Abstract
TRPS1, the gene mutated in human "Tricho-Rhino-Phalangeal syndrome," encodes a multi zinc-finger nuclear regulator of chondrocyte proliferation and differentiation. Here, we have identified a new function of Trps1 in controlling mitotic progression in chondrocytes. Loss of Trps1 in mice leads to an increased proportion of cells arrested in mitosis and, subsequently, to chromosome segregation defects. Searching for the molecular basis of the defect, we found that Trps1 acts as regulator of histone deacetylation. Trps1 interacts with two histone deacetylases, Hdac1 and Hdac4, thereby increasing their activity. Loss of Trps1 results in histone H3 hyperacetylation, which is maintained during mitosis. Consequently, chromatin condensation and binding of HP1 is impaired, and Trps1-deficient chondrocytes accumulate in prometaphase. Overexpression of Hdac4 rescues the mitotic defect of Trps1-deficient chondrocytes, identifying Trps1 as an important regulator of chromatin deacetylation during mitosis in chondrocytes. Our data provide the first evidence that the control of mitosis can be linked to the regulation of chondrocyte differentiation by epigenetic consequences of altered Hdac activity.
Introduction
During endochondral ossification the skeletal elements are preformed as cartilage templates. Chondrocytes inside the cartilage anlagen initially proliferate and, starting from the center, differentiate into hypertrophic chondrocytes, which are subsequently replaced by bone and bone marrow. As bone growth is dependent on chondrocyte proliferation, the proliferation rate, the size of the proliferating region, and the rate of hypertrophic differentiation, which is linked to cell cycle exit, are tightly controlled by a complex network of transcription factors . Citation1During recent years, cell cycle progression has been intensively studied in chondrocytes, and many regulators have been identified, most of which are linked to S-phase entry.Citation2 In contrast, less is known about the genes controlling the transition into G2-phase or progression through mitosis. Both, DNA replication and chromatin segregation are dependent on DNA–histone interactions, which are, among others, regulated by histone-modifying enzymes, histone acetyltransferases (Hats), and histone deacetylases (Hdacs).Citation3 These enzymes modulate chromatin condensation by reversible acetylation of lysine residues on histone tails. As cells enter mitosis, the chromatin needs to be condensed into structures of higher order. Several recent studies using either synthetic inhibitors or genetic inactivation of Hdacs demonstrated that DNA condensation requires histone deacetylases.Citation4,Citation5 Consequently, cells with impaired Hdac function activate the mitotic checkpoint and stay arrested in prometaphase.Citation6,Citation7 Eventually, the cells override the mitotic checkpoint and resume mitosis with misaligned chromosomes, generating aneuploid cells.Citation8,Citation9
Tricho-Rhino-Phalangeal syndrome (TRPS) is an autosomal dominant human disorder characterized by dysplastic hair, short stature, and malformation of the skeleton.Citation10 TRPS1, the gene mutated in this disease, encodes a multi zinc-finger transcription factor with nine predicted zinc-finger (ZF) motifs.Citation11 While no function has been assigned to ZF1–3, yet, ZF4–6 and ZF8–9, which belong to the Ikaros class, have been shown to mediate interaction with other proteins.Citation12-Citation15 The seventh ZF belongs to the GATA family and is required for DNA binding.Citation11 During mouse skeletal development, Trps1 is expressed in proliferating chondrocytes showing strong expression levels at the border of proliferating and hypertrophic chondrocytes.Citation16 Homozygous Trps1-/- mutants are characterized by a reduced skeletal sizeCitation17 and distinct defects in chondrocyte differentiation, which are, at least partially, based on deregulation of Runx2.Citation18,Citation19 In addition Trps1-/- mutants show a reduced proliferation rate, which has been linked to mechanisms regulating S-phase entry.Citation14,Citation20
In this study we show that deletion of Trps1 leads to an enrichment of cells in mitosis and chromosome segregation defects. We demonstrate that Trps1 interacts with at least two Hdac proteins, Hdac1 and Hdac4. Loss of Trps1 results in hyperacetylation of histone H3 in vitro and in vivo, identifying Trps1 as a positive regulator of Hdac function. Analysis of mitotic cells revealed that the hyperacetylation of H3 is maintained during mitosis, leading to decreased HP1 binding. Finally we show that the accumulation of mitotic cells can be rescued by Hdac4 overexpression.
Results
Trps1 acts on two distinct steps of cell cycle progression
Trps1-deficient (Trps1-/-) mice display a reduced size of the skeletal elements, which is, at least in part, due to a decreased chondrocyte proliferation rate.Citation14,Citation19,Citation20 To characterize cell cycle progression, we analyzed chondrogenic precursor cells isolated from E12.5 limb mesenchyme (primary chondrocytes) by flow cytometry after labeling cells in S-phase with bromodesoxyuridine (BrdU) and DNA with 7-aminoactinomycin (7-AAD) (). In agreement with previous reports,Citation14,Citation19,Citation20 we found that the proportion of cells in S-phase was decreased from 20.5% in wild-type cultures to 14.4% in cultures of Trps1-/- mutants (). Surprisingly, although a lower percentage of Trps1-/- cells replicated their DNA, the proportion of cells in G2/M-phase was significantly increased to 20.4%, while in wild-type cultures, 16.7% of chondrocytes were in G2/M-phase. To support a role of Trps1 in regulating cell cycle progression we knocked-down Trps1 in HEK293 EBNA cells using siRNA, which resulted in a reduction of Trps1 protein to 36% (). In agreement with our data obtained in primary chondrocytes, siRNA-mediated knockdown of Trps1 in HEK293 EBNA reduced the numbers of cells in S-phase and increased the number of cells in G2/M-phase in HEK293 EBNA cells ().
Figure 1. Trps1 regulates two independent steps of the cell cycle. (A–C) Representative flow cytometry after labeling primary chondrocytes from wild-type (A) and Trps1-/- (B) mice with BrdU and 7-AAD identifies significantly decreased numbers of cells in S-phase, while the proportion of cells in G2/M-phase is increased in Trps1-/- mutants. (C) Statistic evaluation, n =6; p*S-phase = 0.001, p*G2/M-phase = 0.0021). Western blot (D and E) and flow cytometry (F) of HEK293 EBNA cells transfected with Trps1 specific or unspecific (mock) siRNA. The level of Trps1 protein was quantified in relation to histone H3 protein (n =6; p* = 0.000066 in (E). (F) Similar to Trps1-/- mutants, knockdown of Trps1 in HEK293 EBNA cells decreases the number of cells in S-phase and increases the number of cells in G2/M-phase (n =3; p* = 0.007 in (F). (G and H) Statistical evaluation derived from flow cytometric analyses of Aphidicolin- and Nocodazole-treated Trps1-/- (red) and wild-type (blue) cells compared with control cells. (G) During Aphidicolin treatment, Trps1-/- and wild-type chondrocytes are similarly arrested prior to S-phase. One h after release from Aphidicolin 5.8% wild-type chondrocytes have entered S-phase, while few Trps1-/- cells progress into S-phase (n =3; p* < 0.05). (H) Trps1-/-−/− cells are less sensitive to Nocodazole induced mitotic block (n =6; p* < 0.005).
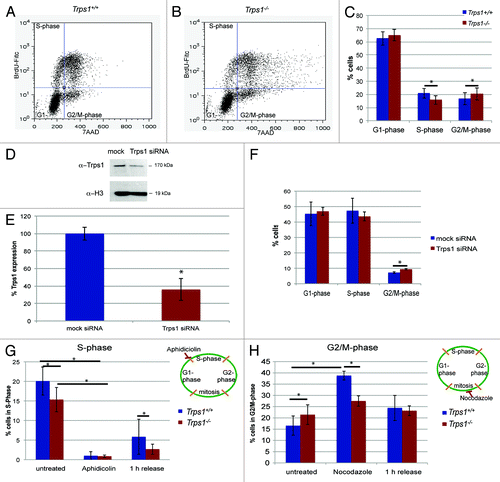
We next asked if Trps1 regulates S- and G2/M-phase progression by independent mechanisms. Treatment with Aphidicolin, which arrests cells prior to S-phase,Citation21 reduced the percentage of cells in S-phase to 0.99% and 0.88% in cultures of wild-type and Trps1-/- mice, respectively. Upon release of the Aphidicolin block, chondrocytes of both genotypes resumed proliferation. Interestingly, whereas 5.8% of wild-type cells can be found in S-phase 1 h after the release from Aphidicolin, only 2.7% of Trps1-/- chondrocytes re-enter S-phase, revealing a slower progression from G1 into S-phase in Trps1-/- mutants (). In contrast to its effect on S-phase, Aphidicolin treatment did not affect the increased proportion of Trps1-/- cells in G2/M-phase (Fig. S1A).
