Abstract
SIRT1 is an NAD+-dependent deacetylase that counteracts multiple disease states associated with aging and may underlie some of the health benefits of calorie restriction. Understanding how SIRT1 is regulated in vivo could therefore lead to new strategies to treat age-related diseases. SIRT1 forms a stable complex with DBC1, an endogenous inhibitor. Little is known regarding the biochemical nature of SIRT1-DBC1 complex formation, how it is regulated and whether or not it is possible to block this interaction pharmacologically. In this study, we show that critical residues within the catalytic core of SIRT1 mediate binding to DBC1 via its N-terminal region, and that several carboxamide SIRT1 inhibitors, including EX-527, can completely block this interaction. We identify two acetylation sites on DBC1 that regulate its ability to bind SIRT1 and suppress its activity. Furthermore, we show that DBC1 itself is a substrate for SIRT1. Surprisingly, the effect of EX-527 on SIRT1-DBC1 binding is independent of DBC1 acetylation. Together, these data show that protein acetylation serves as an endogenous regulatory mechanism for SIRT1-DBC1 binding and illuminate a new path to developing small-molecule modulators of SIRT1.
Introduction
The protein deacetylase SIRT1 regulates a variety of physiological processes, including fat mobilization,Citation1 muscle differentiation,Citation2 and glucose and insulin homeostasisCitation3,Citation4. As such, it has emerged as an attractive drug target for the treatment of many age-related pathologies,Citation5 including type 2 diabetes,Citation6,Citation7 cancer,Citation8,Citation9 and Alzheimer disease.Citation10 The cellular activity of SIRT1 is regulated by multiple mechanisms including levels of the co-substrate NAD+,Citation11 the endogenous inhibitor nicotinamide (NAM),Citation12 and by sumoylationCitation13 and phosphorylation.Citation14,Citation15 Two protein binding partners, active regulator of SIRT1 (AROS)Citation16 and deleted in breast cancer 1 (DBC1)Citation17,Citation18 have also been shown to activate and repress SIRT1, respectively.
DBC1 (KIAA1967) was initially identified as a candidate tumor suppressor gene found in a region frequently deleted in breast cancers.Citation19 However, refined deletion analysis and expression studies revealed that in fact, DBC2, was the candidate tumor suppressor.Citation19 Subsequently, DBC1 has been shown to facilitate apoptosis following its cleavage by caspasesCitation20 and to interact in a ligand-independent fashion with ERα to suppress breast cancer cell apoptosis in the absence of hormone.Citation21,Citation22 In addition to inhibiting SIRT1,Citation17,Citation18 DBC1 regulates NAD+ levels via a c-MYC-NAMPT-DBC1-SIRT1 feedback loop,Citation23 inhibits HDAC3Citation24 and SUV39H1,Citation25 regulates nuclear receptor Rev-erbα stability and function,Citation26 and is a component of the DBIRD complex involved in alternative mRNA splicing.Citation27
DBC1 is thought to bind to the catalytic core of SIRT1 and inhibit SIRT1 enzymatic activityCitation17,Citation18 via a leucine zipper domain (LZ)Citation18 or an N-terminal region.Citation25 Recently, a structured C-terminal domain in SIRT1, known as the ESA region, was shown to be required for its enzymatic activityCitation5,Citation28. It has been proposed that DBC1 inhibits SIRT1 activity by competing with and preventing binding of ESA with the catalytic domain.Citation28 Indeed, overexpression of DBC1 results in repression of SIRT1 activity, concomitant with increased levels of p53,Citation17 FOXO,Citation18 and HSF1Citation29 acetylation. In mice, knockout of DBC1 results in increased SIRT1 activity in several tissues, protection from liver steatosis and inflammation,Citation30 and the “browning” of white-adipose tissue (WAT).Citation31
The SIRT1-DBC1 interaction is dynamically regulated under starvation conditionsCitation30 and in breast cancer cells.Citation32 Activation of the cAMP/PKA pathway results in an AMPK-dependent dissociation of the SIRT1-DBC1 complex,Citation33 possibly by AMPK-mediated phosphorylation of key residues on SIRT1 or DBC1.Citation33 Conversely, the DBC1-SIRT1 interaction is enhanced during DNA damage and oxidative stress by ATM-mediated phosphorylation of Thr454, which plays an important role in cell fate determination following genotoxic stress.Citation34
Activating SIRT1 is viewed as a promising path to treating and preventing a variety of age-related disorders.Citation35 Toward this end, direct allosteric activators of SIRT1 (STACs) have been developed.Citation5,Citation7 Another potential way to activate SIRT1 would be to find small molecules that could specifically interfere with DBC1-mediated repression of SIRT1. Aside from molecules that act on proteins that post-translationally modify DBC1, and thereby influence the binding of SIRT1 to DBC1,Citation33 no direct small-molecule regulators of the SIRT1-DBC1 complex have thus far been reported.
Here, we identify several critical residues within the catalytic core of SIRT1 required for complex formation with DBC1, we show that DBC1 is acetylated on two critical residues that mediate its binding to SIRT1, and we demonstrate that DBC1 is a substrate for SIRT1 deacetylation. Additionally, we show that carboxamide-based scaffolds such as EX-527Citation36,Citation37 interfere with the ability of DBC1 to bind SIRT1 in cells. We demonstrate that the dose at which EX-527 blocks SIRT1-DBC1 binding is comparable to its IC50 for SIRT1 in vitro, suggesting that the compound acts directly on SIRT1, but that this effect is independent of DBC1 acetylation status. These findings provide insight into the formation and regulation of the SIRT1-DBC1 complex and pinpoint pharmacological scaffolds that could potentially be used to regulate the complex for the treatment of age-related diseases.
Results
We first set out to define the key structural regions in SIRT1 and DBC1 required for their interaction. Two regions on DBC1 have previously been implicated in binding to the catalytic core region of SIRT1 ().Citation17,Citation18 One report indicated that DBC1 binds to SIRT1 via the leucine zipper motif (LZ) (residues 243 to 264),Citation18 and a subsequent report indicated that residues 1–240 of DBC1 are important for the interaction.Citation25 We performed an immunoprecipitation of overexpressed Flag-tagged DBC1 (Fl-DBC1) from 293T cells, and found that while full-length DBC1 protein interacted strongly with SIRT1, a mutant DBC1 lacking the N-terminal residues 1–240 did not (), confirming the latter findings.
