Abstract
Renal cell carcinomas (RCCs) are frequently occurring genitourinary malignancies in the aged population. A morphological characteristic of RCCs is an irregular nuclear shape, which is used to index cancer grades. Other features of RCCs include the genetic inactivation of the von Hippel-Lindau gene, VHL, and p53 genetic-independent inactivation. An aberrant nuclear shape or p53 suppression has not yet been demonstrated. We examined the effect of progerin (an altered splicing product of the LMNA gene linked to Hutchinson Gilford progeria syndrome; HGPS) on the nuclear deformation of RCCs in comparison to that of HGPS cells. In this study, we showed that progerin was suppressed by pVHL and was responsible for nuclear irregularities as well as p53 inactivation. Thus, progerin suppression can ameliorate nuclear abnormalities and reactivate p53 in response to genotoxic addition. Furthermore, we found that progerin was a target of pVHL E3 ligase and suppressed p53 activity by p14/ARF inhibition. Our findings indicate that the elevated expression of progerin in RCCs results from the loss of pVHL and leads to p53 inactivation through p14/ARF suppression. Interestingly, we showed that progerin was expressed in human leukemia and primary cell lines, raising the possibility that the expression of this LMNA variant may be a common event in age-related cancer progression.
Introduction
Although cancer incidence is obviously increased in the aged population, a molecular mechanism that links the aging process and cancer has not yet been clearly demonstrated. A multistep carcinogenesis model has been proposed to explain aging and tumor formation.Citation1 According to this model, several types of genetic mutations (including mutations in Ras, Smad4, p53, etc.) are collectively required for transformation into a fully tumorigenic cell over a long period of time. However, in inherited cancers, such as Li-Fraumeni and von Hippel-Lindau (VHL) syndromes,Citation2,Citation3 tumor formation often occurs within the third decade of life, suggesting that the inactivation of certain gene(s) can accelerate tumor formation.
The recent finding that p53 function declines during the aging processCitation4 has led to another model to explain age-associated cancer incidence. Because p53 is a tumor suppressor, the reduction of p53 function may lead to accelerated mutation and cancer development. These results imply that age-induced cellular changes and cancer susceptibility would be promoted by the suppression of tumor suppressor genes, such as p53 or VHL.
Renal cell carcinomas (RCCs) are well known age-related cancers.Citation5 In addition, RCCs share common features, including nuclear irregularities and resistance to ionizing radiation treatment. However, p53 mutations, which lead to IR resistance in other cancers, occur with a very low incidence in RCCs.Citation6 These features led us to speculate that there is a novel mechanism that can suppress p53 function in RCCs.
One of the most frequently detected genetic events in RCCs (over 70% of primary cancers) is the mutation of the von Hippel Lindau gene, VHL.Citation2,Citation7 As an E3 ligase, pVHL blocks HIF-1α activity by promoting proteasome-dependent degradation.Citation8 Because HIF-1α induces vascular endothelial cell growth factor (VEGF), erythropoietin (EPO), and other pro-angiogenic factors in response to hypoxia,Citation9,Citation10 the activation of HIF-1α is required in advancing tumors, which is located in an avascular condition. Although angiogenesis is thought to be important at the late stage of cancerCitation11 and is able to supply enough oxygen and nutrients, it is puzzling why VHL mutation is an early event in RCCs, considering that the kidney possesses well-organized blood vessels. In fact, VHL deletion is not detected in other types of invasive cancers.Citation12 Thus, we speculate that pVHL may have other targets relevant to RCC formation in the early stage of cancer.
A-type lamins are nuclear membrane proteins encoded by the LMNA locus.Citation13,Citation14 Genetic mutations of LMNA occur in several different human diseases, including Hutchinson Gilford progeria syndrome.Citation15,Citation16 The most common HGPS mutant allele, G608G, does not change an amino acid but produces a novel splicing donor site, leading to a smaller Lamin A product, termed progerin.Citation15,Citation16 One of the well-defined features of HGPS is the nuclear deformation, which is also observed in aged normal fibroblasts.Citation17
Given that the incidence of RCCs is dramatically increased in the aging population, p53 function declines without genetic mutation in aging cells and that VHL is frequently mutated at the early stage of RCCs, we proposed the hypothesis that the loss of pVHL would be related with aging-related gene expression, which can suppress p53 function. To explore this hypothesis, we focused on the nuclear irregularity of RCCs, which resembles the nuclear deformation observed in Hutchinson-Gilford progeroid syndrome.Citation15,Citation16 Moreover, it has been reported that progerin, a causal gene of HGPS, is expressed in aged cells.Citation17 Here, we demonstrate the connection between progerin and the nuclear irregularity of RCCs cells. In addition, we reveal that progerin can suppress p53 function through the inactivation of p14/ARF.