To investigate the role of Trps1 in regulating mitotic progression, primary chondrocytes of Trps1-/- and wild-type mice were treated with Nocodazole, a microtubuli-destabilizing agent, and analyzed by flow cytometry.Citation22 As expected, Nocodazole treatment increased the proportion of wild-type chondrocytes in G2/M-phase (from 16.7 to 36.2%). In contrast, in cultures of Trps1-/- chondrocytes, in which cells are already enriched in G2/M-phase without treatment, only a moderate increase from 20.4 to 25.5% was observed (). After releasing the Nocodazole block, wild-type cells resumed proliferation and progressed from G2/M to G1 and S-phase, whereas the Trps1-/- cells were less sensitive to the release and remained in G2/M-phase (; Fig. S1B). Similar effects, an increased proportion of cells in G2/M-phase and a reduced sensitivity toward Nocodazole-induced activation of the mitotic spindle checkpoint, can be observed for mutations in components of the mitotic checkpoint or regulators of DNA condensation.Citation9,Citation23 Trps1 might thus regulate mitotic progression of chondrocytes independent of its role in controlling S-phase entry by acting on either of these mechanisms.
Trps1 deficiency impairs progression into metaphase during mitosis
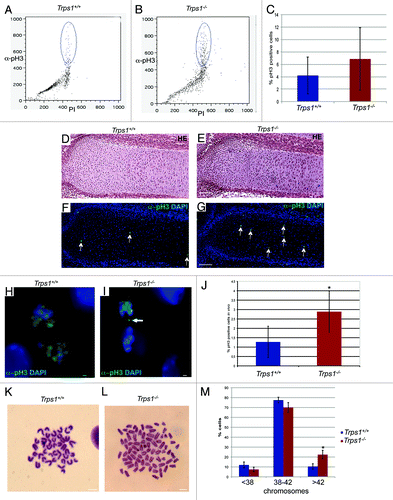
As loss of Trps1 leads to an accumulation of cells in G2/M-phase, we next investigated the phosphorylation of histone H3 at serine 10 (pH3), which serves as specific marker for mitotic cells.Citation24 Flow cytometry of cells labeled with an α-pH3 antibody revealed that 3.5% wild-type chondrocytes in culture were pH3-positive (), while in cultures of Trps1-/- mutants, the proportion of mitotic cells was increased to 7.3% (). To examine if loss of Trps1 leads to a similar increase in mitotic cells in vivo, we quantified pH3-positive cells in the region of proliferating chondrocytes of E16.5 mouse ulnae (). Comparable to our in vitro data, the number of pH3-positive cells was increased to 2.9% in Trps1-/- mutants, while 1.3% pH3-positive cells can be found in wild-type littermates (). Trps1 thus activates mitotic progression in vitro and in vivo. Interestingly, at high magnification, irregular chromatin distribution could be observed in a subset of pH3-positive, Trps1-/- chondrocytes, likely reflecting disturbed chromosome segregation (, arrow). We next investigated chromosome spreads of E12.5 primary chondrocyte cultures from Trps1-/- () and wild-type mice (), and found the proportion of aneuploid cells with high chromosome numbers to be increased from 10.5% in wild-type mice to 22% in Trps1-/- mutants ().
Figure 2.Trps1-/- chondrocytes display aberrant mitosis. (A–C) Representative flow cytometry of primary chondrocyte cultures from wild-type (A) and Trps1-/- mice (B) stained with α-pH3 antibody and Propidium Iodide (PI) revealed increased numbers of pH3-positive cells in Trps1-/- mutants. (C) Statistic evaluation of pH3 measurement (n =5; p* = 0.018). (D and E) Hematoxylin-Eosin (HE) staining of paraffin sections of E16.5 wild-type and Trps1-/- ulnae showing the region of proliferating chondrocytes. Parallel sections (F and G) were used for immunofluorescent detection of pH3-positive, mitotic cells (green) in wild-type (F and H) and Trps1-/- mice (G–I). (J) The statistic evaluation of the pH3-positive cells in vivo revealed increased numbers of mitotic cells (green, arrows) in Trps1-/- mutants. (n =5; p* = 0.025). (H and I) DAPI counterstaining of this sections showed asymmetric chromatin segregation in mitotic chondrocytes of Trps1-/- mutants (I, arrow). (K and L) Giemsa stained chromosome spreads of wild-type (K) and Trps1-/- mice (L) revealed increased numbers of aneuploid cells in Trps1-/- mutants. (M) Statistic evaluation of chromosome numbers in mitotic cells (n =4, 50 mitoses each; p* = 0.0028). Scale bar: 100 µm, (F and G), 10 µm, (I and J).
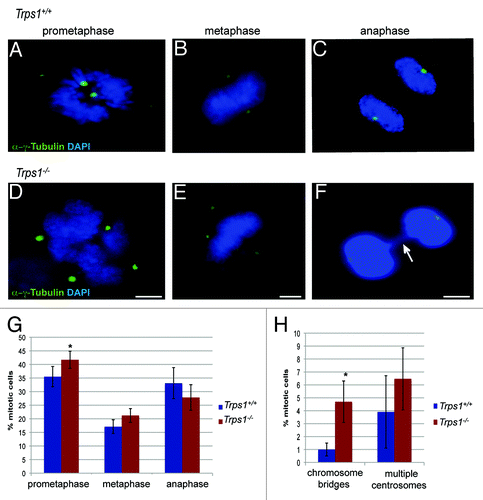
To further characterize the mitotic defect, we determined the percentage of primary chondrocytes in distinct phases of mitosis labeled with DAPI to stain DNA and an α-y-tubulin antibody to detect centrosomes (). In cultures of wild-type chondrocytes, 35.5% mitotic cells could be found in prometaphase. In contrast, in Trps1-/- cultures the proportion of chondrocytes in prometaphase was significantly increased to 41.7%, whereas the number of cells in the other mitotic phases was not significantly altered compared with wild-type cells (). Close inspection of DAPI stained cells revealed irregular chromatin condensation in Trps1-/- cells (see ). We analyzed chromosome segregation in anaphase and found chromosome bridges in 4.7% Trps1-/- mutant cells, whereas we detected chromosome bridges in only 0.5% of wild-type chondrocytes (). Furthermore, our analysis revealed that 6.6% of mitotic Trps1-/- cells displayed multiple centrosomes compared with 2.8% cells in wild-type cultures ().
Figure 3. Loss of Trps1 arrests primary chondrocytes in prometaphase. (A–F) Immunofluorescent staining of primary chondrocytes from wild-type (A–C) and Trps1-/- (D–F) mice with an α-γ-tubulin antibody(green) counterstained with DAPI (blue). Statistic quantification reveals an enrichment of cells in prometaphase (n =4; p* = 0.036) (G) and increased numbers of cells with chromosome bridges or multiple centrosomes (H), arrow in (F) indicating chromosome segregation defects in Trps1-/- mutants (n =4; p* = 0.029). Scale bar: 10 µm.