Figure 1. Regions on SIRT1 and DBC1 involved in complex formation. (A) Schematic of SIRT1 domains illustrating the putative DBC1 binding region and the ESA region. (B) Co-immunoprecipitation of full-length DBC1 or a truncated version lacking amino acids 1–242 with SIRT1 from 293-T cells. (C) Co-immunoprecipitation of wild type SIRT1 and SIRT1 point mutants with DBC1 from 293-T cells. (D) In vitro activity of SIRT1, SIRT1-P291Y and SIRT1-E416A purified from cells using the BIOMOL assay. Ten µM EX-527 was used as a negative control; mean + s.d. shown (n = 3). (E) Co-immunoprecipitation of DBC1 and the corresponding SIRT1 variants from (D) from 293-T cells.
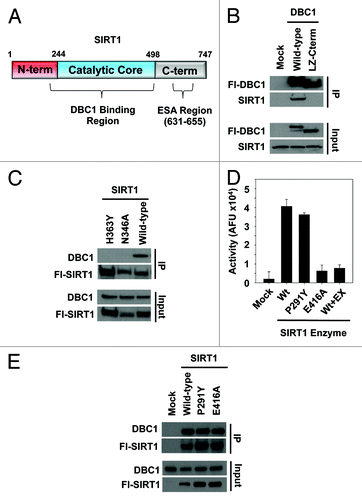
Given that the catalytic core region of SIRT1 binds to DBC1,Citation17,Citation18,Citation25 we tested the hypothesis that catalytic activity of SIRT1 is required for complex formation with DBC1. We first performed immunoprecipitation experiments with two catalytic mutants of SIRT1. Mutation of the catalytic histidine (H363Y) or a critical NAD-binding residue (N346A), which have previously been shown to result in catalytically inactive SIRT1,Citation10,Citation38 abolished SIRT1 binding to DBC1 in cells (). We next generated two additional point mutants, SIRT1-P291Y and SIRT1-E416A, to further test this hypothesis. P291 lies within a semi-conserved flexible loop region adjacent to NAD+ binding residues in SIRT1,Citation39 while E416 lies directly within the peptide binding site and has been shown to make direct contact with the acetylated substrate.Citation40 While the H363Y and N346A SIRT1 mutations () were designed to directly disrupt catalysis and NAD+ binding,Citation38 P291Y and E416A were designed to alter the loop regionCitation39 and weaken/inhibit acetyl-substrate binding to SIRT1,Citation40 respectively. We purified these mutant proteins from cell extracts and tested their deacetylase activity using an in vitro assay (). We found that while the catalytic activity of SIRT1-P291Y was only slightly decreased relative to wild-type SIRT1, the activity of SIRT1-E416A was dramatically reduced, comparable to SIRT1 activity in the presence of saturating amounts of EX-527, a potent SIRT1 inhibitorCitation36 (). As expected, both wild-type SIRT1 and SIRT1-P291Y bound DBC1 in cells (). DBC1 was also bound by SIRT1-E416A, with slightly reduced affinity relative to wild-type SIRT1, normalized to the amount of protein immunoprecipitated ().
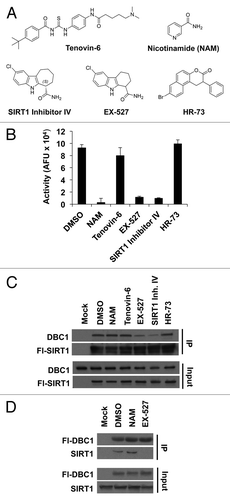
To further test the requirement of SIRT1 catalytic activity for DBC1 binding, we next tested a panel of SIRT1 inhibitors employing multiple different inhibition mechanisms for their effect on the SIRT1-DBC1 complex. Several classes of small molecules have been reported to bind and non-competitively inhibit SIRT1 (reviewed inCitation41), including nicotinamide (NAM),12N-[[[4-[[5-(dimethylamino)-1-oxopentyl]amino]phenyl]amino]thioxomethyl]-4-(1,1-dimethylethyl)-benzamide (tenovin-6), 8-Bromo-1,2-dihydro-2-phenyl-3H-naphtho[2,1-b]pyran-3-one (HR-73),Citation42 6-chloro-2,3,4,9-tetrahydro-1H-carbazole-1-carboxamide (EX-527)Citation36,Citation43 and the structurally related molecule (6S)-2-chloro-5,6,7,8,9,10-hexahydro-cyclohepta[b]indole-6-carboxamide (SIRT1 inhibitor IV)Citation36,Citation43 (). We first validated the compounds using an in vitro SIRT1 assay. We observed that while NAM, EX-527 and SIRT1 inhibitor IV displayed robust inhibition of SIRT1 in vitro, tenovin-6 had a weaker effect (consistent with its higher IC50),Citation44,Citation45 and HR73Citation42 had no effect under the conditions used (). Strikingly, we found that EX-527 and SIRT1 inhibitor IV dramatically reduced the levels of the SIRT1-DBC1 complex (), whereas NAM did not. The ability of EX-527 to inhibit SIRT1-DBC1 binding and the lack of an effect for NAM were consistent in reciprocal co-immunoprecipitation experiments ().
Figure 2. Effect of various SIRT1 inhibitors on SIRT1-DBC1 complex formation. (A) Chemical structures of SIRT1 inhibitors. (B) SIRT1 activity in the absence or presence of various inhibitors; mean + s.d. shown (n = 3). (C) Co-immunoprecipitation of Flag-wild-type SIRT1 with DBC1 from 293-T cells following 24 h treatment with the indicated compounds. (D) Co-immunoprecipitation of Flag-DBC1 and SIRT1 from 293-T cells in the presence of the indicated compounds (24 h treatment). Compound doses used in panels (B, C, and D) were NAM (20 mM), Tenovin-6 (25 µM), EX-527 (10 µM), SIRT1 Inhibitor IV (10 µM) and 15 µM HR-73.
Our SIRT1 mutant and inhibitor data were suggestive of an enzyme-substrate relationship between SIRT1 and DBC1. To explore this possibility, we first mapped post-translational modifications on DBC1 both in the presence and absence of the SIRT1 inhibitor EX-527 using LC-MS/MS. We identified seven phosphorylation sites () and four acetylation sites on DBC1 () under normal growth conditions. Phosphorylation on S640 of DBC1 was detected only in cells treated with EX-527 (). Interestingly, all four of the acetylation sites lie within the N-terminal domain in DBC1 (), which was previously shown,Citation25 and confirmed in this study (), to be involved in binding to SIRT1.
