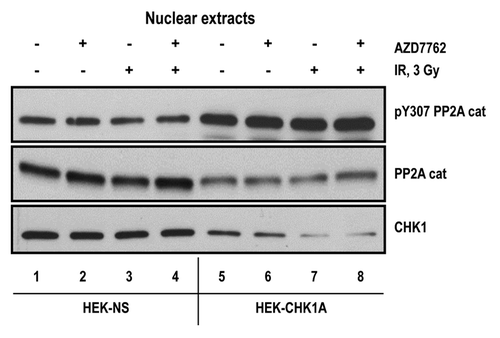
To examine the regulatory relationship between CHK1, ATM and PP2A, we analyzed the subcellular distribution of the catalytic subunit of PP2A in HEK-NS and HEK-CHK1A cells. As the activity of our proteins of interest (CHK1, ATM and PP2A), at least in the context of the DNA damage response, is associated with the nuclear fraction, we examined the status of PP2A catalytic subunit in the nuclear extracts from non-silencing and CHK1-depleted cells. While the bulk of the PP2A catalytic subunit was found in the cytoplasmic fractions (data not shown), we found that CHK1-depleted HEK-CHK1A cells have a significantly lower level of nuclear PP2A catalytic subunit (, middle panel). Furthermore, CHK1-deficient HEK-CHK1A cells also exhibited increased levels of the Y307-phosphorylated form of PP2A catalytic subunit despite having lower total levels (). Phosphorylation of tyrosine 307 has been reported to have an inhibitory effect on PP2A catalytic function.Citation18,Citation19 Interestingly, inhibition of CHK1 kinase activity with the CHK1 inhibitor AZD7762 did not have the same effect on the nuclear localization of PP2A, as did depletion of the levels of the CHK1 protein (), suggesting that there may be protein-protein interactions involving CHK1 that are critical in the regulation of PP2A nuclear activity. Therefore, these observations support reduced PP2A activity in the nuclei of CHK1-depleted cells, providing a plausible explanation for augmented ATM and CHK1 phosphorylation detected in these cells. Furthermore, at 24 h post-irradiation, when ATM phosphorylation is approaching basal levels (), treatment with the phosphatase inhibitor okadaic acid (1 nM) resulted in an enhancement of ATM phosphorylation in the CHK1-deficient cells compared with the control cell line HEK-NS (Fig. S3).
Figure 6. Status of the PP2A catalytic subunit in nuclear extracts from CHK1-depleted cells. Subcellular fractionation was performed as described in “Materials and Methods.” For all samples, nuclear extracts were prepared from equal numbers of cells. The cells were treated with 10 nM AZD7762 1 h prior to irradiation with 3 Gy γ-IR and harvested at 2 h post-IR. Antibodies are against the catalytic subunit of PP2A (PP2A cat; total or phosphorylated on Y307).
CHK1 and ATM interactions in human mammary epithelial cells
Previously, we reported that ATM-deficient human mammary epithelial cell cultures (HME-CC) exhibit only an attenuated G2-checkpoint following irradiation.Citation13 This prompted us to examine CHK1 status and function in ATM-depleted cells. Interestingly, CHK1 mRNA levels were reduced by roughly 30% in ATM-depleted cells (), and this correlated with a comparable decrease in CHK1 protein expression level (, compare open and gray bars).
Figure 7. CHK1 expression and phosphorylation status in human mammary epithelial cell lines. To analyze CHK1 expression levels, the cells were cultured under normal growth conditions to a maximum of 70–80% confluency. At harvesting, the cell pellets for each sample were divided into two tubes, one for total RNA extraction and another one for protein extraction. The extractions were from at least three biological replicates for each indicated cell line. (A) CHK1 mRNA abundance was measured by qRT-PCR as described in “Materials and Methods.” (B) CHK1 protein abundance was analyzed by western blot. The graph is from the quantified and normalized data from at least three biological replicates. The wild-type expression level (LacZ-NS cell line) is set to 100% with the values obtained for all other cell lines recalculated relative to the wild-type control. (C) HME-CC cells were seeded 44–48 h prior to treatment with 3 Gy γ-IR and harvested at 2 h post-exposure.
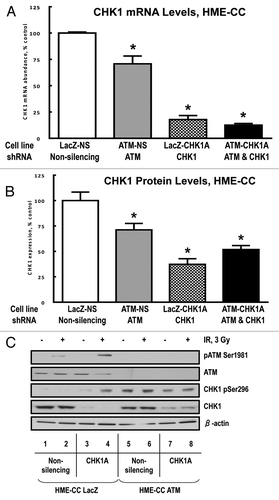
Stable transduction of wild-type (LacZ) and ATM-deficient HME-CC cells with the CHK1A lentiviral construct resulted in a depletion of CHK1 mRNA of 63% and 48%, respectively (). Interestingly, the doubly depleted HME-CC ATM-CHK1A cells exhibited somewhat higher CHK1 protein levels compared with their HME-CC ATM-proficient/CHK1-deficient counterpart, LacZ-CHK1A cells (, compare checkered and black bars). It is possible that the higher residual level of CHK1 protein in the doubly deficient cell line is required for survival of these cells with reduced levels of ATM, supporting our hypothesis of a compensatory relationship between ATM and CHK1. Furthermore, we expected this functional redundancy to be reflected in the ability of cells to arrest in G2/M following genotoxic exposures.
Therefore, we examined the G2/M checkpoint signaling response to IR-induced damage in HME-CC cells deficient in ATM, CHK1 or both ATM and CHK1. Comparison of wild-type HME-CC LacZ-NS and HME-CC LacZ-CHK1A cells showed that their behavior is similar to that of their HEK-293T counterparts (HEK-NS and HEK-CHK1A). ATM phosphorylation was elevated in a DNA damage-dependent manner in the control HME-CC LacZ-NS cells and hyper-phosphorylated in the CHK1-depleted HME-CC LacZ-CHK1A cells. Interestingly, in ATM-depleted HME-CC cells, with either normal or depleted levels of CHK1 (ATM-NS or ATM-CHK1A), CHK1 phosphorylation on Ser296 was uniformly elevated, irrespective of treatment with IR ().