Trps1 acts as a positive regulator of Hdac activity
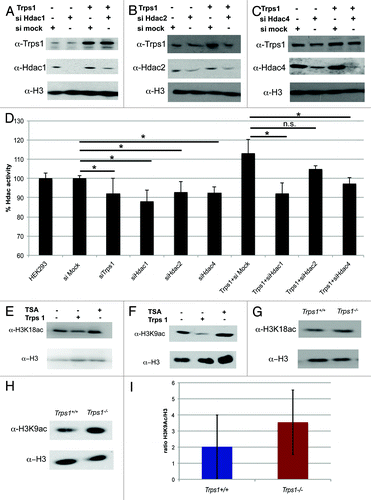
So far, our experiments showed that loss of Trps1 leads to a decreased proportion of cells in S-phase combined with increased numbers of cells in mitosis. A similar phenotype has been observed after inhibition of histone deacetylases (Hdacs) in established cell lines.Citation25,Citation26 To test if inhibition of Hdac activity induces a comparable mitotic arrest in primary chondrocytes, we treated wild-type and Trps1-/- chondrocytes with the Hdac inhibitor Trichostatin A (TSA). In accordance with earlier studies,Citation27,Citation26 TSA treatment reduced the proportion of primary chondrocytes in S-phase from 23.7 to 11.5%, while the percentage of cells in G2/M-phase was increased from 12.2 to 21.1% (). Hdac inhibition thus induced a similar cell cycle defect in wild-type chondrocytes as observed in Trps1-/- mutants. We next asked if Trps1 might interact with Hdac proteins. Hdac4 expression overlaps with that of Trps1 in proliferating and prehypertrophic chondrocytes, interacts, and represses Runx2 similar to Trps1 and, therefore, seemed to be a good candidate for such an interaction.Citation29-Citation31 First, we confirmed the endogenous expression of both proteins in the chondrogenic cell line, ATDC5 (, input). After immunoprecipitation using either an α-Hdac4 or α-Trps1 antibody, western blot analysis demonstrated an interaction of Trps1 with Hdac4 in a protein complex (). Next, we asked if other Hdacs might similarly interact with Trps1. Hdac1 and Hdac2 are ubiquitously expressed and share redundant functions in most tissues.Citation32 Co-expression of Flag-tagged Hdac1 or Flag-tagged Hdac2 protein with Trps1 in HEK293 EBNA cells followed by immunoprecipitation with either an α-Flag or an α-Trps1 antibody revealed an interaction of Trps1 with Hdac1 () but not with Hdac2 (), demonstrating the specificity of the interaction. As Trps1 interacts with Hdac1, but not with the highly homologous Hdac2 protein, we ask which protein domain mediates the interaction with Trps1. Surprisingly, immunoprecipitation of GFP-tagged deletion constructs of Hdac1 and Hdac2 and Flag-tagged Trps1 revealed that the N-terminal region, containing the conserved Hdac domain but not the C-terminal domain of Hdac1, is required for the interaction with Trps1 (Fig. S2C, E, and F). In contrast the N-terminal region of Hdac2 did not interact with Trps1 (Fig. S2D).
Figure 4. Trps1 interacts with Hdac1 and Hdac4 and increases their activity. (A) Immunoprecipitation with an α-Hdac4 or α-Trps1 antibody followed by western blot analysis demonstrates an interaction of the endogenous proteins in ATDC5 cells. (B and C) Overexpression of Trps1 and Flag-tagged Hdac1 (B) or Hdac2 (C) in HEK293 EBNA cells, followed by immunoprecipitation with an α-Flag οr α-Trps1 antibody identifies an interaction of Trps1 with Hdac1, but not Hdac2. (In, input; Sn, supernatant; IP, immunopreciptiation). (D and E) Trps1 and Hdac4 were overexpressed in HEK293 EBNA cells and Hdac activity was analyzed using a fluorometric assay. (D) Western blot analysis confirms overexpression of both proteins. (E) Overexpression of Trps1 and Hdac4 increases Hdac activity by 9% and 15%, respectively, compared with an empty vector. Co-transfection of Hdac4 and Trps1 augments Hdac activity by 20%. TSA treatment reduced Hdac activity to 88%, representing loss of all class 1 and class 2 Hdac activity in the protein extracts (blue brackets); (n =3; p* < 0.05). (F) Analysis of Hdac activity in protein extracts of E16.5 limbs revealed reduced Hdac activity in Trps1-/- mutants compared with wild-type mice in vivo (n =4; p* < 0.03). (G) Primary chondrocytes were treated with TSA to block Hdac activity, and cell cycle distribution was measured by flow cytometry. TSA treatment decreases the numbers of cells in S- and increases the proportion of cells in G2/M-phase (n =3; p*S-phase < 0.004, p*G2/M-phase = 0.035).
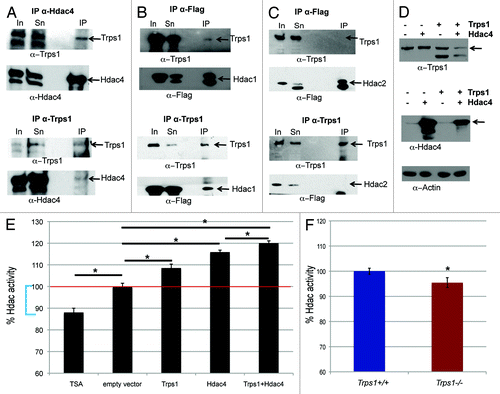
To elucidate if the Trps1-Hdac interaction regulates the enzymatic Hdac activity, we used a fluorometric activity assay to measure global histone deacetylase activity. Treatment of HEK293 EBNA cells with TSA, which inhibits all class I and class II Hdacs, reduced the enzymatic activity to 88%. As expected, overexpression of Hdac4 enhanced the total cellular Hdac activity by 15% compared with control cells (). Similarly Hdac activity was also increased by 9% after Trps1 overexpression. Moreover, co-expression of Trps1 and Hdac4 further increased the Hdac activity compared with either single transfection (by 20%, ), indicating that Trps1 acts as an activator of Hdac function in vitro.
To test if Trps1 has a similar activating function in vivo, we analyzed Hdac activity in protein extracts of E16.5 Trps1-/- and wild-type limbs. Compared with wild-type extracts, Trps1-/- mutants displayed a significant reduction of total Hdac activity to 95% (), reflecting about 45% of total class 1 and class II activity. Trps1 seems thus to act as a positive regulator of Hdac activity in vivo.
As Trps1 can interact with two different Hdac proteins, we investigated whether the increased deacetylase activity could be attributed to a specific Hdac homolog. To decrease endogenous Hdac activity, we transfected HEK293 EBNA cells with siRNA against Hdac1, Hdac2, Hdac4, and Trps1. The knockdown efficiency of the respective siRNAs was confirmed by western blot analysis ( and ). As expected, we detected a decreased Hdac activity of 8–12% of the empty vector control after transfection of either individual Hdac siRNAs (). Similarly, knockdown of Trps1 decreased the total Hdac activity (), substantiating our in vivo data (). In parallel, we co-expressed each siRNA together with a Trps1-overexpression construct (). While overexpression of Trps1 increased Hdac activity, concurrent knockdown of either Hdac1 or Hdac4 decreased the Trps1-dependent elevated Hdac activity (). In contrast, knockdown of Hdac2, which does not interact with Trps1, had a less severe effect on the Trps1-mediated activation (). These data strongly indicate that the interaction of Trps1 with Hdac1 and Hdac4 increases the total deacetylase activity.
Figure 5. Trps1 activates histone deacetylases in vitro and in vivo. (A–D) Trps1 overexpression constructs were transfected into HEK293 EBNA cells together with siRNA against Hdac1 (A), Hdac2 (B) and Hdac4 (C) or an unspecific siRNA (mock) as indicated. The expression levels were verified by western blotting. (D) Knockdown of Trps1, Hdac1, 2 and 4 decreases Hdac activity, while overexpression of Trps1 increases Hdac activity in a fluorometric assay. Cotransfection of Trps1 with siRNA against Hdac4 or Hdac1 reduced the increased Hdac activity, while siRNA against Hdac2 had only subtle effects (n =3; p* < 0.05, n.s.: not significant). (E and F) Overexpression of Trps1 in HEK293 EBNA cells increased the acetylation of H3K9 (F) and H3K18 (E) compared with untransfected control cells. (G–I) Western blot analysis of E16.5 limb extracts from Trps1-/- and wild-type mice showed decreased levels of H3K18 (G) and H3K9 (H and I) acetylation in Trps1-/- mutants (n =5).