Figure 3. Post-translational modifications on DBC1 and deacetylation by SIRT1. (A) Schematic of DBC1 domains illustrating the position of acetylation (blue) and phosphorylation (green) sites. (B) Table indicating phosphorylation sites on Flag-DBC1 purified from 293-T cells, identified by LC/MS-MS following 24-h treatment with either DMSO or EX-527 (10 µM). (C) Semi-quantitative LC/MS-MS comparison of acetylation on immunoprecipitated DBC1 from 293-T cells following 24- hour treatment with DMSO or EX-527 (10 µM). (D) Acetylation analysis of immunoprecipitated Flag-DBC1 from 293-T cells treated for 24 h with NAM (20 mM), EX-527 (10 µM) or TSA (1 µM). (E) SIRT1 deacetylation of a native peptide corresponding to Ac-K215 on DBC1, measured using the PNC1-OPT assay; mean ± s.d. is shown for each point (n = 3).
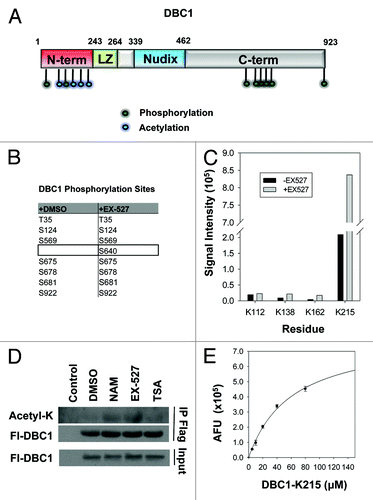
Semi-quantitative analysis of the mass spectrometry data revealed that lysine acetylation at three sites, K138, K162, and K215, but not K112, was induced by ~4-fold in the presence of EX-527, indicating that they are likely deacetylated by SIRT1 (). Treatment of cells with the pan-sirtuin inhibitor nicotinamide or the SIRT1-specific inhibitor EX-527 resulted in hyperacetylation of DBC1, while treatment with the HDAC inhibitor Trichostatin A (TSA) had little effect (). Furthermore, we found that SIRT1 could directly deacetylate a synthetic peptide corresponding to acetyl-K215, the most abundant acetylated site on DBC1 ().
We next set out to test the role of the two most abundant acetylation sites, K112 and K215, in SIRT1-DBC1 complex formation. We generated deacetyl-mimetic (K− > R) and acetyl-mimetic (K− > Q) substituted variants corresponding to each lysine position. We found that mutation of K112 or K215 to arginine did not affect the ability of DBC1 to interact with SIRT1 () but that mutation of either of these residues to glutamine substantially reduced the levels of SIRT1-DBC1 complex (). Consistent with a role for DBC1 in suppressing SIRT1 activity,Citation17,Citation18 stable overexpression of DBC1 resulted in induction of several NFκB target genes, including TNFα, IRF2 and NFκß2.Citation46 (). However, these effects were diminished in cells overexpressing an equal amount of DBC1-K112Q or DBC1-K215Q ().
Figure 4. Effects of DBC1 acetylation on SIRT1 binding and sub-cellular localization. (A) Co-immunoprecipitation of Flag-wild-type DBC1 or deacetyl-mimetics with endogenous SIRT1 in stably transduced U2OS cells. (B) Co-immunoprecipitation of Flag-wild-type DBC1 or acetyl-mimetics with endogenous SIRT1 in stably transduced U2OS cells. (C) Effect of wild-type DBC1 and acetyl-mimetics on the transcription NFκB- regulated genes in stably transduced U2OS cells; mean + s.e. is shown (n = 3). *Denotes statistical significance (p < 0.05; t-test) for empty vs. DBC1-Wt; +denotes statistical significance (p < 0.05; t-test) for DBC1-Wt vs. DBC1–112Q; and #denotes statistical significance (p < 0.05; t-test) for DBC1-Wt vs. DBC1–215Q. (D) Top: Sequence of putative NLS with K215 indicated with a blue-filled circle, and positively charged residues indicated in blue, Bottom: localization of GFP, wild-type DBC1 and acetyl-mimetics in transiently transfected 293-T cells.
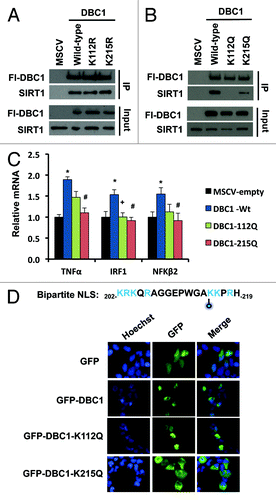
Upon inspection of the acetylation sites, we noticed that that K215 lies within an N-terminal region on DBC1 with dense positive charge (), which was previously implicated in the regulation of DBC1 localization.Citation20 Bioinformatic analysis of the DBC1 protein sequence using MyHits Motif Scan revealed that K215 does indeed fall within a putative bipartite nuclear localization sequence (, top). Thus, we hypothesized that acetylation at K215 may prevent nuclear accumulation of DBC1 and thereby prevent its binding to SIRT1. Consistent with this, GFP-DBC1-K215Q displayed cytoplasmic localization (, bottom), whereas GFP-DBC1 and GFP-K112Q-DBC1 were predominately nuclear.
Next we tested whether hyperacetylation of DBC1 could be the mechanism by which EX-527 blocks the SIRT1-DBC1 interaction. As shown in , EX-527 was still able to block SIRT1 binding to DBC1 when all four acetylated lysine residues on DBC1 had been mutated to arginine (K4R) to mimic deacetylation. Thus, EX-527 inhibits SIRT1-DBC1 complex formation independent of DBC1 acetylation status.
Figure 5. Mechanism and dose-dependence of EX-527 and C28. (A) Co-immunoprecipitation of Flag-wild-type DBC1or a non-acetylated DBC1 mutant protein (DBC1–4KR) with endogenous SIRT1 in the presence or absence of EX-527 (10 µM) following 24 h treatment. (B) Validation of the luciferase complementation assay in 293 cells using different titers of SIRT1 virus and non-binding negative control proteins; mean + s.e. is shown (n = 2). Dose-response effect of 24 h treatment with (C) EX-527 or (D) C28 (structure depicted in inset) on the SIRT1-DBC1 interaction and a control protein binding pair (Bach1-Mafk) in 293 cells; mean ± s.e. is shown (n = 2).