Results
Elevated expression of progerin in Renal Cell Carcinomas
Because the nuclear irregularities of RCCs and the nuclear deformation of HGPS appear to be similar, we examined the nuclear morphology of RCC cell lines by staining with Lamin A/C antibody. Consistent with previous studies,Citation5 the human RCC cell lines UMRC2 (C2) and Caki-2 showed the similar nuclear morphology with HGPS cells (; Fig. S1A). Because the nuclear irregularity (so called nuclear deformation) of the HGPS cells resulted from elevated progerin expression, we checked the expression of progerin in the RCC cell line using RT-PCR. Although HGPS and aged normal cells showed a high level of progerin expression,Citation17 other human cancer cell lines did not show a distinguishable difference at the transcription level (). In contrast, protein expression exhibited a dramatic difference between some types of RCC (Caki-2, C2, A498, and A704), and non-RCC cell lines (A549 and HCT116) or other types of RCC cell lines (C2V and ACHN; ). Concerning human tumor cell lines, progerin expression displayed a mutually exclusive pattern with pVHL expression (). Indeed, A549 and HCT116 did not show the nuclear irregularity or nuclear deformation (data not shown). To address the relevance between progerin expression and the nuclear irregularity of RCC, we stained the Lamin A/C in the RCC cell lines and found that VHL-wild-type-ACHN showed the normal nuclear morphology, whereas A498 and A704 showed the nuclear irregularity (Fig. S1B). However, the nuclear morphology of RCC was not related to the aging of patients but was related to the genetic status of VHL (Fig. S1B and C). Since we observed the increase of progerin by treatment of nocodazole or colcemide (our unpublished data), we examined the expression of progerin in several kinds of cell lines after treatment with nocodazole or colcemide. As we expected, progerin expression was increased in nocodazole- or colcemide-treated C2, Caki-2, HGPS, and A498 cells but not in ACHN (Fig. S1D and E). To address the role of progerin in the nuclear irregularity of RCC, we generated si-RNA against progerin as previously describedCitation18 () and checked its effect in HGPS cells. As we expected, si-progerin ameliorated the nuclear deformation (; Fig. S2B). Reversely, progerin-transfection into 293 could induce nuclear deformation (). We next examined the nuclear morphology of RCC cell lines. Progerin knockdown rescued the defect of the nuclear morphology in RCC cell lines (; Fig. S2C and D). Finally, we checked the effect of a farnesyltransferase inhibitor (FTI) on the nuclear morphology because it can ameliorate the nuclear deformation in HGPS cells.Citation14 As we expected, FTI could block the nuclear defect in RCC cell lines (Fig. S2E). These results suggest that the nuclear irregularity of HGPS and RCC cells is commonly induced by progerin expression.
Figure 1. Nuclear irregularity of RCCs is responsible for progerin. (A) The irregularity of the nuclear membrane in human RCC cell line UMRC2 (C2). C2 cells were fixed and stained with Lamin A/C (LMA; green). Nuclear DNA was stained with DAPI (blue). Left panel shows the nuclear morphology of normal fibroblasts (29-y-old healthy subject). (B) Nuclear deformation in HGPS (HS). Cells were also stained with Lamin A/C (LMA; green) and DAPI. (C) Transcriptional expression of progerin (PG) in human cancer cell lines. Expression of progerin was determined by RT-PCR in RCC cell lines (Caki-2, C2, C2V, ACHN, A498, and A704) and non-RCC cell lines (A549 and HCT116) or normal fibroblasts (81-y-old and 29-y-old subjects) compared with HGPS cells (positive control). Because progerin is partially deleted from Lamin A (intra-deletion of 150 bp; arrow), the smaller Lamin A PCR product is progerin (arrowhead). We also confirm that this small band is progerin by cloning-sequencing method. In addition, we showed the relative expression of progerin regarding GAPDH as a graph using a densitometer (see below). (D) Progerin (PG) expression at the translational level in RCC cell lines. WB analysis showed the expression of progerin in C2, Caki-2, A704, and A498 cell lines, but not in A549 and HCT116 at the translation level. HGPS and normal fibroblasts were used for positive and negative controls for PG. We also showed the expression of pVHL and Lamin A/C. Actin was used as a loading control. (E) Si-progerin (Si-PG) ameliorates the nuclear deformation of HGPS (HS). HGPS cells were stained with Lamin A/C after transfection with si-progerin (Si-PG). Si-C indicates scrambled si-RNA. In contrast, progerin transfection induces nuclear deformation in 293 cells (bottom). (F) Si-progerin (Si-PG) ameliorates the nuclear deformation of RCCs. C2 and Caki-2 cells were transfected with si-progerin for 24 h and stained with Lamin A/C and DAPI.