Histone H3 is hyperacetylated during mitosis in Trps1-/- chondrocytes
As outlined above, histones have to be hypoacetylated during prometaphase to facilitate chromatin condensation.Citation5 We thus tested if histone H3 could be a physiological target of the Trps1-Hdac complex. First, we analyzed the acetylation status of histone H3 at lysine 9 (H3K9ac) and lysine 18 (H3K18ac) after overexpression of Trps1 in HEK293 EBNA cells by western blotting. As a control, we blocked Hdac activity with TSA, which, as expected, resulted in enhanced H3 acetylation (). In contrast, overexpression of Trps1 resulted in decreased levels of H3K9 and H3K18 acetylation (). To determine if altered H3 acetylation contributes to the Trps1-/- phenotype, we analyzed the acetylation level of H3K9 and H3K18 in limb extracts of wild-type and Trps1-/- mice (). Strikingly, Trps1-/- mutants display reproducibly higher acetylation levels of H3 at both lysine residues (), strongly supporting the hypothesis that Trps1 acts as a regulator of Hdac activity in vivo.
We next asked if the increased H3 acetylation levels were maintained during mitosis and investigated H3K9 acetylation at distinct mitotic stages in primary chondrocyte cultures after immunofluorescent labeling with an α-H3K9ac antibody. As described for other cell types,Citation6 H3K9 acetylation decreases during prophase and prometaphase, leading to reduced acetylation levels in metaphases of wild-type chondrocytes (). In contrast, in Trps1-/- metaphases, H3K9 acetylation appeared to be increased compared with that in wild-type cells (). To quantify the differences in H3K9 acetylation in mitotic cells, we measured the mean fluorescent intensity of acetylated H3K9 after immunofluorescent labeling in relation to DAPI-stained DNA in 100 mitotic cells per culture of 5 embryos per genotype. In mitotic wild-type chondrocytes, the ratio of acetylated H3K9 to DAPI was 3.55, while it was significantly increased to 4.53 in Trps1-/- cells ().
Figure 6. Acetylation of H3K9 is increased in mitotic, Trps1-/- cells. (A–F and M) Immunofluorescent labeling of primary chondrocyte from wild-type (A–C) and Trps1-/- (D–F) mice using α-H3K9ac and α-y-Tubulin antibodies and DAPI counterstaining revealed increased H3K9 acetylation in metaphases of Trps1-/- mutants (D–F). (M) Quantification of the mean fluorescence intensity of H3K9 acetylation in relation to DAPI (n =5; 100 mitoses per animal; p* = 0.000003). (G–L) Immunofluorescent staining of wild-type (G–I) and Trps1-/- chondrocytes (J–L) with an α-H3K9acS10p antibody revealed an increased level of acetylated pH3 in mitotic, Trps1-/- cells. Scale bar: 10 µm. (M–R) Immunofluorescent labeling with an α-HP1β antibody as marker for pericentric heterochromatin showed reduced staining in Trps1-/- cells (P–R) compared with wild-type chondrocytes (M–O). (A) Quantification of the mean fluorescence intensity of H3K9 acetylation in mitotic cells in relation to DAPI (n =5; 100 mitoses per animal; p* = 0.000003). (B) Quantification of the mean fluorescence intensity of Hβ1 staining in mitotic cells in relation to DAPI (n =4; 30 mitoses per animal; p* = 0.00004)
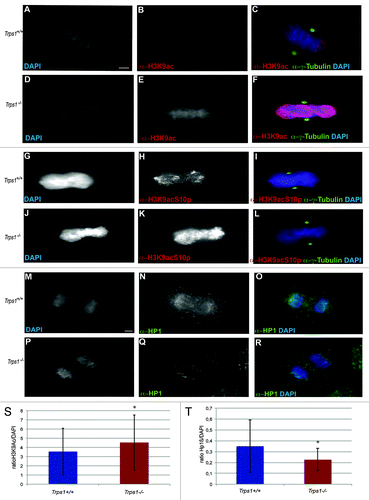
Since deacetylation of H3K9 is normally linked to the phosphorylation of H3, we tested whether pH3-positive cells also maintained H3K9 acetylation.Citation4 Using an antibody that specifically detects only double-modified (acetylated and phosphorylated) histone H3 (α-H3K9AcS10p),Citation33 we found only low levels of double-modified histone H3 in wild-type chondrocyte cultures (), whereas increased levels of acetylated pH3 could be detected in mitotic chondrocytes of Trps1-/- mutants ().
Another step that is linked to chromatin compaction is the binding of the heterochromatin protein 1 (HP1) to pericentric, heterochromatic regions. Quantification of chromatin-associated HP1β in relation to DAP1 revealed significantly reduced levels in anaphases of Trps1-/- chondrocytes () compared with wild-type cells (). Taken together, our in situ investigations of mitotic primary chondrocytes strongly supported a role of Trps1 in regulating chromatin deacetylation and condensation during mitosis.
We next aimed to quantify histone acetylation in mitotic cells by an independent method. To enrich mitotic cells, we treated Trps1-/- and wild-type primary chondrocytes with Nocodazole and examined the H3K9 acetylation level. We found increased levels of H3K9 acetylation in Trps1-/- chondrocytes in treated and untreated cultures (Fig. S3A). However, as the enrichment of mitotic chondrocyte by Nocodazole treatment is not very effective (Fig. S1B), the higher acetylation level might just reflect acetylation differences in other cell cycle stages.
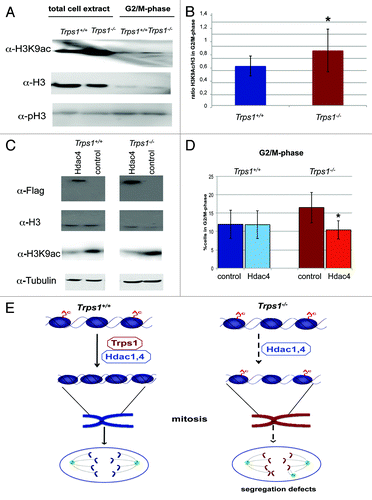
To further enrich the population of chondrocytes in G2/M-phase, we used Vybrant DyeCycleGreen, which incorporates into DNA in living cells, thereby allowing the subsequent isolation of mitotic chondrocytes according to their DNA content by FACS (Fig. S3B and C). We isolated cells in G2/M-phase from three Trps1-/- and wild-type mice per experiment. Flow cytometric re-analysis of a subset of isolated G2/M-phase cells confirmed an enrichment to about 90% in both genotypes (Fig. S3D and E). We next quantified acetylated H3K9 in relation to total H3 protein by western blotting (). We found the ratio of acetylated H3K9 to total H3 to be significantly increased from 0.6 in wild-type cells to 0.83 in Trps1-/- chondrocytes (), demonstrating that the hyperacetylated chromatin is maintained during mitosis.
Figure 7. Overexpression of Hdac4 rescues the G2/M-phase delay in Trps1-/- chondrocytes. (A and B) The level of H3K9ac in FACS sorted G2/M-phase cells after staining with DyeCycle green was quantified by western blot analysis (A). Acetylation is increased in unsorted (total cell extract) and isolated, G2/M-phase Trps1-/- chondrocytes. α-pH3 detection confirms enrichment of G2/M-phase cells. (B) The level of H3K9 acetylation in relation to total H3 is increased in sorted G2/M-phase cells (n =3; p* < 0.05). (C and D) Wild-type and Trps1-/- primary chondrocytes were transfected with a Flag-tagged Hdac4 and the overexpression was confirmed by western blotting (C). Cell cycle progression was analyzed by flow cytometry after BrdU and 7-AAD labeling. Hdac4 overexpression decreases H3K9 acetylation in Trps1-/- chondrocytes similar to wild-type levels, while H3 levels were not altered. (D) Hdac4 overexpression rescued the increased proportion of G2/M-phase cells in Trps1-/- mutants, but had little effect on wild-type chondrocytes (n =3; p* < 0.05). (E) Interaction of Trps1 with Hdac1 and Hdac4, or a complex of both, increases the histone deacetylase activity on histone H3. Loss of Trps1 reduces Hdac activity, thereby leading to hyperacetylated chromatin and disturbed chromatin condensation. Subsequently, chromosome segregation is impaired.