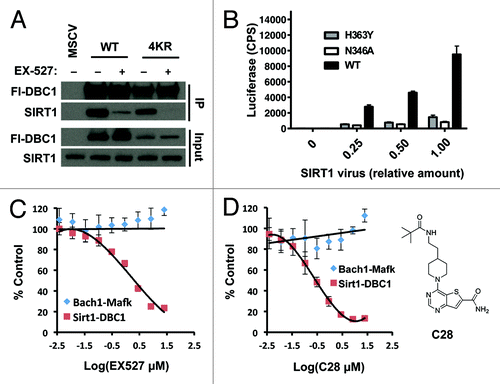
To provide a complementary assay of SIRT1-DBC1 complex formation in living cells, we developed a split-luciferase complementation binding assay that employs two catalytically inactive portions of the luciferase enzyme.Citation47,Citation48 We fused the N-terminal fragment of luciferase to SIRT1, and the C-terminal portion of luciferase to DBC1, such that upon formation of a SIRT1 and DBC1 complex, the two luciferase fragments would be brought into close proximity, forming a functional luciferase enzyme via complementation. BacMam transduction of the system into 293-T cells enabled real-time monitoring of SIRT1-DBC1 binding. Initially, to verify the functionality and specificity of our system, we transfected our DBC1-fusion component with the corresponding luciferase complement linked to wild-type SIRT1, SIRT1-H363Y, or SIRT1-N346A (see ). Consistent with the immunoprecipitation data, robust complementation was observed between the wild-type SIRT1 and DBC1 fusions, while the signal using the non-binding SIRT1 mutants was severely reduced ().
Next, we applied the system to quantitatively assess the dose-response relationship of two carboxamide inhibitors on SIRT1-DBC1 complex formation in cells, namely EX-527 and a recently described Thieno[3,2-d]pyrimidine-6-carboxamide SIRT1 inhibitor called C28.Citation37 Titrations of inhibitor spanning several orders in magnitude were performed on cells expressing either SIRT1/DBC1 luciferase-component fusions, or Bach1/Mafk-luciferase component fusions. Bach1 and Mafk have previously been reported to interact in numerous cells typesCitation49 and served as a negative control in these experiments. Treatment of cells with either EX-527 or C28 had no effect on expression levels of the various fusion proteins (Fig. S1A). Both EX-527 and C28 decreased the SIRT1-DBC1 luciferase signal in a dose-dependent manner, with an efficacy that parallels their IC50 values for SIRT1 inhibition in vitroCitation36,Citation37 (). The inhibitors had little effect on the Bach1-Mafk control luciferase complementation pair ().
Discussion
In the present work, we provide insight into the structural features of SIRT1 and DBC1 that mediate their binding, we identify DBC1 acetylation as a post-translation mechanism that regulates DBC1 localization and association with SIRT1, and we describe small molecules that perturb the DBC1-SIRT1 interaction in vivo. Our results support a SIRT1-DBC1 interaction model in which structural features within the catalytic core of SIRT1, specifically in the vicinity of the NAD+-binding region, play a critical role in mediating the interaction with the N terminus of DBC1. Several lines of evidence presented here suggest an enzyme/substrate or catalytic interaction between SIRT1 and DBC1. First, point mutations that target the catalytic core or the NAD+-binding pocket of SIRT1 both reduce DBC1 binding. Additionally, carboxamide small-molecule inhibitors of SIRT1, such as EX-527, SIRT1 inhibitor IV, and C28 all prevent binding of DBC1 to SIRT1. However, we also show that nicotinamide, used at a dose that almost completely inactivates SIRT1 in vitro, does not inhibit SIRT1-DBC1 complex formation in cells. The particular mode by which NAM inhibits SIRT1, namely by product inhibition via the C-pocket in the catalytic core,Citation12,Citation39 could be a distinguishing factor.
We also show for the first time that DBC1 is acetylated on several lysine residues within its N terminus. We present evidence that these sites can serve as substrates for SIRT1 deacetylation, and influence DBC1 subcellular localization and SIRT1 binding. It is interesting to speculate that the acetylation of DBC1 may act as a rheostat to fine-tune SIRT1 activity. In our model, low SIRT1 activity would result in accumulation of acetyl-DBC1, blocking the ability of DBC1 to bind SIRT1 and, in turn, increasing SIRT1 activity through relief of DBC1 inhibition. Conversely, high SIRT1 activity would result in the deacetylation of DBC1, leading to complex formation and inhibition of SIRT1 in a negative feedback loop. However, our results suggest that the basal levels of acetylation on DBC1 are quite low in the cell line used, and that acetylation is most likely induced by physiological stimuli yet to be identified. Moreover, residues K112 and K215 may indeed mediate distinct responses, as suggested by their different susceptibilities to SIRT1-deacetylation and the different localization of the acetyl-mimetic mutants. It will be interesting to identify whether these post-translational modifications also affect other DBC1 interactors, such as SUV39H1Citation25 and HDAC3.Citation24 Exploring if and how the acetylation sites interface with previously reported phosphorylation sites on DBC1 may also provide valuable information about the control of DBC1 by multiple intersecting pathwaysCitation50,Citation51.
In this study, we also identify three carboxamide-based molecules with the ability to reduce formation of the SIRT1-DBC1 complex in cells, namely EX-527, SIRT1 inhibitor IV and C28. These molecules have previously been shown to act directly on SIRT1,Citation36,Citation37 raising the possibility that the effects of these compounds on the SIRT1-DBC1 complex could be due to a direct interaction with SIRT1. The correlation between the dose at which EX-527 and C28 block formation of the SIRT1-DBC1 complex and their respective IC50 values for SIRT1 in vitroCitation36,Citation37 supports this possibility. Recently, a report describing the co-crystallization of EX-527 bound to the catalytic core domain of SIRT1 was published.Citation52
Given their considerable potential for treating age-related diseases, the identification and characterization of SIRT1 activators remains an active area of investigation.Citation53 Allosteric substrate-specific SIRT1 activators have been described,Citation7,Citation54 and compounds such as iso-NAM can activate SIRT1 by relieving NAM inhibition.Citation55 Potent substrate-agnostic direct SIRT1 activators, however, remain elusive. Our results suggest that it may be possible to create such compounds by relieving DBC1 inhibition. For example, carboxamide derivatives of EX-527 with the ability to bind to SIRT1 and inhibit DBC1-binding, while not inhibiting SIRT1 catalytic activity, may be feasible and would present a novel therapeutic strategy for SIRT1 activation.