Analysis of G2/M DNA damage checkpoint arrest following IR exposure in these cell lines at 2 h post-irradiation showed that ATM-deficiency caused only a small attenuation in the HME-CC ATM-NS cells, with a RMI of 10.8% vs. 2.97% in LacZ-NS (). Depletion of CHK1 by 63% of the wild-type protein level in the HME-CC LacZ-CHK1A cells resulted in a much larger attenuation of the G2/M checkpoint, with a RMI of 33.4%. Additionally, the combination of both deficiencies in the HME-CC ATM1-CHK1A cells appeared to be additive in character, yielding a 45.03% RMI value (). Furthermore, at 4 h post-irradiation, the control HME-CC LacZ-NS cell line still demonstrated a modest G2/M checkpoint arrest (RMI 30.9%), whereas all the HME-CC cell lines deficient in either ATM, CHK1 or both were not significantly different from one another, completely exiting the G2/M cell cycle arrest ().
Figure 8. ATM and CHK1 cooperation is required for both the early checkpoint response and recovery of the mitotic indices post-irradiation. HME-CC cells were seeded 44–48 h prior to treatment with 3 Gy γ-IR and harvested at the indicated time points post exposure. The samples were processed for flow cytometry analysis as described in “Materials and Methods.” Graphed data represents the relative mitotic values for 2 h (A) and 4 h post exposure (B). For experiments in HEK293 cells, HEK-NS and HEK-CHK1A cell lines were seeded and allowed to recover for 18 h. ATM inhibitor Ku55933(10 µM) or DMSO (vehicle) were added to the cell culture media 1 h prior to irradiation with 3 Gy γ-IR. All samples were collected at the indicated time points and processed for flow cytometry analysis as described in “Materials and Methods.” (C) Relative mitotic indices following irradiation ± Ku55933 in HEK-NS and HEK-CHK1A cells.
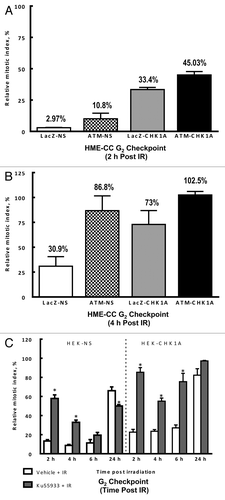
To summarize, G2/M checkpoint analysis in HME-CC cell lines indicated that both ATM and CHK1 contribute and cooperate in enforcing the G2/M checkpoint following DNA damage, and their intact functionality is also important for a properly paced exit from cell cycle arrest. The non-synergistic, additive character of ATM and CHK1 with regard to G2/M checkpoint responses in these cell lines can be reasonably attributed to residual CHK1 protein (50% of wild-type expression level) detected in the doubly deficient cells.
This prompted us to analyze the contribution of ATM to G2/M checkpoint responses in the HEK-293 set of cell lines, HEK-NS and HEK-CHK1A, whose level of CHK1 protein depletion exceeded 90% (). For this purpose, we pretreated both cell lines with either vehicle or 10 µM Ku55933 for 1 h, followed by irradiation at 3 Gy. The samples were collected at 2, 4, 6 and 24 h after irradiation, and the relative mitotic indices were calculated and plotted. Pre-treatment with the ATM inhibitor Ku55933 (10 µM) for 1 h prior to irradiation had no significant effect on RMIs compared with the vehicle control. ATM inhibition followed by irradiation at 2 h post-treatment led to a large attenuation of the G2/M checkpoint in the HEK-NS cells (RMI of 57.9%) (), whereas in the HEK-CHK1A cells this checkpoint was essentially abrogated (RMI of 85.4%) (). Interestingly, the non-silencing cell line with wild-type levels of CHK1 gradually but progressively generated a G2/M checkpoint arrest even in the presence of ATM inhibition over the next 4 h; thus, by 6 h post-exposure, the RMI values did not differ significantly from those obtained with IR treatment in the absence of ATM inhibition. In the case of the CHK1-depleted cells treated with the ATM inhibitor, a modest drop in the RMI value was observed following IR treatment at 4 h; however, this weak G2/M checkpoint arrest could not be sustained in the same manner as in the CHK1-proficient cells, reverting to an almost completely abrogated G2/M checkpoint arrest by 6 h following IR-exposure (). Moreover, at 24 h post-treatment, the CHK1-depleted cells seemed to have completely exited the G2 arrest when ATM was inhibited prior to irradiation. Exit from the G2/M checkpoint at 24 h in the non-silencing cell line was incomplete, and ATM inhibition of these CHK1-proficient cells resulted in even further suppression of cell cycle resumption at 24 h post-IR (). It should be noted that in HEK293 cells the p53 pathway is dysfunctional,Citation20 and therefore observations for HEK-NS and HEK-CHK1A cell lines represent responses to irradiation under conditions of CHK1 depletion, p53 dysfunction and ATM inhibition. Human mammary epithelial cell lines, however, express wild-type p53 and are able to mount p53-mediated responses to UVCitation21 but have a weak G1/S checkpoint following irradiation, most likely due to an alteration in the p16-p53 regulatory circuit.Citation22,Citation23 Taken together, these observations suggest that CHK1 and ATM contribute independently to the early stages of the G2/M checkpoint activation, and that CHK1-proficiency is required for an efficient enforcement of the G2/M checkpoint in the absence of ATM at later time points. Also, CHK1 is needed for preventing a premature exit from the checkpoint and may be compensating for impaired ATM function by imposing a delay in cell cycle resumption (, HEK-NS cell line). ATM inhibition in CHK1-deficient cells showed only a mild tendency toward such a delay, reflecting, perhaps, the residual CHK1 kinase activity present in this cell line ().