Hdac4 overexpression rescues the G2/M-phase delay in Trps1-/- chondrocytes
To further support our hypothesis that the effect of Trps1 on mitotic progression is mediated by Hdac activity, we overexpressed Trps1 and Hdac4 in Trps1-/- and wild-type primary chondrocytes and analyzed the level of H3 acetylation and cell cycle distribution by flow cytometry (; Fig. S4). Overexpression of Flag-tagged Trps1 rescued the proportion of cells in G2/M-phase in Trps1-/- chondrocytes, but had no effect on wild-type cells (Fig. S4B). As expected, overexpression of Flag-tagged Hdac4 reduced the acetylation level of H3K9 in Trps1-/- cells to a similar level as in wild-type cells (). Interestingly, the increased Hdac activity had no obvious effect on the proportion of cells in G2/M-phase in wild-type chondrocytes. In contrast, in Trps1-/- chondrocytes, overexpression of Hdac4 reduced the increased proportion of chondrocytes in G2/M-phase to similar levels as in wild-type chondrocytes (), strongly supporting the hypothesis that Trps1 regulates mitosis by activating Hdac activity.
Discussion
During mitosis, the accurate condensation and subsequent distribution of chromosomes requires distinct epigenetic modifications. Here, we demonstrate that Trps1, a multi zinc-finger protein that has previously been described as a transcriptional regulator of chondrocyte proliferation and differentiation, plays a critical role in regulating progression through mitosis by increasing Hdac activity. Our experiments show that loss of Trps1 leads to an accumulation of mitotic chondrocytes. This effect is especially surprising, as the proliferation rate, as measured by the number of cells in S-phase, is reduced in Trps1-/- mutants.
One critical step required for the condensation of chromatin during mitosis is the deacetylation of histones.Citation6,Citation7,Citation34 Searching for the basis of the mitotic defect in Trps1-/- chondrocytes, we have identified Trps1 as a positive regulator of Hdac activity. We demonstrate that Trps1 interacts with two Hdac proteins, Hdac1 and Hdac4, thereby increasing their deacetylase activity. Furthermore, Trps1-/- chondrocytes are characterized by increased H3 acetylation, which is maintained during mitosis. We hypothesize that the increased histone deacetylase activity in Trps1-/- mutants leads to chromatin condensation defects, decreased HP1 binding of mitotic chromatin, and, subsequently, to the mitotic defects observed in Trps1-/- chondrocytes ().
Trps1 as regulator of mitotic progression
During recent years, many lines of evidence, mostly based on synthetic Hdac inhibitors and knockdown experiments, have demonstrated the importance of Hdac function in regulating mitosis.Citation5,Citation35 Chromatin hyperacetylation interferes with its compaction, leading to the activation of the spindle attachment checkpoint and an arrest of mitosis in prometaphase.Citation9,Citation26,Citation27 In agreement with this hypothesis, most studies show that inhibition of Hdac function results in less condensed chromatin during prometaphase in combination with histones that are hyperacetylated at various positions.Citation5,Citation36 Furthermore, several lines of evidence suggest a specific role of H3K9 deacetylation in centromeric regions to allow trimethylation at this position. H3K9 trimethylation supports binding of the heterochromatin binding protein 1 (HP1) to centromeres (Li et al. 2006; Park et al. 2011) and the subsequent recruitment of the chromosomal passenger complex (CPC),Citation37,Citation38 which regulates kinetochore formation and mitotic spindle attachment. Our results provide evidence that Trps1 acts as a regulator of mitosis: Trps1-/- mutants show increased numbers of pH3-positive mitotic cells, which are especially enriched in prometaphase, indicating defects in chromatin compaction. In addition, as observed for other regulators of mitotic progression including Hdacs, Trps1-/- primary chondrocytes are less sensitive to a Nocodazole-induced block of the spindle attachment checkpoint. Consequently, chromosome segregation is impaired. Most importantly, we not only found increased acetylation at H3K9 in Trps1-/- mutant cells, but also decreased levels of chromatin-associated HP1 protein, indicating that in addition to impaired chromatin condensation, disturbed CPC assembly might contribute to the observed chromosome segregation phenotype.
Recently, first evidence has been obtained that besides their role in histone deacetylation, Hdacs might regulate mitosis directly by deacetylating components of the mitotic spindle.Citation39 If the decreased Hdac activity of Trps1-/- mutants also impairs the deacetylation of such regulators needs to be investigated in additional studies.
Trps1 as regulator of Hdac activity
Having identified a link between Trps1 and Hdac proteins, it is important to decipher at which level this interaction regulates Hdac activity. Theoretically, Trps1 might regulate the transcription of Hdacs, recruit Hdacs to the chromatin by stabilizing DNA contact with its DNA binding GATA zinc finger domain or directly act on the enzymatic activity of the protein. Using a fluorometric activity assay, we found increased Hdac activity after overexpression of Trps1, while loss of Trps1 decreases Hdac activity in vivo. Thus our results strongly support a role of Trps1 in regulating the enzymatic activity of Hdacs.
The role of individual Hdac family members in regulating mitotic progression has not been analyzed in detail. Inactivation of Hdac4 has been linked to chromosome segregation and stability defects in HeLa cells Citation40 but no mitotic defects have been described for loss of Hdac1 or Hdac4 in vivo, yet. So far only targeted deletion of Hdac3 has been shown to result in mitotic defects in mouse fibroblasts in vivo,Citation8,Citation41 mimicking those we have observed in Trps1-/- mice. First analyses did not provided strong evidence for a direct interaction of Trps1 and Hdac3 (not shown). However, it is interesting to note that Hdac4, which lacks histone deacetylase activity, Citation42,Citation43 interacts with both Hdac1 and Hdac3, thereby increasing their histone deacetylase activity.Citation42,Citation43 The increased Hdac activity that has been observed after Hdac4 overexpression in these studies has been attributed to the recruitment of either Hdac1 or Hdac3.Citation42,Citation43 As we found altered histone H3 acetylation after overexpressing either Hdac4 or Trps1, we propose that Trps1 regulates Hdac activity by recruiting or stabilizing Hdac-containing protein complexes. As the mitotic phenotype of Trps1-/- mutants is relatively mild, one might speculate that some Hdacs are generally active in most cells, whereas others are regulated by cell type specific interaction partners like Trps1. Investigating mitotic progression in other tissues that express Trps1 will be important to support this hypothesis.
The role of the Trps1-Hdac interaction for the skeletal phenotype
Besides the newly identified defects in mitotic progression loss of Trps1 leads to several distinct alterations in chondrocyte development. The previously described roles of Trps1 in regulating proliferation rate and hypertrophic differentiation likely contribute to many aspects of the human skeletal phenotype. However, various aspects, like the reduced region of low proliferating chondrocytes at the ends of the skeletal elements, the increased region of prehypertrophic cells,Citation14,Citation19 and the disturbed cell cycle exit require additional explanations. In this respect, the identified delay of mitotic progression might reduce the number of cycling cells, thereby contributing to the growth defects observed in mutant mice and in TRPS patients.
Independent of its function during mitosis, the interaction of Trps1 and Hdacs might regulate other steps of chondrocyte proliferation and differentiation. During hypertrophic differentiation, Trps1 deficiency disturbs the highly synchronized transition from proliferating into hypertrophic chondrocytes, and BrdU-positive cells are found intermingled with hypertrophic cells.Citation14 One might speculate that, similar to what has been shown during neuronal differentiation,Citation45 disturbed mitotic progression impairs cell cycle exit, which is linked to hypertrophic differentiation in wild-type mice. As chondrocyte hypertrophy contributes significantly to longitudinal bone growth, impaired differentiation might thus contribute to a reduced skeleton size.
In addition, the interaction might directly regulate the hypertrophic differentiation program. The onset of hypertrophic differentiation is controlled by the tightly balanced interaction of a large number of activating and repressing transcription factors, including Runx2 and Hdac4, which interact with Trps1.Citation14,Citation19,Citation31 Runx2 is required to initiate hypertrophic differentiation,Citation46 whereas Hdac4 inhibits hypertrophy by repression of Runx2 expression in proliferating chondrocytes.Citation19,Citation47,Citation48 As the expression of these factors overlap with that of Trps1 in the region where chondrocytes switch from proliferation to hypertrophy, Trps1 might fine-tune the expression of Runx2 by modulating Hdac4 activity, thereby regulating hypertrophic differentiation and bone growth.