Materials and Methods
Plasmids and mutagenesis
Full-length and truncated Flag-hDBC1 sequences were cloned into the XhoI and EcoRI sites of MSCV-puro to generate retroviral expression constructs, and into pEGFP-C3 for GFP imaging experiments. Site directed mutagenesis of SIRT1 and DBC1 constructs was performed using the Quickchange II XL Site Directed Mutagenesis kit (Stratagene).
Chemical reagents
All chemicals were purchased from Sigma Aldrich, with the following exceptions: EX-527 (Tocris), SIRT1 inhibitor IV (Santa Cruz), Tenovin-6 (Santa Cruz). Compound C28 was synthesized and provided by Sirtris Pharmaceuticals.
Cell culture
293-T and U20S cells were cultured in high glucose DMEM media with pyruvate (Gibco) supplemented with 10% FBS + 1× pen/strep +1× glutamine (Gibco).
Transient transfection
Transfections were performed using FuGENE HD according to the manufacturer’s instructions.
Retroviral production, infection and selection of stable cell lines
GP2–293 or Phoenix cells were co-transfected with plasmids encoding VSV-G, Gag-Pol and MSCV-puro using FuGENE HD transfection reagent, according to the manufacturer’s instructions. Twenty hours after transfection the media was replaced with fresh DMEM + 10% FBS + 1× pen/strep + 1× glutamine. Subsequently, virus was harvested between 48 and 72 h post-transfection. Infection was performed by exposing cells to filtered viral supernatants in the presence of 5 ug/mL polybrene (Sigma). Stably transduced colonies were selected 48 h later using puromycin (0.5 ug/mL).
Cell lysis/western blotting/antibodies
Cells were lysed in Triton-X lysis buffer (1% Triton-X, 50 mM Tris pH = 8, 150 mM NaCl) supplemented with protease inhibitors (Roche) for 40 min at 4°C. Cellular debris was cleared by centrifugation (14000 rpm × 10 min). SDS-PAGE loading buffer was added, and samples were subjected to electrophoresis and western blot. The following antibodies were used for detection: rabbit α-DBC1 (Bethyl Labs), rabbit α-Sir2/SIRT1 (Epitomics), rabbit α-acetylated lysine (Cell Signaling), and rabbit α-Flag (Sigma).
Co-immunoprecipitation
Clarified cell lysate was prepared as described above, and Flag-M2 agarose beads (Sigma) were added to the resulting supernatant. Immunoprecipitation was allowed to proceed for 2 h at 4 °C with gentle rotation. Subsequently, beads were washed 3× with lysis buffer. Protein was eluted from the agarose beads using either 3× FLAG peptide (Sigma) or 2× SDS-PAGE sample buffer.
In vitro measurement of SIRT1 activity on p53-FdL
Enzymatic activity of SIRT1 was assayed using the BIOMOL SIRT1-Fluor de Lys Assay. Approximately 1 µg each of wild-type or mutant protein was added to a reaction containing 200 μM β-NAD+ and 100 μM p53-FdL substrate for the assay, in the presence or absence of indicated compounds. The assay was performed according to the manufacturer’s instructions, and readings were taken on a VictorCitation3 fluorometer.
Analysis of post-translational modifications by LC-MS/MS
Stable Flag-DBC1 cell lines were washed briefly with PBS (Gibco) following treatment and lysed in Triton-X Lysis buffer (1% Triton-X, 50 mM Tris pH = 8, 150 mM NaCl) supplemented with Roche protease inhibitor cocktail, 10 μM EX-527, 20 mM NAM, 1 μM TSA and 1X Sigma phosphatase inhibitor cocktails 2 and 3 for 40 min at 4°C with gentle rotation. Cellular debris was cleared by centrifugation (14000 rpm × 10 min). Flag-M2 agarose beads (Sigma) were added to the resulting supernatant, and immunoprecipitation was allowed to proceed for 2 h at 4°C with gentle rotation. Subsequently, beads were washed 3× with lysis buffer. Protein was eluted from the agarose beads using 3X FLAG peptide (Sigma) according to the manufacturer’s instructions. Eluate was separated on an SDS-PAGE gel and then silver stained. Darkly stained bands were cut out and subject to analysis by LC-MS/MS.
In vitro measurement of SIRT1 activity on native DBC1 peptide
Deacetylation of the native K215 DBC1-derived peptide (Ac-KQRAGGEPWGAK(Ac)KPRHDLPPYR-NH2; synthesized by Peptide 2.0) was measured using the PNC1-OPT assay (commercially available as the Millipore SIRTainty assay), as previously described.Citation5,Citation56 Briefly, 1 µg of SIRT1 and PNC1 were used per reaction in the presence of 1 mM β-NAD+, and the indicated peptide concentration. Reactions were performed in Dulbecco’s PBS buffer (Gibco) supplemented with 1 mM DTT, and developed and read as previously described.Citation56
Gene expression analysis/qRT-PCR
RNA from U2OS cells was extracted using an RNeasy kit (Qiagen) according to the manufacturer’s instructions and was quantified using the NanoDrop 1000 spectrophotometer (Thermo Scientific). cDNA was synthesized with the iSCRIP cDNA synthesis kit (BioRad) using 200 ng of RNA. Quantitative RT-PCR reactions were performed using 1 µM of primers and LightCycler® 480 SYBR Green Master (Roche) on a LightCycler® 480 detection system (Roche). Calculations were performed by a comparative method (2−ΔΔCT) using 18S RNA as an internal control. The following sets of primers were used: TNFα - 5′ACTTTGGAGTGATCGGCC′3 (fwd) and 5′GCTTGAGGGTTTGCTACAAC′3 (rev), IRF1 - 5′AGTGATCTGTACAACTTCCAGG′3 (fwd) and 5′CCTTCCTCATCCTCATCTGTTG′3 (rev), NFκβ2 -
5′ GAAGCCAGTCATCTCCCAG′3 (fwd) and CATCTTTCTGCACCTTGTCAC (rev), 18S - 5′CGGCTACCACATCCAAGGAA′3 (fwd) and 5′GCTGGAATTACCGCGGCT ′3 (rev).