Discussion
Previous work with ATM-deficient cell lines showed that ATM depletion alone results in an attenuated G2/M checkpoint in response to irradiation and suggested that the compensatory mechanism included increased CHK1 phosphorylation on Ser345 compared with wild-type cells. This hyperactivation was significantly reduced and correlated with an abrogated G2/M checkpoint when IR was combined with DNA-PKcs inhibition, indicating a potential contribution from increased CHK1 signaling to checkpoint enforcement in ATM-deficient cells.Citation13 Furthermore, Jiang et al. showed that under conditions of p53-deficiency, ATM suppression results in sensitization of tumor cells to genotoxic treatments, and ATM-deficient cancer cells are “addicted” to DNA-PKcs and require it for survival after double-stranded DNA breaks.Citation24
The current report illustrates that a high level of CHK1 depletion (90% at the protein level) resulted in significantly decreased survival rates following irradiation and heightened radiosensitivity as measured in clonogenic assays (). In the absence of DNA damage, both CHK1-depleted and ATM-deficient cells displayed a hyper-proliferative phenotype, while the combined ATM/CHK1 depletion was growth-inhibitory (data not shown). ATM activation levels and dynamics were altered in CHK1-depleted cells: ATM phosphorylation on Ser1981 was higher in CHK1-deficient cells, peaking at a later time point compared with CHK1-proficient, HEK-NS cells, although by 24 h, a resolution of this response was observed by western blot analysis. This was somewhat surprising, because other studies investigating CHK1 depletion effects have reported persistent ATM activation in the absence of CHK1.Citation25 This apparent contradiction could potentially be explained by different detection thresholds for western blot and immunofluorescence. Both our experiments and data published by other groups for other genotoxic treatments show that ATM is hyperactivated under CHK1 deficiency (either by depletion or inhibition) conditions. Furthermore, transient CHK1 depletion showed a more dramatic increase in ATM activation following IR (compare ). In transiently depleted cells, this effect was largely attributable to ongoing ATM auto-phosphorylation events, although a fraction clearly persisted even in the presence of ATM inhibitor Ku55933 (), suggesting a possible phosphatase involvement. Multiple serine-threonine phosphatases have been reported to dephosphorylate ATM and CHK1 (reviewed in ref. Citation15), among which are PP1, PP2A and Wip1. We focused our attention on PP2A phosphatase as a plausible candidate that could mediate the enhanced phosphorylation effects, specifically because it has been previously reported to share a feedback regulatory relationship with CHK1.Citation14 Leung-Pineda and co-authors examined the phosphorylation of CHK1 by ATR and the involvement of CHK1 kinase in modulating its own signaling responses by negatively regulating PP2A activity. It is plausible that CHK1 kinase activity controls the frequency and/or timing of dephosphorylation events by PP2A via a regulatory phosphorylation, and the potential for such an interaction was recently documented in a report identifying a PP2A specific sequence as a strong target for CHK1 phosphorylation.Citation26 Lee et al. reported that in HeLa cells treated with doxorubicin PP2A co-localized with γ-H2AX foci and was involved in dephosphorylation of Polo-like kinase 1 (Plk1), allowing accumulation of cells in G2 phase. ATM or CHK1 depletion blocked mitotic Plk1 dephosphorylation following doxorubicin treatment.Citation27 The data reported in our study indicate that CHK1 depletion altered the subcellular localization for the PP2A catalytic subunit, which appeared diminished in the nuclear fraction of CHK1-deficient cells compared with their wild-type counterparts. Additionally, the higher level of PP2A inhibitory phosphorylation on tyrosine-307 in the nuclear extracts derived from CHK1-deficient cells is in good agreement with the observed ATM and CHK1 hyper-phosphorylation states. Taken together, these observations suggest that CHK1 may be involved in controlling the duration of cell cycle arrest by modulating both PP2A activity and subcellular localization. Interestingly, CHK1 abundance (both mRNA and protein) seem to be influenced by ATM status, with ATM-depleted cells exhibiting a small, but statistically significant CHK1 downregulation. This correlates with heightened CHK1 auto-phosphorylation () in ATM cells and a checkpoint response that is only modestly attenuated (, ATM-NS). Combined CHK1 and ATM depletion was additive in early checkpoint responses (), whereas checkpoint resolution at 4 h post-irradiation was of comparable magnitude in all deficient cell lines (, compare RMI values). It should also be noted that ATM depletion and ATM inhibition, although similar, are not entirely equivalent with regard to checkpoint responses. For example, inhibition of ATM kinase activity in HEK-NS cells prior to irradiation led to a large attenuation of the G2/M checkpoint at 2 h post-IR (RMI of ~60%, ); however a strong compensatory response was seen after 6 h (, HEK-NS). ATM inhibition also resulted in a delayed recovery of mitotic indices at 24 h post-irradiation in CHK1-proficient cells, whereas in cells stably depleted for ATM (human mammary epithelial cell lines), the checkpoint exit did not seem to be hindered. This observed dissimilarity between ATM inhibition with Ku55933 and stable ATM depletion via shRNA in terms of recovery of mitotic indices at 24 h post-irradiation could be due to differences in the composition and stoichiometry of ATM-containing molecular complexes, CHK1 dosage effects, ATM inhibitor metabolic washout and subsequent relief of ATM inhibition or a combination of all these factors. Future studies would be needed to compare the molecular consequences of ATM depletion and ATM inhibition.
CHK1 depletion in combination with ATM inhibition resulted in a complete abrogation of the early G2/M checkpoint (see , HEK-CHK1A, 2 h time point). Although a temporary reduction in RMI was seen at 4 h post-IR in CHK1-depleted and ATM-inhibited cells, this compensatory response could not be sustained. Additionally, the delayed recovery in mitotic rates seen at 24 h in cells that were ATM inhibited prior to IR was completely absent in CHK1-depleted cells, suggesting that both ATM and CHK1 contribute to and modulate the checkpoint exit. In summary, reciprocal regulation within the ATM-CHK1-PP2A circuit provides a mechanism whereby compensatory responses may be elicited following genotoxic stress (). Cells with wild-type levels of ATM and CHK1 control the magnitude and frequency of phosphorylation events for these kinases, and PP2A dephosphorylation is part of this “balancing act” (). A functional feedback loop between PP2A and CHK1Citation14 ensures that under normal growth conditions ATM and CHK1 phosphorylation stays below detection levels. In cells depleted for CHK1, the PP2A-CHK1 feedback loop is disrupted, resulting in hyper-phosphorylation of PP2A substrates, including ATM and CHK1 (). Heightened ATM auto-phosphorylation enforces the G2/M checkpoint, counteracting the CHK1 deficiency. ATM depletion in human mammary epithelial cell cultures resulted in a small (30%) reduction in CHK1 mRNA and protein levels, but enhanced CHK1 autophosphorylation on Ser296 ( and ), which could explain the unexpectedly modest effect on the G2/M checkpoint. Furthermore, the inability to reach a higher level of CHK1-depletion in cells with an ATM-deficient background is likely an adaptive response, suggesting that cellular survival in the absence of ATM requires at least 50% of CHK1 protein expression. Additionally, an examination of cell viability at late time points following irradiation showed that the control cell line HME-CC LacZ-NS, as well as the cell lines with single deficiencies in either CHK1 or ATM (LacZ-CHK1A and ATM-NS) exhibited a growth delay after IR at 3 Gy (Fig. S4, panels A–C). The double-deficient cell line HME-CC ATM-CHK1A, however, did not appear to slow down during the first 24 h post-treatment, with viable cell numbers that closely matched those in the unirradiated control samples. However, at 48 h post-IR the double-deficient cell line appears to have halted cell growth (Fig. S4D). We anticipate that this synthetic growth inhibition seen with the double-deficient cell line would likely be even more prominent when evaluated in clonogenic assays for a larger variety of irradiation doses. It would also be of interest to evaluate whether ATM deficiency will sensitize the cells to CHK1 inhibition.