Conclusion
Our data provide first evidence that Trps1 plays a critical role in fine-tuning progression through mitosis in chondrocytes. By interacting with at least two Hdac proteins, Trps1 regulates the global acetylation of histones in chondrocytes. During mitosis, Trps1-dependent Hdac function is required for maximal chromatin deacetylation. Accordingly, Trps1 deficiency leads to hyperacetylated chromatin and, subsequently, impaired mitotic progression. Although we cannot rule out an additional function of Trps1 in regulating other mitotic factors, the close similarity of the phenotypes in combination with the rescued proportion of chondrocytes in G2/M-phase after Hdac4 overexpression in Trps1-/- mutants strongly indicates that Trps1 regulates mitosis by increasing Hdac activity ().
To our knowledge, this is the first report of a proposed transcription factor impacting on the global histone acetylation status in chondrocytes. The interaction of Trps1 with different histone deacetylases extends its function beyond that of a classical transcription factor towards an epigenetic chromatin modifier. Based on these data, aspects of the Trps1-/- phenotype might have to be re-evaluated, taking the regulation of Hdac function into account. Furthermore, many transcription factors interact with histone-modifying enzymes, and it will be important to evaluate whether such interactions similarly influence global chromatin acetylation.
Material and Methods
Transgenic mice
Heterozygous Trps1+/- miceCitation17 were maintained on a C57Bl/6J genetic background. All animal studies were undertaken according to institutional guidelines of the University Duisburg-Essen and approved by the animal welfare committee (§15). Mouse husbandry was approved by the city of Essen (Az: 32-2-11-80-71/203) in accordance with § 11 (1) 1a of the “Tierschutzgesetz” and by the animal welfare committee (§15). Genotyping PCR was performed on genomic tail DNA as described previously.Citation14
Cell culture and transfection
HEK293 EBNA cells were grown in DMEM high glucose medium with 10% FCS (Invitrogen) and transfected with 2 µg expression plasmid or 50 ng siRNA with Lipofectamine transfection reagent (Invitrogen) according to the manufacturer’s instructions. Human HDAC2 and HDAC4 siRNACitation49 were obtained from Dharmacon. Predesigned siRNA against human TRPS1 and HDAC1 (esiRNA) were obtained from Sigma Aldrich. Cells were lysed 24 h after plasmid transfection or 48 h after siRNA transfection. ATDC5 cells were cultivated in DMEM/F12 (Invitrogen) with 5% FCS. Primary chondrocytes were isolated as chondrogenic precursor cells from E12.5 limb buds as described previouslyCitation50 and cultured as monolayer. For all analyses, cells were passaged once to avoid dedifferentiation. Transfection of ATDC5 cells and primary chondrocytes was performed with the Lonza transfection device using 2 µg plasmid DNA and transfection solution T. After 24 h, the cells were lysed and analyzed. Histone deacetylase activity was inhibited by treatment with the histone deacetylase inhibitor Trichostatin A (TSA, 50 µM, Calbiochem). To inhibit cell cycle progression, primary chondrocytes were treated with 1 µg/ml Nocodazole (Calbiochem) or 1.25 µg/ml Aphidicolin (Calbiochem) for a period of 18 h. Blocking was released by medium replacement. DMSO (Sigma Aldrich), or ethanol treatment was used as negative control.
Western blot analysis and co-immunoprecipitation
Cells were lysed in ice-cold lysis buffer supplemented with Dithiothreitol and protease inhibitor cocktail (Roche Diagnostics), triturated 10 times through a G21 syringe and centrifuged at 5000 g for 1 min. Co-immunoprecipitation was performed with Protein A or Protein G-Sepharose beads (Roche) and 2.5 µg of each of the following antibodies: α-Flag M2 (Stratagene), α-GFP (Abcam), α-Hdac1 (Milipore), α-Hdac2, and α-Hdac4 (Santa Cruz Biotechnology); mouse IgG (Milipore), rabbit IgG (Calbiochem), α-Trps1Citation14 overnight. Complexes were dissolved in Laemmli buffer, separated on an SDS-PAGE and blotted on nitrocellulose membrane (Bio-Rad Laboratories). Antibodies were obtained as following: α-H3, α-H3K9ac, α-pH3 (Abcam), goat α-mouse HRP (Santa Cruz Biotechnology), rabbit α-goat and swine α-rabbit HRP coupled secondary antibodies (Dako). Chemiluminescence (ECL, Pierce) was detected on X-ray films (Kodak) or a Bioanalyser (BioRad). To quantify the signal intensity, pictures were taken using the Fusion SL imaging system (Vilbert Loumart). Signal intensity was measured with the Bio1D Advanced analysis software (Vilbert Loumart). The ratio of signal intensities of acetylated H3K9 was normalized against total H3.
Hdac activity assay
Protein concentration was determined in a Bradford assay (Calbiochem), 5 µg of cell lysates or limb extracts were used for the activity assay (Calbiochem) and performed according to the manufacturer’s instructions. Fluorescence was measured on a GeniosPro ELISA reader (Tecan) and analyzed with the Magellan software (Tecan). The Hdac activity in untransfected HEK293 EBNA cells or wild-type limb extracts was set to 100%. All measurements were performed in triplicates per probe. For statistical analysis, lysates from at least 3 independent mice, overexpression or siRNA experiments were used. Unpaired Student t test was used to evaluate statistical significance.
Flow cytometry and cell cycle analysis
For flow cytometry, primary chondrocytes and HEK293 EBNA cells were labeled with 10 µM BrdU for 2 h and with the FITC BrdU Flow kit (BD Bioscience) according to the manufacturer’s protocol. DNA was counterstained with 7-aminoactinomycin (7-AAD). Ten thousand cells were analyzed for each sample. Fluorescence was measured on a FACSCalibur flow cytometer with the CellQuest Pro software and analyzed with FlowJo software (BD Bioscience). An IgG isotype control (BD Bioscience) was used as negative control. Statistic analysis was performed from at least 4 Trps1-/- and wild-type mice from different litters. To analyze cell cycle progression, cells were labeled with BrdU for 1 h either at the end of inhibitor treatment or during the release from the inhibitor. Mitotic cells were detected with an Alexa568-labeled α-pH3 antibody (Becton Dickinson) according to the manufacturers protocol. DNA was counterstained with 0.5 µg/µl propidium iodide (BD Bioscience). For sorting of G2/M-phase cell, 2 × 107 chondrocytes were stained with Vybrant DyeCycle Green (Invitrogen) according to the manufacturers protocol. Cells with high DNA content were sorted on a FACSCantoII flow cytometer with a 100 µm nozzle and analyzed with the FACSDiva software (BD Bioscience). Statistic significance was evaluated by a Wilcoxon-Mann-Whitney test using the R analysis software.
Immunohistochemical and immunofluorescent staining
Paraffin sections from E16.5 mice were microwaved in citrate buffer and incubated with an α-pH3 antibody (Upstate Biotechnology). Cell cultures were fixed in methanol before staining with the following antibodies: α-H3K9ac, (Abcam), α-pH3 (Upstate Biotechnology), α-H3K9acS10p, (Cell Signaling), α-HP1β (Abcam), α-y-Tubulin (Sigma Aldrich), and detected with an Alexa568 or Alexa488 goat anti-rabbit or Alexa488 rabbit α-mouse secondary antibodies (Invitrogen). DNA was counterstained with 5 mg/ml DAPI (Roth). The mean fluorescent intensity was determined using the Metamorph imaging software (Visitron Imaging Systems). To quantify the mitotic defects, 100 mitotic cells in each of 4 independent chondrocyte cultures from Trps1-/- and wild-type mice were analyzed.
Chromosome preparation
1× 107 chondrocytes from Trps1-/- and wild-type mice were treated with 0.008 mg/ml Colcemid in growth medium for 2 h. Cells were swollen in 0.075 M potassium chloride, fixed in methanol: acetic acid (3:1) and spotted on humid slides. Slides were stained in 5% Giemsa solution (Sigma), and chromosome numbers were scored at 630-fold magnification. Chromosome preparations were prepared from 4 different Trps1-/- and wild-type mice, and 200 chromosome spreads were counted for statistic evaluation. Significant differences in chromosome numbers were evaluated by Wilcoxon-Mann-Whitney test using the R analysis software.
Imaging and statistic analysis
Fluorescence pictures were taken on a Zeiss Axiovert 200 microscope with a Spot 23.0 camera (Diagnostik Instruments) and Metamorph imaging software (Visitron Imaging Systems) using the same settings per experiment. Statistic significance was assessed by an unpaired, two-tailed student’s t-tests or Wilcoxon test using R software package, depending on the experimental set-up. The number of experiments or mutants is given in the figure legends. P values < 0.05 were regarded significant. Standard deviation for each parameter is given as error bar in each histogram.