GFP imaging
Following transfection of GFP-tagged plasmids, cells were treated with cell permeable Hoechst 33342 stain (Invitrogen) for 1 h according to the manufacturer’s instructions, and washed for 1 h prior to being examined using a Nikon TiE fluorescent microscope.
Luciferase complementation system
The BacMam/luciferase complementation system was set-up as previously described.Citation47,Citation48 HEK293 cells were transduced with BacMam viruses: SIRT1.luc2N at the percentages indicated (0.5% for the EX-527 dose-response) and DBC1.luc2C fixed at 0.5%. The cells were plated into 96-well plates at 100 μL of 2 × 105 cells/mL in each well and incubated with virus overnight. The following day, 100 μL of Steady Glo Luciferin reagent (Promega) was added to each well and the plate was read on a BMGLabtech Pherastar reader.
Li-Cor protein blot of luciferase-fusion proteins
Cells transduced with Sirt1.lucN and DBC1.lucC bacmams were lysed in RIPA buffer containing HALT protease and phosphatase inhibitors (Thermo Pierce). Total cell lysates were loaded onto a 4–12% gel and blotted with anti-luciferase antibody (Promega) and IR800-labeled anti-goat secondary antibody (Jackson Labs). Bands were imaged on a Li-Cor instrument.
Additional material
Download Zip (105 KB)Acknowledgments
This work was supported by grants from the Glenn Foundation for Medical Research, the Ellison Medical Foundation, The Juvenile Diabetes Foundation, The United Mitochondrial Disease Foundation, NIA/NIH grants to DAS and by an NSERC PGS-D Fellowship to BPH.
Disclosure of Potential Conflicts of Interest
CL, JSD, JLE, and GPV are employees of Sirtris, a GSK company, and QL and TLGD are employees of GSK. DAS is a consultant and is an inventor on patents licensed to Sirtris, a GSK company.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/25268
References
- Picard F, Kurtev M, Chung N, Topark-Ngarm A, Senawong T, Machado De Oliveira R, et al. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004; 429:771 - 6; http://dx.doi.org/10.1038/nature02583; PMID: 15175761
- Fulco M, Cen Y, Zhao P, Hoffman EP, McBurney MW, Sauve AA, et al. Glucose restriction inhibits skeletal myoblast differentiation by activating SIRT1 through AMPK-mediated regulation of Nampt. Dev Cell 2008; 14:661 - 73; http://dx.doi.org/10.1016/j.devcel.2008.02.004; PMID: 18477450
- Biason-Lauber A, Böni-Schnetzler M, Hubbard BP, Bouzakri K, Brunner A, Cavelti-Weder C, et al. Identification of a SIRT1 mutation in a family with type 1 diabetes. Cell Metab 2013; 17:448 - 55; http://dx.doi.org/10.1016/j.cmet.2013.02.001; PMID: 23473037
- Bordone L, Cohen D, Robinson A, Motta MC, van Veen E, Czopik A, et al. SIRT1 transgenic mice show phenotypes resembling calorie restriction. Aging Cell 2007; 6:759 - 67; http://dx.doi.org/10.1111/j.1474-9726.2007.00335.x; PMID: 17877786
- Hubbard BP, Gomes AP, Dai H, Li J, Case AW, Considine T, et al. Evidence for a common mechanism of SIRT1 regulation by allosteric activators. Science 2013; 339:1216 - 9; http://dx.doi.org/10.1126/science.1231097; PMID: 23471411
- Imai S, Kiess W. Therapeutic potential of SIRT1 and NAMPT-mediated NAD biosynthesis in type 2 diabetes. Front Biosci 2009; 14:2983 - 95; http://dx.doi.org/10.2741/3428; PMID: 19273250
- Milne JC, Lambert PD, Schenk S, Carney DP, Smith JJ, Gagne DJ, et al. Small molecule activators of SIRT1 as therapeutics for the treatment of type 2 diabetes. Nature 2007; 450:712 - 6; http://dx.doi.org/10.1038/nature06261; PMID: 18046409
- Han J, Hubbard BP, Lee J, Montagna C, Lee HW, Sinclair DA, et al. Analysis of 41 cancer cell lines reveals excessive allelic loss and novel mutations in the SIRT1 gene. Cell Cycle 2013; 12:263 - 70; http://dx.doi.org/10.4161/cc.23056; PMID: 23255128
- Firestein R, Blander G, Michan S, Oberdoerffer P, Ogino S, Campbell J, et al. The SIRT1 deacetylase suppresses intestinal tumorigenesis and colon cancer growth. PLoS One 2008; 3:e2020; http://dx.doi.org/10.1371/journal.pone.0002020; PMID: 18414679
- Kim D, Nguyen MD, Dobbin MM, Fischer A, Sananbenesi F, Rodgers JT, et al. SIRT1 deacetylase protects against neurodegeneration in models for Alzheimer’s disease and amyotrophic lateral sclerosis. EMBO J 2007; 26:3169 - 79; http://dx.doi.org/10.1038/sj.emboj.7601758; PMID: 17581637
- Sauve AA. Pharmaceutical strategies for activating sirtuins. Curr Pharm Des 2009; 15:45 - 56; http://dx.doi.org/10.2174/138161209787185797; PMID: 19149602
- Bitterman KJ, Anderson RM, Cohen HY, Latorre-Esteves M, Sinclair DA. Inhibition of silencing and accelerated aging by nicotinamide, a putative negative regulator of yeast sir2 and human SIRT1. J Biol Chem 2002; 277:45099 - 107; http://dx.doi.org/10.1074/jbc.M205670200; PMID: 12297502
- Yang Y, Fu W, Chen J, Olashaw N, Zhang X, Nicosia SV, et al. SIRT1 sumoylation regulates its deacetylase activity and cellular response to genotoxic stress. Nat Cell Biol 2007; 9:1253 - 62; http://dx.doi.org/10.1038/ncb1645; PMID: 17934453