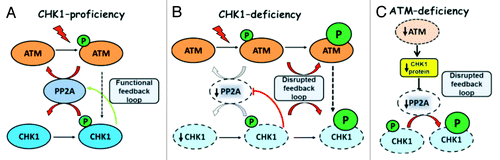
Figure 9. Compensatory regulation within the ATM-CHK1-PP2A circuit. (A) Physiological phosphorylation of ATM and CHK1 is kept in balance by PP2A-mediated dephosphorylation events and a functional CHK1-PP2A feedback regulation loop. No phosphorylation is detectable under normal growth conditions and represents a response to genotoxins. (B) CHK1-depletion disrupts the feedback loop to PP2A and allows for the accumulation of phosphorylated ATM and CHK1 molecules that exceed the proportion of phosphorylated molecules in wild-type cells. Enhanced ATM phosphorylation can enforce the G2/M checkpoint in spite of impaired CHK1 function. (C) ATM deficiency in human mammary epithelial cells is accompanied by a CHK1 insufficiency at the protein expression level. The disruption in the PP2A-CHK1 feedback loop leads to higher CHK1 phosphorylation that may compensate for the lack of ATM.
CHK1 inhibitors are being actively tested as promising therapeutic agents for a variety of cancer types, with CHK1 status in different cancers providing further support for this approach. For example, Bertoni et al. reported that colon and endometrial cancer samples display frameshift mutations in the CHK1 gene, and these are often accompanied by microsatellite instability.Citation28 In stomach tumors with microsatellite instability, CHK1 mutations occurred with a relatively low frequency (9% of tumors).Citation29 Tumors observed in germline Chk1+/− mice did not appear to lose the second allele of Chk1,Citation30 suggesting that Chk1 is a haploinsufficient tumor suppressor. CHK1 is involved in both normal mammary gland development and breast carcinogenesis.Citation31,Citation32 For instance, CHK1 transcript abundance was increased in histologic grade 3 tumors and in tumors in which the expression of estrogen receptor (ER) and progesterone receptor (PR) was lost.Citation33 Grade 3 breast carcinomas with a triple-negative ER-/PR-/HER-2-phenotype also displayed significantly increased CHK1 mRNA levels compared with other grade 3 tumors.Citation33 Furthermore, Tort and co-authors showed that CHK1 gene inactivation is uncommon in human lymphomas; however, loss of CHK1 protein expression was observed in a subset of aggressive lymphomas and it occurred alternatively to ATM gene alterations.Citation34 This observation once again highlights the potential for a compensatory relationship between CHK1 and ATM. Moreover, the radiosensitive phenotype of ATM-deficient cells could be rescued by CHK1 overexpression through a remediation of the G2/M checkpoint.Citation35
Reports published to date showed that targeting CHK1 alone, as well as combining its inhibition with other antineoplastic agents are both worthwhile approaches.Citation36-Citation40 It is becoming increasingly clear, however, that treatment strategies taking into account the individual tumor genetic make-up (or at least exploiting its known deficiencies or vulnerabilities) have a higher likelihood of success, hence the interest in researching potential compensatory relationships within the DNA damage response pathways. Published studies have highlighted the importance of assessing CHK1 expression status prior to administering a particular anti-neoplastic treatment regimen. For example, Ren and co-authors showed that cells with high levels of CHK1 were highly sensitive to microtubule-destabilizing drugs.Citation41 Although embryonically lethal, CHK1 depletion in some tissues may not necessarily result in a discernible phenotype: Greenow et al. reported that inducible CHK1 deletion from somatic epithelial cells in the adult mouse liver was tolerated well, whereas the small intestine cells displayed high levels of apoptosis following CHK1 ablation.Citation42
Therefore, identification of pathways that may compensate for CHK1 inhibition/depletion is of critical importance for designing efficacious and low-toxicity anticancer treatment regimens. Several reports illustrate this concept both in cell line systems, as well as in experimental tumor models. For example, use of Src and CHK1 inhibitors in multiple myeloma cells showed synergistic pro-apoptotic and anti-angiogenic responses.Citation43 Simultaneous inhibition of CHK1 and Mad2, p53, p21 or CHK2 also increased cell killing compared with CHK1 inhibitor alone, whereas the 14-3-3 protein inhibition did not alter this response following CHK1 inhibition.Citation44 Selective CHK1 inhibition in conjunction with WEE1 depletion had anti-proliferative effects.Citation45 Lu et al. showed that PARP-1-null cells exhibited a stronger G2/M checkpoint through a compensatory CHK1 hyperactivation and could be rendered highly sensitive to ionizing radiation if CHK1 was depleted by siRNA.Citation46 A study by Jurvansuu and co-authors reported that CHK1-mediated G2/M arrest is transient, and it is maintained through an additional mechanism involving the p53-dependent transcriptional repression of mitotic proteins.Citation47 Interestingly, CHK1 heterozygosity caused abnormal mammary gland development, without an increase in tumor incidence in p53-proficient mice; however, deletion of one copy of p53 in these animals (and consequently double heterozygous status for CHK1 and p53) synergistically stimulated tumorigenesis without rescuing the abnormal developmental defects. In mice with complete ablation of p53 and one CHK1 gene copy (p53−/− CHK1+/− genotypes), an inhibition of neoplastic transformation was observed.Citation32
Cross-talk between different branches of DNA damage response pathways has become widely appreciated in recent years. Redundancy within DNA damage response pathways and circuits provides alternative genome integrity preservation mechanisms. It also represents a source of survival mechanisms for aggressive tumors. Some anti-neoplastic regimens, although efficient in eliminating most of the cancerous cells, may inadvertently select for these aggressive phenotypes. CHK1 is traditionally regarded as part of the canonical ATR-CHK1 pathway, with its activation by ATM seen as an example of cross-talk. Studies in fission yeast suggest the possibility that ATM may be directly responsible for a portion of Chk1 activation.Citation48 Furthermore, CHK1 inhibitor treatments lead to increased ATM/ATR-dependent CHK1 phosphorylation (Ser345).Citation49 Consequently, blocked CHK1 kinase activity activates the negative feedback CHK1-PP2A regulatory loop and CHK1 hyperphosphorylation.Citation14 Combined gemcitabine and CHK1 inhibitor (AZD7762) treatment also results in an increased CHK1 Ser345 phosphorylation, both as a consequence of genotoxic stress and via a PP2A-mediated contribution.Citation50 In our experimental system, CHK1 depletion alone (transient and stable) resulted in higher basal levels of ATM autophosphorylation. Our findings suggest that these effects are at least partly attributable to altered PP2A subcellular localization (depletion of the nuclear fraction) and increased inhibitory phosphorylation on Y307, secondary to diminished CHK1 abundance. With limited PP2A access to ATM, its activation is exacerbated in response to DNA damage, and thus leads to a compensatory enforcement of the G2/M checkpoint, which appears attenuated instead of abrogated. Conversely, stable ATM depletion creates a state of CHK1-insufficiency through downregulation of CHK1 protein, which, in turn, leads to augmented CHK1 phosphorylation on Ser296 (auto-phosphorylation). The modest attenuation of the G2/M checkpoint in ATM-depleted cells is the biological consequence of CHK1 hyperactivation, whether it results from ATR,Citation14 DNA-PKcsCitation13 or both. Abrogation of the G2/M checkpoint through combined CHK1 depletion and ATM inhibition highlights a strategy whereby cross-talk and alternative pathway switching is blocked, removing the potential compensatory signaling with regard to checkpoint responses following DNA damage. It also emphasizes the potential use of CHK1 inhibitor treatments in tumors harboring ATM mutations, as well as combined ATM and CHK1 inhibitor regimens for cancers characterized by high CHK1 expression.