Additional material
Download Zip (828.2 KB)Acknowledgment
We would like to thank V Schneider for helpful discussion and reading of the manuscript, D Hoffmann for critical evaluation of the statistical analysis, FJ Kaiser for sharing expression plasmids for Hdac1-Flag and Hdac2-Flag, and A Westendorf for sorting of mitotic cells. This work was supported by the DFG graduate program GRK 1431 to AV.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25267
Related Research Data
References
- Wuelling M, Vortkamp A. Transcriptional networks controlling chondrocyte proliferation and differentiation during endochondral ossification. Pediatr Nephrol 2010; 25:625 - 31; http://dx.doi.org/10.1007/s00467-009-1368-6; PMID: 19949815
- Beier F. Cell-cycle control and the cartilage growth plate. J Cell Physiol 2005; 202:1 - 8; http://dx.doi.org/10.1002/jcp.20111; PMID: 15389526
- Haberland M, Montgomery RL, Olson EN. The many roles of histone deacetylases in development and physiology: implications for disease and therapy. Nat Rev Genet 2009; 10:32 - 42; http://dx.doi.org/10.1038/nrg2485; PMID: 19065135
- Kruhlak MJ, Hendzel MJ, Fischle W, Bertos NR, Hameed S, Yang XJ, et al. Regulation of global acetylation in mitosis through loss of histone acetyltransferases and deacetylases from chromatin. J Biol Chem 2001; 276:38307 - 19; PMID: 11479283
- Park JA, Kim AJ, Kang Y, Jung YJ, Kim HK, Kim KC. Deacetylation and methylation at histone H3 lysine 9 (H3K9) coordinate chromosome condensation during cell cycle progression. Mol Cells 2011; 31:343 - 9; http://dx.doi.org/10.1007/s10059-011-0044-4; PMID: 21359677
- Cimini D, Mattiuzzo M, Torosantucci L, Degrassi F. Histone hyperacetylation in mitosis prevents sister chromatid separation and produces chromosome segregation defects. Mol Biol Cell 2003; 14:3821 - 33; http://dx.doi.org/10.1091/mbc.E03-01-0860; PMID: 12972566
- Robbins AR, Jablonski SA, Yen TJ, Yoda K, Robey R, Bates SE, et al. Inhibitors of histone deacetylases alter kinetochore assembly by disrupting pericentromeric heterochromatin. Cell Cycle 2005; 4:717 - 26; http://dx.doi.org/10.4161/cc.4.5.1690; PMID: 15846093
- Ishii S, Kurasawa Y, Wong J, Yu-Lee LY. Histone deacetylase 3 localizes to the mitotic spindle and is required for kinetochore-microtubule attachment. Proc Natl Acad Sci USA 2008; 105:4179 - 84; http://dx.doi.org/10.1073/pnas.0710140105; PMID: 18326024
- Magnaghi-Jaulin L, Eot-Houllier G, Fulcrand G, Jaulin C. Histone deacetylase inhibitors induce premature sister chromatid separation and override the mitotic spindle assembly checkpoint. Cancer Res 2007; 67:6360 - 7; http://dx.doi.org/10.1158/0008-5472.CAN-06-3012; PMID: 17616695
- Momeni P, Glöckner G, Schmidt O, von Holtum D, Albrecht B, Gillessen-Kaesbach G, et al. Mutations in a new gene, encoding a zinc-finger protein, cause tricho-rhino-phalangeal syndrome type I. Nat Genet 2000; 24:71 - 4; http://dx.doi.org/10.1038/71717; PMID: 10615131
- Malik TH, Shoichet SA, Latham P, Kroll TG, Peters LL, Shivdasani RA. Transcriptional repression and developmental functions of the atypical vertebrate GATA protein TRPS1. EMBO J 2001; 20:1715 - 25; http://dx.doi.org/10.1093/emboj/20.7.1715; PMID: 11285235
- Kaiser FJ, Möröy T, Chang GT, Horsthemke B, Lüdecke HJ. The RING finger protein RNF4, a co-regulator of transcription, interacts with the TRPS1 transcription factor. J Biol Chem 2003; 278:38780 - 5; http://dx.doi.org/10.1074/jbc.M306259200; PMID: 12885770
- Kaiser FJ, Brega P, Raff ML, Byers PH, Gallati S, Kay TT, et al. Novel missense mutations in the TRPS1 transcription factor define the nuclear localization signal. Eur J Hum Genet 2004; 12:121 - 6; http://dx.doi.org/10.1038/sj.ejhg.5201094; PMID: 14560312
- Wuelling M, Kaiser FJ, Buelens LA, Braunholz D, Shivdasani RA, Depping R, et al. Trps1, a regulator of chondrocyte proliferation and differentiation, interacts with the activator form of Gli3. Dev Biol 2009; 328:40 - 53; http://dx.doi.org/10.1016/j.ydbio.2009.01.012; PMID: 19389374
- Kaiser FJ, Tavassoli K, Van den Bemd GJ, Chang GT, Horsthemke B, Möröy T, et al. Nuclear interaction of the dynein light chain LC8a with the TRPS1 transcription factor suppresses the transcriptional repression activity of TRPS1. Hum Mol Genet 2003; 12:1349 - 58; http://dx.doi.org/10.1093/hmg/ddg145; PMID: 12761050
- Kunath M, Lüdecke HJ, Vortkamp A. Expression of Trps1 during mouse embryonic development. Mech Dev 2002; 119:Suppl 1 S117 - 20; http://dx.doi.org/10.1016/S0925-4773(03)00103-5; PMID: 14516672
- Malik TH, Von Stechow D, Bronson RT, Shivdasani RA. Deletion of the GATA domain of TRPS1 causes an absence of facial hair and provides new insights into the bone disorder in inherited tricho-rhino-phalangeal syndromes. Mol Cell Biol 2002; 22:8592 - 600; http://dx.doi.org/10.1128/MCB.22.24.8592-8600.2002; PMID: 12446778
- Napierala D, Garcia-Rojas X, Sam K, Wakui K, Chen C, Mendoza-Londono R, et al. Mutations and promoter SNPs in RUNX2, a transcriptional regulator of bone formation. Mol Genet Metab 2005; 86:257 - 68; http://dx.doi.org/10.1016/j.ymgme.2005.07.012; PMID: 16140555
- Napierala D, Sam K, Morello R, Zheng Q, Munivez E, Shivdasani RA, et al. Uncoupling of chondrocyte differentiation and perichondrial mineralization underlies the skeletal dysplasia in tricho-rhino-phalangeal syndrome. Hum Mol Genet 2008; 17:2244 - 54; http://dx.doi.org/10.1093/hmg/ddn125; PMID: 18424451
- Suemoto H, Muragaki Y, Nishioka K, Sato M, Ooshima A, Itoh S, et al. Trps1 regulates proliferation and apoptosis of chondrocytes through Stat3 signaling. Dev Biol 2007; 312:572 - 81; http://dx.doi.org/10.1016/j.ydbio.2007.10.001; PMID: 17997399
- Pedrali-Noy G, Spadari S, Miller-Faurès A, Miller AO, Kruppa J, Koch G. Synchronization of HeLa cell cultures by inhibition of DNA polymerase alpha with aphidicolin. Nucleic Acids Res 1980; 8:377 - 87; http://dx.doi.org/10.1093/nar/8.2.377; PMID: 6775308
- Decordier I, Cundari E, Kirsch-Volders M. Survival of aneuploid, micronucleated and/or polyploid cells: crosstalk between ploidy control and apoptosis. Mutat Res 2008; 651:30 - 9; http://dx.doi.org/10.1016/j.mrgentox.2007.10.016; PMID: 18242119
- Liu X, Zheng H, Qu CK. Protein tyrosine phosphatase Shp2 (Ptpn11) plays an important role in maintenance of chromosome stability. Cancer Res 2012; 72:5296 - 306; http://dx.doi.org/10.1158/0008-5472.CAN-12-1495; PMID: 22890240
- Gillespie DA, Walker M. Mitotic index determination by flow cytometry. Subcell Biochem 2006; 40:355 - 8; PMID: 17623920