- Sasaki T, Maier B, Koclega KD, Chruszcz M, Gluba W, Stukenberg PT, et al. Phosphorylation regulates SIRT1 function. PLoS One 2008; 3:e4020; http://dx.doi.org/10.1371/journal.pone.0004020; PMID: 19107194
- Kang H, Jung JW, Kim MK, Chung JH. CK2 is the regulator of SIRT1 substrate-binding affinity, deacetylase activity and cellular response to DNA-damage. PLoS One 2009; 4:e6611; http://dx.doi.org/10.1371/journal.pone.0006611; PMID: 19680552
- Kim EJ, Kho JH, Kang MR, Um SJ. Active regulator of SIRT1 cooperates with SIRT1 and facilitates suppression of p53 activity. Mol Cell 2007; 28:277 - 90; http://dx.doi.org/10.1016/j.molcel.2007.08.030; PMID: 17964266
- Zhao W, Kruse JP, Tang Y, Jung SY, Qin J, Gu W. Negative regulation of the deacetylase SIRT1 by DBC1. Nature 2008; 451:587 - 90; http://dx.doi.org/10.1038/nature06515; PMID: 18235502
- Kim JE, Chen J, Lou Z. DBC1 is a negative regulator of SIRT1. Nature 2008; 451:583 - 6; http://dx.doi.org/10.1038/nature06500; PMID: 18235501
- Hamaguchi M, Meth JL, von Klitzing C, Wei W, Esposito D, Rodgers L, et al. DBC2, a candidate for a tumor suppressor gene involved in breast cancer. Proc Natl Acad Sci USA 2002; 99:13647 - 52; http://dx.doi.org/10.1073/pnas.212516099; PMID: 12370419
- Sundararajan R, Chen G, Mukherjee C, White E. Caspase-dependent processing activates the proapoptotic activity of deleted in breast cancer-1 during tumor necrosis factor-alpha-mediated death signaling. Oncogene 2005; 24:4908 - 20; http://dx.doi.org/10.1038/sj.onc.1208681; PMID: 15824730
- Trauernicht AM, Kim SJ, Kim NH, Boyer TG. Modulation of estrogen receptor alpha protein level and survival function by DBC-1. Mol Endocrinol 2007; 21:1526 - 36; http://dx.doi.org/10.1210/me.2007-0064; PMID: 17473282
- Trauernicht AM, Kim SJ, Kim NH, Clarke R, Boyer TG. DBC-1 mediates endocrine resistant breast cancer cell survival. Cell Cycle 2010; 9:1218 - 9; http://dx.doi.org/10.4161/cc.9.6.11010; PMID: 20237431
- Menssen A, Hydbring P, Kapelle K, Vervoorts J, Diebold J, Lüscher B, et al. The c-MYC oncoprotein, the NAMPT enzyme, the SIRT1-inhibitor DBC1, and the SIRT1 deacetylase form a positive feedback loop. Proc Natl Acad Sci USA 2012; 109:E187 - 96; http://dx.doi.org/10.1073/pnas.1105304109; PMID: 22190494
- Chini CC, Escande C, Nin V, Chini EN. HDAC3 is negatively regulated by the nuclear protein DBC1. J Biol Chem 2010; 285:40830 - 7; http://dx.doi.org/10.1074/jbc.M110.153270; PMID: 21030595
- Li Z, Chen L, Kabra N, Wang C, Fang J, Chen J. Inhibition of SUV39H1 methyltransferase activity by DBC1. J Biol Chem 2009; 284:10361 - 6; http://dx.doi.org/10.1074/jbc.M900956200; PMID: 19218236
- Chini CC, Escande C, Nin V, Chini EN. DBC1 (Deleted in Breast Cancer 1) modulates the stability and function of the nuclear receptor Rev-erbα. Biochem J 2013; 451:453 - 61; http://dx.doi.org/10.1042/BJ20121085; PMID: 23398316
- Close P, East P, Dirac-Svejstrup AB, Hartmann H, Heron M, Maslen S, et al. DBIRD complex integrates alternative mRNA splicing with RNA polymerase II transcript elongation. Nature 2012; 484:386 - 9; http://dx.doi.org/10.1038/nature10925; PMID: 22446626
- Kang H, Suh JY, Jung YS, Jung JW, Kim MK, Chung JH. Peptide switch is essential for Sirt1 deacetylase activity. Mol Cell 2011; 44:203 - 13; http://dx.doi.org/10.1016/j.molcel.2011.07.038; PMID: 22017869
- Raynes R, Pombier KM, Nguyen K, Brunquell J, Mendez JE, Westerheide SD. The SIRT1 modulators AROS and DBC1 regulate HSF1 activity and the heat shock response. PLoS One 2013; 8:e54364; http://dx.doi.org/10.1371/journal.pone.0054364; PMID: 23349863
- Escande C, Chini CC, Nin V, Dykhouse KM, Novak CM, Levine J, et al. Deleted in breast cancer-1 regulates SIRT1 activity and contributes to high-fat diet-induced liver steatosis in mice. J Clin Invest 2010; 120:545 - 58; http://dx.doi.org/10.1172/JCI39319; PMID: 20071779
- Qiang L, Wang L, Kon N, Zhao W, Lee S, Zhang Y, et al. Brown remodeling of white adipose tissue by SirT1-dependent deacetylation of Pparγ. Cell 2012; 150:620 - 32; http://dx.doi.org/10.1016/j.cell.2012.06.027; PMID: 22863012
- Kim JE, Lou Z, Chen J. Interactions between DBC1 and SIRT 1 are deregulated in breast cancer cells. Cell Cycle 2009; 8:3784 - 5; http://dx.doi.org/10.4161/cc.8.22.10055; PMID: 19855164
- Nin V, Escande C, Chini CC, Giri S, Camacho-Pereira J, Matalonga J, et al. Role of deleted in breast cancer 1 (DBC1) protein in SIRT1 deacetylase activation induced by protein kinase A and AMP-activated protein kinase. J Biol Chem 2012; 287:23489 - 501; http://dx.doi.org/10.1074/jbc.M112.365874; PMID: 22553202
- Yuan J, Luo K, Liu T, Lou Z. Regulation of SIRT1 activity by genotoxic stress. Genes Dev 2012; 26:791 - 6; http://dx.doi.org/10.1101/gad.188482.112; PMID: 22465953
- Donmez G, Guarente L. Aging and disease: connections to sirtuins. Aging Cell 2010; 9:285 - 90; http://dx.doi.org/10.1111/j.1474-9726.2010.00548.x; PMID: 20409078