Materials and Methods
Chemicals and reagents
Blasticidin S was purchased from Life Technologies (Catalog #R210-01), Ku55933 from Selleck Chemicals (Catalog #S1092) and AZD7762 from Axon Medchem (Catalog #Axon 1399). Non-targeting siRNA control (Catalog #D001210-01) and SmartPool CHK1 siRNA (Catalog #L003255-00) were purchased from Thermo Scientific. Viral particles for CHK1 shRNA pGIPZ constructs (Catalog numbers V2LHS_112996, V3LHS_637954 and V3LHS_644862) and the non-silencing shRNA pGIPZ control (Catalog #RHS4348) were obtained from Open Biosystems. Lipofectamine2000 was purchased from Life Technologies (Catalog #11668-019).
Cell culture and experimental treatments
Human embryonic kidney cell line HEK-293T was grown in DMEM (high glucose), supplemented with 10% FBS, 0.1 mM MEM non-essential amino acids, 6 mM L-glutamine and 1 mM MEM sodium pyruvate. Human mammary epithelial cell cultures (HME-CC) HME-CC-LacZ (normal) and HME-CC-ATM1 (stable lentiviral ATM knockdown) were grown as described previously (Arlander et al., 2008). All cell lines were cultured at 37°C in a 5% CO2 incubator. For experimental treatments, exponentially growing cells were seeded at 50–70% confluency and allowed to attach overnight before being exposed to γ-radiation using a 137Cs source. Unless otherwise indicated, inhibitors were added 1 h prior to irradiation, and cells were harvested at different time points following the DNA-damaging treatment. In experiments where cells were exposed to IR first, the inhibitors were added to the culture media 30 min later.
RNA interference
Exponentially growing cells were seeded at 50% confluency and allowed to attach overnight. Transfections were performed in 6-well dishes with 300 pmoles of non-targeting siRNA or CHK1 siRNA per well with Lipofectamine 2000 according to the manufacturer's instructions. All subsequent DNA-damaging and inhibitor treatments were administered at 48 h post-transfection and harvested at 2 h post-irradiation.
Stable cell line generation
HEK-293 cells were transduced with either a non-silencing shRNA pGIPZ viral particle preparation or with one of the CHK1 shRNA constructs (A, B or C) at multiplicities of infection (MOI’s) of 0, 1, 2 and 5. Puromycin (2 μg/ml) was added to the media 48 h later, and the cells were monitored under the microscope for turbo-GFP expression (Fig. S1). Within 3 d from adding the selective drug, no cells were left in the MOI = 0 plates. At this time cells transduced with pGIPZ constructs were split and stored as early passage stocks. For maintenance purposes, the puromycin concentration in the cell culture media was reduced to 1 μg/ml.
Nuclear and cytoplasmic cell fractionation
For cellular fractionation experiments, the cells were counted, and the extraction procedure was performed with volumes proportional to the cell number in each sample and the NE-PER® cell fractionation kit from Thermo Scientific according to the manufacturer’s instructions. Protein concentrations were confirmed in BCA protein quantification assays (Thermo Scientific). The quality of cellular fractions was verified by western blotting for strictly nuclear or cytoplasmic proteins.
Western blotting
Treated or control cells were harvested by trypsinization, washed 1× with ice-cold PBS and lysed in RIPA buffer supplemented with phosphatase and protease inhibitors (Thermo Scientific). Cell lysates were sonicated for 15 sec, mixed with sample dilution buffer and denatured by heating at 98°C for 5–10 min. Aliquots representing equal amounts of protein from each lysate were separated on SDS-PAGE gels and analyzed by western blot analysis. Antibodies to phospho-CHK1 (Ser317, Ser345 and Ser296) were from Cell Signaling Technology (Catalog numbers 8191S, 2341S and 2349S). Antibody to CHK1 was from Santa Cruz Biologicals (Catalog #sc-8408). The antibodies to ATM, phospho-ATM (Ser1981), PP2A and phospho-PP2A Tyr307 were from Epitomics (Catalog numbers 1549-1, 2152-1, 1512-1 and 1155-1). Hsp90 antibody was from Life Technologies (Catalog #37-9400). Equivalent loading and protein transfer were confirmed by Ponceau stain and western blot with the loading control antibody from Sigma (β-actin). Primary antibodies were detected with a peroxidase-conjugated secondary antibody and enhanced chemiluminescence according to the manufacturer's instructions (Pierce). Quantitation of bands in western blots was performed with the ImageQuant TL v.2005 software (GE Healthcare).