- Nakajima H, Kim YB, Terano H, Yoshida M, Horinouchi S. FR901228, a potent antitumor antibiotic, is a novel histone deacetylase inhibitor. Exp Cell Res 1998; 241:126 - 33; http://dx.doi.org/10.1006/excr.1998.4027; PMID: 9633520
- Zhang X, Zhang Z, Chen G, Zhao M, Wang D, Zhang X, et al. FK228 induces mitotic catastrophe in A549 cells by mistargeting chromosomal passenger complex localization through changing centromeric H3K9 hypoacetylation. Acta Biochim Biophys Sin (Shanghai) 2010; 42:677 - 87; http://dx.doi.org/10.1093/abbs/gmq077; PMID: 20817931
- Shin HJ, Baek KH, Jeon AH, Kim SJ, Jang KL, Sung YC, et al. Inhibition of histone deacetylase activity increases chromosomal instability by the aberrant regulation of mitotic checkpoint activation. Oncogene 2003; 22:3853 - 8; http://dx.doi.org/10.1038/sj.onc.1206502; PMID: 12813458
- Towers M, Mahood R, Yin Y, Tickle C. Integration of growth and specification in chick wing digit-patterning. Nature 2008; 452:882 - 6; http://dx.doi.org/10.1038/nature06718; PMID: 18354396
- Arnold MA, Kim Y, Czubryt MP, Phan D, McAnally J, Qi X, et al. MEF2C transcription factor controls chondrocyte hypertrophy and bone development. Dev Cell 2007; 12:377 - 89; http://dx.doi.org/10.1016/j.devcel.2007.02.004; PMID: 17336904
- Dy P, Wang W, Bhattaram P, Wang Q, Wang L, Ballock RT, et al. Sox9 directs hypertrophic maturation and blocks osteoblast differentiation of growth plate chondrocytes. Dev Cell 2012; 22:597 - 609; http://dx.doi.org/10.1016/j.devcel.2011.12.024; PMID: 22421045
- Vega RB, Matsuda K, Oh J, Barbosa AC, Yang X, Meadows E, et al. Histone deacetylase 4 controls chondrocyte hypertrophy during skeletogenesis. Cell 2004; 119:555 - 66; http://dx.doi.org/10.1016/j.cell.2004.10.024; PMID: 15537544
- Brunmeir R, Lagger S, Seiser C. Histone deacetylase HDAC1/HDAC2-controlled embryonic development and cell differentiation. Int J Dev Biol 2009; 53:275 - 89; http://dx.doi.org/10.1387/ijdb.082649rb; PMID: 19412887
- McManus KJ, Hendzel MJ. The relationship between histone H3 phosphorylation and acetylation throughout the mammalian cell cycle. Biochem Cell Biol 2006; 84:640 - 57; http://dx.doi.org/10.1139/o06-086; PMID: 16936834
- Valls E, Sánchez-Molina S, Martínez-Balbás MA. Role of histone modifications in marking and activating genes through mitosis. J Biol Chem 2005; 280:42592 - 600; http://dx.doi.org/10.1074/jbc.M507407200; PMID: 16199528
- Gabrielli B, Chia K, Warrener R. Finally, how histone deacetylase inhibitors disrupt mitosis!. Cell Cycle 2011; 10:2658 - 61; http://dx.doi.org/10.4161/cc.10.16.16953; PMID: 21811095
- Eot-Houllier G, Fulcrand G, Watanabe Y, Magnaghi-Jaulin L, Jaulin C. Histone deacetylase 3 is required for centromeric H3K4 deacetylation and sister chromatid cohesion. Genes Dev 2008; 22:2639 - 44; http://dx.doi.org/10.1101/gad.484108; PMID: 18832068
- Heit R, Rattner JB, Chan GK, Hendzel MJ. G2 histone methylation is required for the proper segregation of chromosomes. J Cell Sci 2009; 122:2957 - 68; http://dx.doi.org/10.1242/jcs.045351; PMID: 19638412
- Xin H, Yoon HG, Singh PB, Wong J, Qin J. Components of a pathway maintaining histone modification and heterochromatin protein 1 binding at the pericentric heterochromatin in Mammalian cells. J Biol Chem 2004; 279:9539 - 46; http://dx.doi.org/10.1074/jbc.M311587200; PMID: 14665632
- Fadri-Moskwik M, Weiderhold KN, Deeraksa A, Chuang C, Pan J, Lin SH, et al. Aurora B is regulated by acetylation/deacetylation during mitosis in prostate cancer cells. FASEB J 2012; 26:4057 - 67; http://dx.doi.org/10.1096/fj.12-206656; PMID: 22751009
- Cadot B, Brunetti M, Coppari S, Fedeli S, de Rinaldis E, Dello Russo C, et al. Loss of histone deacetylase 4 causes segregation defects during mitosis of p53-deficient human tumor cells. Cancer Res 2009; 69:6074 - 82; http://dx.doi.org/10.1158/0008-5472.CAN-08-2796; PMID: 19622775
- Bhaskara S, Knutson SK, Jiang G, Chandrasekharan MB, Wilson AJ, Zheng S, et al. Hdac3 is essential for the maintenance of chromatin structure and genome stability. Cancer Cell 2010; 18:436 - 47; http://dx.doi.org/10.1016/j.ccr.2010.10.022; PMID: 21075309
- Chan JK, Sun L, Yang XJ, Zhu G, Wu Z. Functional characterization of an amino-terminal region of HDAC4 that possesses MEF2 binding and transcriptional repressive activity. J Biol Chem 2003; 278:23515 - 21; http://dx.doi.org/10.1074/jbc.M301922200; PMID: 12709441
- Fischle W, Dequiedt F, Hendzel MJ, Guenther MG, Lazar MA, Voelter W, et al. Enzymatic activity associated with class II HDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol Cell 2002; 9:45 - 57; http://dx.doi.org/10.1016/S1097-2765(01)00429-4; PMID: 11804585
- Jones P, Altamura S, De Francesco R, Gallinari P, Lahm A, Neddermann P, et al. Probing the elusive catalytic activity of vertebrate class IIa histone deacetylases. Bioorg Med Chem Lett 2008; 18:1814 - 9; http://dx.doi.org/10.1016/j.bmcl.2008.02.025; PMID: 18308563
- Ruggiero R, Kale A, Thomas B, Baker NE. Mitosis in neurons: Roughex and APC/C maintain cell cycle exit to prevent cytokinetic and axonal defects in Drosophila photoreceptor neurons. PLoS Genet 2012; 8:e1003049; http://dx.doi.org/10.1371/journal.pgen.1003049; PMID: 23209426
- Yoshida CA, Yamamoto H, Fujita T, Furuichi T, Ito K, Inoue K, et al. Runx2 and Runx3 are essential for chondrocyte maturation, and Runx2 regulates limb growth through induction of Indian hedgehog. Genes Dev 2004; 18:952 - 63; http://dx.doi.org/10.1101/gad.1174704; PMID: 15107406
- Jensen ED, Nair AK, Westendorf JJ. Histone deacetylase co-repressor complex control of Runx2 and bone formation. Crit Rev Eukaryot Gene Expr 2007; 17:187 - 96; http://dx.doi.org/10.1615/CritRevEukarGeneExpr.v17.i3.20; PMID: 17725488
- Jeon EJ, Lee KY, Choi NS, Lee MH, Kim HN, Jin YH, et al. Bone morphogenetic protein-2 stimulates Runx2 acetylation. J Biol Chem 2006; 281:16502 - 11; http://dx.doi.org/10.1074/jbc.M512494200; PMID: 16613856
- Mottet D, Pirotte S, Lamour V, Hagedorn M, Javerzat S, Bikfalvi A, et al. HDAC4 represses p21(WAF1/Cip1) expression in human cancer cells through a Sp1-dependent, p53-independent mechanism. Oncogene 2009; 28:243 - 56; http://dx.doi.org/10.1038/onc.2008.371; PMID: 18850004
- Stanton LA, Sabari S, Sampaio AV, Underhill TM, Beier F. p38 MAP kinase signalling is required for hypertrophic chondrocyte differentiation. Biochem J 2004; 378:53 - 62; http://dx.doi.org/10.1042/BJ20030874; PMID: 14594450