- Solomon JM, Pasupuleti R, Xu L, McDonagh T, Curtis R, DiStefano PS, et al. Inhibition of SIRT1 catalytic activity increases p53 acetylation but does not alter cell survival following DNA damage. Mol Cell Biol 2006; 26:28 - 38; http://dx.doi.org/10.1128/MCB.26.1.28-38.2006; PMID: 16354677
- Disch JS, Evindar G, Chiu CH, Blum CA, Dai H, Jin L, et al. Discovery of Thieno[3,2-d]pyrimidine-6-carboxamides as Potent Inhibitors of SIRT1, SIRT2, and SIRT3. J Med Chem 2013; 56:3666 - 79; http://dx.doi.org/10.1021/jm400204k; PMID: 23570514
- Finnin MS, Donigian JR, Pavletich NP. Structure of the histone deacetylase SIRT2. Nat Struct Biol 2001; 8:621 - 5; http://dx.doi.org/10.1038/89668; PMID: 11427894
- Avalos JL, Bever KM, Wolberger C. Mechanism of sirtuin inhibition by nicotinamide: altering the NAD(+) cosubstrate specificity of a Sir2 enzyme. Mol Cell 2005; 17:855 - 68; http://dx.doi.org/10.1016/j.molcel.2005.02.022; PMID: 15780941
- Cosgrove MS, Bever K, Avalos JL, Muhammad S, Zhang X, Wolberger C. The structural basis of sirtuin substrate affinity. Biochemistry 2006; 45:7511 - 21; http://dx.doi.org/10.1021/bi0526332; PMID: 16768447
- Blum CA, Ellis JL, Loh C, Ng PY, Perni RB, Stein RL. SIRT1 modulation as a novel approach to the treatment of diseases of aging. J Med Chem 2011; 54:417 - 32; http://dx.doi.org/10.1021/jm100861p; PMID: 21080630
- Pagans S, Pedal A, North BJ, Kaehlcke K, Marshall BL, Dorr A, et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol 2005; 3:e41; http://dx.doi.org/10.1371/journal.pbio.0030041; PMID: 15719057
- Napper AD, Hixon J, McDonagh T, Keavey K, Pons JF, Barker J, et al. Discovery of indoles as potent and selective inhibitors of the deacetylase SIRT1. J Med Chem 2005; 48:8045 - 54; http://dx.doi.org/10.1021/jm050522v; PMID: 16335928
- Medda F, Russell RJ, Higgins M, McCarthy AR, Campbell J, Slawin AM, et al. Novel cambinol analogs as sirtuin inhibitors: synthesis, biological evaluation, and rationalization of activity. J Med Chem 2009; 52:2673 - 82; http://dx.doi.org/10.1021/jm8014298; PMID: 19419202
- Pirrie L, McCarthy AR, Major LL, Morkūnaitė V, Zubrienė A, Matulis D, et al. Discovery and validation of SIRT2 inhibitors based on tenovin-6: use of a ¹H-NMR method to assess deacetylase activity. Molecules 2012; 17:12206 - 24; http://dx.doi.org/10.3390/molecules171012206; PMID: 23079492
- Yeung F, Hoberg JE, Ramsey CS, Keller MD, Jones DR, Frye RA, et al. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBO J 2004; 23:2369 - 80; http://dx.doi.org/10.1038/sj.emboj.7600244; PMID: 15152190
- Luker KE, Smith MC, Luker GD, Gammon ST, Piwnica-Worms H, Piwnica-Worms D. Kinetics of regulated protein-protein interactions revealed with firefly luciferase complementation imaging in cells and living animals. Proc Natl Acad Sci USA 2004; 101:12288 - 93; http://dx.doi.org/10.1073/pnas.0404041101; PMID: 15284440
- Remy I, Michnick SW. A highly sensitive protein-protein interaction assay based on Gaussia luciferase. Nat Methods 2006; 3:977 - 9; http://dx.doi.org/10.1038/nmeth979; PMID: 17099704
- Toki T, Katsuoka F, Kanezaki R, Xu G, Kurotaki H, Sun J, et al. Transgenic expression of BACH1 transcription factor results in megakaryocytic impairment. Blood 2005; 105:3100 - 8; http://dx.doi.org/10.1182/blood-2004-07-2826; PMID: 15613547
- Stokes MP, Rush J, Macneill J, Ren JM, Sprott K, Nardone J, et al. Profiling of UV-induced ATM/ATR signaling pathways. Proc Natl Acad Sci USA 2007; 104:19855 - 60; http://dx.doi.org/10.1073/pnas.0707579104; PMID: 18077418
- Beausoleil SA, Jedrychowski M, Schwartz D, Elias JE, Villén J, Li J, et al. Large-scale characterization of HeLa cell nuclear phosphoproteins. Proc Natl Acad Sci USA 2004; 101:12130 - 5; http://dx.doi.org/10.1073/pnas.0404720101; PMID: 15302935
- Zhao X, Allison D, Condon B, Zhang F, Gheyi T, Zhang A, et al. The 2.5 Å crystal structure of the SIRT1 catalytic domain bound to nicotinamide adenine dinucleotide (NAD+) and an indole (EX527 analogue) reveals a novel mechanism of histone deacetylase inhibition. J Med Chem 2013; 56:963 - 9; http://dx.doi.org/10.1021/jm301431y; PMID: 23311358
- Baur JA. Biochemical effects of SIRT1 activators. Biochim Biophys Acta 2010; 1804:1626 - 34; http://dx.doi.org/10.1016/j.bbapap.2009.10.025; PMID: 19897059
- Howitz KT, Bitterman KJ, Cohen HY, Lamming DW, Lavu S, Wood JG, et al. Small molecule activators of sirtuins extend Saccharomyces cerevisiae lifespan. Nature 2003; 425:191 - 6; http://dx.doi.org/10.1038/nature01960; PMID: 12939617
- Sauve AA, Moir RD, Schramm VL, Willis IM. Chemical activation of Sir2-dependent silencing by relief of nicotinamide inhibition. Mol Cell 2005; 17:595 - 601; http://dx.doi.org/10.1016/j.molcel.2004.12.032; PMID: 15721262
- Yang Y, Hubbard BP, Sinclair DA, Tong Q. Characterization of murine SIRT3 transcript variants and corresponding protein products. J Cell Biochem 2010; 111:1051 - 8; http://dx.doi.org/10.1002/jcb.22795; PMID: 20677216