Flow cytometry
For determination of DNA content and synthesis, cells were treated with an EdU Click-iT reaction for DNA synthesis and exposed to 4',6-diamidino-2-phenylindole (DAPI) for DNA content. The proportion of the cell population in mitosis was evaluated by assaying the cells for protein expression of phosphorylated histone H3. Typically, cells were harvested 2 h following irradiation and fixed in 4% paraformaldehyde (BioLegend Fixation Buffer, 420801), diluted with PBS and stored at 4°C. For time course experiments, the cells were harvested at the indicated times post-irradiation. To determine the percentage of the population synthesizing DNA during a 2 h period prior to fixation, the manufacturer’s instructions for the Click-iT EdU AF647 Flow Cytometry Assay kit (Invitrogen C10424) were followed, except that Triton X-100 was added to the EdU reaction at a final concentration of 0.2% rather than using the kit’s permeabilization buffer in a separate step. Cells were then rinsed in 1% BSA and stained with phosphorylated histone H3 (phospho-Ser10) antibody (Cell Signaling 9706) in 1% BSA for 2 h at room temperature. Cells were rinsed in BSA and exposed to anti mouse-PE secondary (JIL 115-116-146). Cells were rinsed in BSA and exposed to DAPI for 1 h. Readings were taken using an LSRII flow cytometer (Becton Dickinson) and FACSDiva analysis software. Relative mitotic indices (RMIs) presented in the G2/M checkpoint assays were calculated by dividing the percentage of mitotic cells in the treated sample by the percentage of mitotic cells in its respective untreated sample and expressed as % control. Mean RMIs were then determined from at least three independent experiments and biological replicates.
Cellular assays
The cell proliferation assay was performed with a CellTiter Blue kit from Promega (Catalog #G8081) according to manufacturer’s instructions for a 96-well plate format. Initial measurements were taken prior to treatment to confirm comparable plating efficiencies across replicates and establish the baseline, followed by readings at two more time points, 6 h and 76 h after IR. For each time point, condition and cell line examined, 12 replicates were plated. For cell doubling time calculation, cell viability assessment following irradiation and growth curves, the cells were seeded at low density (30%) and allowed to recover overnight. Untreated or irradiated cells were harvested by trypsinization and counted at different time points, with Trypan Blue exclusion, with an automated cell counter, Cellometer from Nexcelom Biosciences. For the clonogenic assay, exponentially growing cells were seeded at 300–1,000 cells per 100-mm plate, allowed to attach and settle for 48 h before being exposed to ionizing radiation using a 137Cs source. After 10–14 d, cells were stained with 0.2% methylene blue in 50% methanol. Colonies containing >100 cells were counted. The cell fraction surviving treatment was normalized to survival of control cells. Each experimental treatment was performed in quadruplicate.
RNA extraction and qRT-PCR
For gene expression analysis, total RNA was isolated with the Qiagen RNeasy Kit from three biological replicates (Qiagen Inc., Catalog #75142). Two-step qRT-PCR was performed employing SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) and TaqMan Gene Expression Assays (Applied Biosystems), according to the manufacturer’s instructions. Briefly, cDNA was prepared from 0.5 μg of total RNA using SuperScript First-Strand Synthesis System. PCR was then performed with TaqMan Universal PCR Master Mix and TaqMan Gene Expression Assays in an ABI 7500H system (Applied Biosystems). The TaqMan gene expression assays were the following: CHK1, Hs00176236_m1; protein phosphatase 2, catalytic subunit, α isozyme (PPP2CA), Hs00427259_m1; and peptidylprolyl isomerase A (PPIA), Hs04194521_s1. Three biological samples from each experiment were measured and each sample was measured in triplicate. All measurements were normalized to PPIA of the same sample. The fold change of each gene in any tested cell line was compared with that observed in the corresponding wild-type cell line. The results are presented as mean fold change ± standard error of the mean (SEM).
Statistical analysis
All experiments were repeated at least three times to ensure reproducibility across biological replicates. Technical repeats were tested as well within each separate experiment. Statistical analyses were performed with Student's t-test using GraphPrism software version 5.0, with p < 0.05 being considered statistically significant.
| Abbreviations: | ||
| ATM | = | ataxia telangiectasia mutated |
| ATR | = | ATM and Rad 3-related kinase |
| DNA-PK | = | DNA-dependent protein kinase |
| CHK1 | = | checkpoint kinase-1 |
| PP2A | = | protein phosphatase 2 |
| DDR | = | DNA damage response |
| IR | = | ionizing radiation |
| PIKKs | = | phosphoinositide 3-kinase-related kinases |
Additional material
Download Zip (2.1 MB)Acknowledgments
The authors thank Drs Janine Santos and Carl Anderson for their critical reading of the manuscript and helpful comments and the NIEHS Flow Cytometry Core for their assistance with data collection. This research was supported by the Intramural Research Program of the NIH, National Institute of Environmental Health Sciences (Z01ES021157).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/24127
Related Research Data
References
- Martín M, Terradas M, Tusell L, Genescà A. ATM and DNA-PKcs make a complementary couple in DNA double-strand break repair. Mutat Res 2012; 751:29 - 35; http://dx.doi.org/10.1016/j.mrrev.2011.12.006; PMID: 22230547
- Smith J, Tho LM, Xu N, Gillespie DA. The ATM-Chk2 and ATR-Chk1 pathways in DNA damage signaling and cancer. Adv Cancer Res 2010; 108:73 - 112; http://dx.doi.org/10.1016/B978-0-12-380888-2.00003-0; PMID: 21034966
- Kass EM, Jasin M. Collaboration and competition between DNA double-strand break repair pathways. FEBS Lett 2010; 584:3703 - 8; http://dx.doi.org/10.1016/j.febslet.2010.07.057; PMID: 20691183
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008; 18:134 - 47; http://dx.doi.org/10.1038/cr.2007.111; PMID: 18157161
- Sørensen CS, Hansen LT, Dziegielewski J, Syljuåsen RG, Lundin C, Bartek J, et al. The cell-cycle checkpoint kinase Chk1 is required for mammalian homologous recombination repair. Nat Cell Biol 2005; 7:195 - 201; http://dx.doi.org/10.1038/ncb1212; PMID: 15665856
- Enders GH. Expanded roles for Chk1 in genome maintenance. J Biol Chem 2008; 283:17749 - 52; http://dx.doi.org/10.1074/jbc.R800021200; PMID: 18424430
- Ha K, Lee GE, Palii SS, Brown KD, Takeda Y, Liu K, et al. Rapid and transient recruitment of DNMT1 to DNA double-strand breaks is mediated by its interaction with multiple components of the DNA damage response machinery. Hum Mol Genet 2011; 20:126 - 40; http://dx.doi.org/10.1093/hmg/ddq451; PMID: 20940144
- Palii SS, Van Emburgh BO, Sankpal UT, Brown KD, Robertson KD. DNA methylation inhibitor 5-Aza-2′-deoxycytidine induces reversible genome-wide DNA damage that is distinctly influenced by DNA methyltransferases 1 and 3B. Mol Cell Biol 2008; 28:752 - 71; http://dx.doi.org/10.1128/MCB.01799-07; PMID: 17991895
- Shimada M, Niida H, Zineldeen DH, Tagami H, Tanaka M, Saito H, et al. Chk1 is a histone H3 threonine 11 kinase that regulates DNA damage-induced transcriptional repression. Cell 2008; 132:221 - 32; http://dx.doi.org/10.1016/j.cell.2007.12.013; PMID: 18243098
- Bucher N, Britten CD. G2 checkpoint abrogation and checkpoint kinase-1 targeting in the treatment of cancer. Br J Cancer 2008; 98:523 - 8; http://dx.doi.org/10.1038/sj.bjc.6604208; PMID: 18231106
- Dai Y, Grant S. Targeting Chk1 in the replicative stress response. Cell Cycle 2010; 9:1025; http://dx.doi.org/10.4161/cc.9.6.11155; PMID: 20237419
- Ma CX, Janetka JW, Piwnica-Worms H. Death by releasing the breaks: CHK1 inhibitors as cancer therapeutics. Trends Mol Med 2011; 17:88 - 96; http://dx.doi.org/10.1016/j.molmed.2010.10.009; PMID: 21087899
- Arlander SJ, Greene BT, Innes CL, Paules RS. DNA protein kinase-dependent G2 checkpoint revealed following knockdown of ataxia-telangiectasia mutated in human mammary epithelial cells. Cancer Res 2008; 68:89 - 97; http://dx.doi.org/10.1158/0008-5472.CAN-07-0675; PMID: 18172300
- Leung-Pineda V, Ryan CE, Piwnica-Worms H. Phosphorylation of Chk1 by ATR is antagonized by a Chk1-regulated protein phosphatase 2A circuit. Mol Cell Biol 2006; 26:7529 - 38; http://dx.doi.org/10.1128/MCB.00447-06; PMID: 17015476
- Freeman AK, Monteiro AN. Phosphatases in the cellular response to DNA damage. Cell Commun Signal 2010; 8:27; http://dx.doi.org/10.1186/1478-811X-8-27; PMID: 20860841
- Shouse GP, Cai X, Liu X. Serine 15 phosphorylation of p53 directs its interaction with B56γ and the tumor suppressor activity of B56γ-specific protein phosphatase 2A. Mol Cell Biol 2008; 28:448 - 56; http://dx.doi.org/10.1128/MCB.00983-07; PMID: 17967874
- Yan Y, Cao PT, Greer PM, Nagengast ES, Kolb RH, Mumby MC, et al. Protein phosphatase 2A has an essential role in the activation of γ-irradiation-induced G2/M checkpoint response. Oncogene 2010; 29:4317 - 29; http://dx.doi.org/10.1038/onc.2010.187; PMID: 20498628
- Chen J, Martin BL, Brautigan DL. Regulation of protein serine-threonine phosphatase type-2A by tyrosine phosphorylation. Science 1992; 257:1261 - 4; http://dx.doi.org/10.1126/science.1325671; PMID: 1325671
- Chen J, Parsons S, Brautigan DL. Tyrosine phosphorylation of protein phosphatase 2A in response to growth stimulation and v-src transformation of fibroblasts. J Biol Chem 1994; 269:7957 - 62; PMID: 7510677
- Ray S, Anderson ME, Tegtmeyer P. Differential interaction of temperature-sensitive simian virus 40 T antigens with tumor suppressors pRb and p53. J Virol 1996; 70:7224 - 7; PMID: 8794371
- Meyer KM, Hess SM, Tlsty TD, Leadon SA. Human mammary epithelial cells exhibit a differential p53-mediated response following exposure to ionizing radiation or UV light. Oncogene 1999; 18:5795 - 805; http://dx.doi.org/10.1038/sj.onc.1202977; PMID: 10523860
- Troester MA, Hoadley KA, Sørlie T, Herbert BS, Børresen-Dale AL, Lønning PE, et al. Cell-type-specific responses to chemotherapeutics in breast cancer. Cancer Res 2004; 64:4218 - 26; http://dx.doi.org/10.1158/0008-5472.CAN-04-0107; PMID: 15205334
- Zhang J, Pickering CR, Holst CR, Gauthier ML, Tlsty TD. p16INK4a modulates p53 in primary human mammary epithelial cells. Cancer Res 2006; 66:10325 - 31; http://dx.doi.org/10.1158/0008-5472.CAN-06-1594; PMID: 17079452
- Jiang H, Reinhardt HC, Bartkova J, Tommiska J, Blomqvist C, Nevanlinna H, et al. The combined status of ATM and p53 link tumor development with therapeutic response. Genes Dev 2009; 23:1895 - 909; http://dx.doi.org/10.1101/gad.1815309; PMID: 19608766
- Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 2010; 21:739 - 52; http://dx.doi.org/10.1091/mbc.E09-07-0618; PMID: 20053681
- Kim MA, Kim HJ, Brown AL, Lee MY, Bae YS, Park JI, et al. Identification of novel substrates for human checkpoint kinase Chk1 and Chk2 through genome-wide screening using a consensus Chk phosphorylation motif. Exp Mol Med 2007; 39:205 - 12; http://dx.doi.org/10.1038/emm.2007.23; PMID: 17464182
- Lee HJ, Hwang HI, Jang YJ. Mitotic DNA damage response: Polo-like kinase-1 is dephosphorylated through ATM-Chk1 pathway. Cell Cycle 2010; 9:2389 - 98; http://dx.doi.org/10.4161/cc.9.12.11904; PMID: 20581453
- Bertoni F, Codegoni AM, Furlan D, Tibiletti MG, Capella C, Broggini M. CHK1 frameshift mutations in genetically unstable colorectal and endometrial cancers. Genes Chromosomes Cancer 1999; 26:176 - 80; http://dx.doi.org/10.1002/(SICI)1098-2264(199910)26:2<176::AID-GCC11>3.0.CO;2-3; PMID: 10469457
- Menoyo A, Alazzouzi H, Espín E, Armengol M, Yamamoto H, Schwartz S Jr.. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 2001; 61:7727 - 30; PMID: 11691784
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G2/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Lam MH, Liu Q, Elledge SJ, Rosen JM. Chk1 is haploinsufficient for multiple functions critical to tumor suppression. Cancer Cell 2004; 6:45 - 59; http://dx.doi.org/10.1016/j.ccr.2004.06.015; PMID: 15261141
- Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, Vassilopoulos A, et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene 2010; 29:4007 - 17; http://dx.doi.org/10.1038/onc.2010.163; PMID: 20473325
- Verlinden L, Vanden Bempt I, Eelen G, Drijkoningen M, Verlinden I, Marchal K, et al. The E2F-regulated gene Chk1 is highly expressed in triple-negative estrogen receptor-/progesterone receptor-/HER-2- breast carcinomas. Cancer Res 2007; 67:6574 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-06-3545; PMID: 17638866
- Tort F, Hernández S, Beà S, Camacho E, Fernández V, Esteller M, et al. Checkpoint kinase 1 (CHK1) protein and mRNA expression is downregulated in aggressive variants of human lymphoid neoplasms. Leukemia 2005; 19:112 - 7; PMID: 15526025
- Chen P, Gatei M, O’Connell MJ, Khanna KK, Bugg SJ, Hogg A, et al. Chk1 complements the G2/M checkpoint defect and radiosensitivity of ataxia-telangiectasia cells. Oncogene 1999; 18:249 - 56; http://dx.doi.org/10.1038/sj.onc.1202257; PMID: 9926940
- Carrassa L, Damia G. Unleashing Chk1 in cancer therapy. Cell Cycle 2011; 10:2121 - 8; http://dx.doi.org/10.4161/cc.10.13.16398; PMID: 21610326
- Ashwell S. Checkpoint kinase and Wee1 inhibitors as anticancer therapeutics. Elsevier Ltd., 2012:211-34.
- Davies KD, Humphries MJ, Sullivan FX, von Carlowitz I, Le Huerou Y, Mohr PJ, et al. Single-agent inhibition of Chk1 is antiproliferative in human cancer cell lines in vitro and inhibits tumor xenograft growth in vivo. Oncol Res 2011; 19:349 - 63; http://dx.doi.org/10.3727/096504011X13079697132961; PMID: 21936404
- Dent P, Tang Y, Yacoub A, Dai Y, Fisher PB, Grant S. CHK1 inhibitors in combination chemotherapy: thinking beyond the cell cycle. Mol Interv 2011; 11:133 - 40; http://dx.doi.org/10.1124/mi.11.2.11; PMID: 21540473
- Garrett MD, Collins I. Anticancer therapy with checkpoint inhibitors: what, where and when?. Trends Pharmacol Sci 2011; 32:308 - 16; http://dx.doi.org/10.1016/j.tips.2011.02.014; PMID: 21458083
- Ren Q, Liu R, Dicker A, Wang Y. CHK1 affects cell sensitivity to microtubule-targeted drugs. J Cell Physiol 2005; 203:273 - 6; http://dx.doi.org/10.1002/jcp.20222; PMID: 15389625
- Greenow KR, Clarke AR, Jones RH. Chk1 deficiency in the mouse small intestine results in p53-independent crypt death and subsequent intestinal compensation. Oncogene 2009; 28:1443 - 53; http://dx.doi.org/10.1038/onc.2008.482; PMID: 19169280
- Dai Y, Chen S, Shah R, Pei XY, Wang L, Almenara JA, et al. Disruption of Src function potentiates Chk1-inhibitor-induced apoptosis in human multiple myeloma cells in vitro and in vivo. Blood 2011; 117:1947 - 57; http://dx.doi.org/10.1182/blood-2010-06-291146; PMID: 21148814
- Tao Y, Leteur C, Yang C, Zhang P, Castedo M, Pierré A, et al. Radiosensitization by Chir-124, a selective CHK1 inhibitor: effects of p53 and cell cycle checkpoints. Cell Cycle 2009; 8:1196 - 205; http://dx.doi.org/10.4161/cc.8.8.8203; PMID: 19305158
- Davies KD, Cable PL, Garrus JE, Sullivan FX, von Carlowitz I, Huerou YL, et al. Chk1 inhibition and Wee1 inhibition combine synergistically to impede cellular proliferation. Cancer Biol Ther 2011; 12:788 - 96; http://dx.doi.org/10.4161/cbt.12.9.17673; PMID: 21892012
- Lu HR, Wang X, Wang Y. A stronger DNA damage-induced G2 checkpoint due to over-activated CHK1 in the absence of PARP-1. Cell Cycle 2006; 5:2364 - 70; http://dx.doi.org/10.4161/cc.5.20.3355; PMID: 17102615
- Jurvansuu J, Fragkos M, Ingemarsdotter C, Beard P. Chk1 instability is coupled to mitotic cell death of p53-deficient cells in response to virus-induced DNA damage signaling. J Mol Biol 2007; 372:397 - 406; http://dx.doi.org/10.1016/j.jmb.2007.06.077; PMID: 17663993
- Limbo O, Porter-Goff ME, Rhind N, Russell P. Mre11 nuclease activity and Ctp1 regulate Chk1 activation by Rad3ATR and Tel1ATM checkpoint kinases at double-strand breaks. Mol Cell Biol 2011; 31:573 - 83; http://dx.doi.org/10.1128/MCB.00994-10; PMID: 21098122
- Morgan MA, Parsels LA, Zhao L, Parsels JD, Davis MA, Hassan MC, et al. Mechanism of radiosensitization by the Chk1/2 inhibitor AZD7762 involves abrogation of the G2 checkpoint and inhibition of homologous recombinational DNA repair. Cancer Res 2010; 70:4972 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-09-3573; PMID: 20501833
- Parsels LA, Qian Y, Tanska DM, Gross M, Zhao L, Hassan MC, et al. Assessment of Chk1 phosphorylation as a pharmacodynamic biomarker of Chk1 inhibition. Clin Cancer Res 2011; 17:3706 - 15; http://dx.doi.org/10.1158/1078-0432.CCR-10-3082; PMID: 21482692