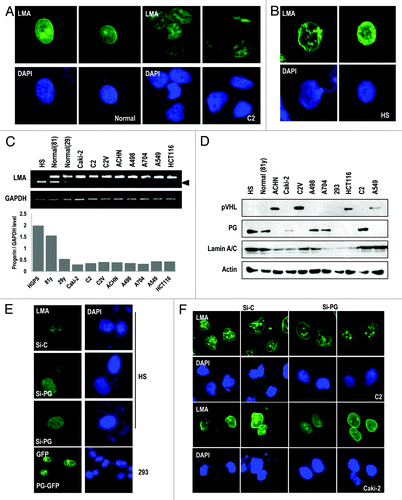
Figure 2. pVHL suppresses progerin expression. (A) pVHL suppresses progerin (PG) expression. Instead, the expression of other Lamin family proteins is not affected by pVHL. C2 cells were co-transfected with pVHL and the indicated GFP-Lamin A (LMA), Lamin B (LMB), Lamin C (LMC), or progerin (PG) vectors for 24 h. Because we used the GFP-fused vectors, the expression of each gene was examined with the GFP antibody. EV indicates empty-vector transfection. (B) Si-VHL induces progerin (PG) expression. C2V cells were transfected with indicating vectors or si-RNA for 24 h. “L. E” and “S. E” indicate longer exposed and shorter exposed results, respectively. (C) Endogenous progerin (PG) is reduced by pVHL overexpression. A704 and A498 cells were transfected with pVHL for 24 h. (D) Si-VHL induces endogenous progerin (PG) expression in C2V. C2V cells were transfected with si-C or si-VHL for 24 h and incubated with Nocodazole (Noc) or colcemide (Col) for 12 h. (E) pVHL suppress the half-life of progerin. 293 cells were transfected with progerin and pVHL or si-VHL for 24 h. After washing, cells were incubated with CHX (de novo protein synthesis inhibitor) for the indicated time. In pVHL-transfected cells, progerin’s half-life was reduced to 3.5 h, whereas si-pVHL extended it up to 24 h. We also provided the graphic analysis data in Figure S4B. (F) Proteasome inhibitor can block pVHL-induced progerin suppression. 293 cells, transfected with indicating vectors, were incubated with ALLN (20 μM) for the indicated time. In pVHL- or EV-transfected cells, progerin expression was increased. However, ALLN did not show this effect in si-pVHL-transfceted cells. (G) pVHL promotes progerin (PG) ubiquitination. 293 cells were co-transfected with HA-Ub, progerin and/or pVHL for 24 h and incubated with ALLN for additional 12 h. Cell lysates were subjected to IP with GFP Ab and WB with HA-Ub. (H) pVHL mutants do not suppress progerin expression. HA-tagged wild-type (WT-VHL) or mutant pVHL (C162F; 162, R167W; 167) were co-transfected with GFP-progerin (GFP-PG) for 24 h. (I) pVHL can ameliorate progerin-induced nuclear deformation. 293 cells were co-transfected with VHL and non-tagged progerin for 24 h. Differently from EV-transfected cells, WT-pVHL blocks progerin-induced nuclear deformation. Cells were fixed with Me-OH and stained with Lamin A/C (green) and HA (red). DAPI staining was used to visualize the DNA.
pVHL regulates progerin expression
In aged cells, it has been reported that progerin is basally expressed without genetic mutation,Citation17 suggesting that progerin is involved in the normal aging process.Citation20 In fact, we observed an increase of progerin expression in aged normal fibroblasts (). However, the reasons why RCC exhibits an elevated expression of progerin have not been addressed. Because pVHL is frequently mutated in RCCs and the genetic status of VHL showed a linkage with the nuclear deformation (; Fig. S1C), we first examined the effect of pVHL on the nuclear irregularity and progerin expression. Compared with the nuclear morphology of C2, C2V (pVHL-stable transfected UMRC2) showed the normal nuclear morphology (Fig. S3A). To examine the relationship between pVHL and progerin, we measured progerin expression in C2 and C2V, and found that C2V showed extremely low progerin expression (Fig. S3B). To confirm this, we measured the expression of progerin after co-transfection of pVHL with progerin or other Lamin family genes (Lamin A, B, and C) and found that VHL expression selectively led to reduced progerin expression (). In contrast, knockdown of pVHL using siRNA (Fig. S3C) increased progerin levels in C2V (Fig. S2B). However, other Lamin family members, including Lamin A, B, and C, were not obviously affected by pVHL expression (). Indeed, overexpression of pVHL reduced endogenous progerin expression in A704 and A498 (). We also obtained a similar result from the A549 cell line, in which pVHL suppressed progerin expression (Fig. S3D and E). To confirm this result, we measured the expression of endogenous progerin in C2V after the knockdown of pVHL. As we expected, knockdown of VHL induced progerin expression (). Because pVHL suppresses HIF-1α and HIF-2α expression,Citation8,Citation21-Citation23 we tested the involvement of HIF-1α and HIF-2α in pVHL-mediated progerin regulation. However, HIF-1α overexpression or knockdown using si-RNA (Fig. S3F) did not significantly alter progerin expression (Fig. S3G and H). In addition, si-HIF2α did not alter the expression of progerin (Fig. S3I). These results indicate that pVHL regulates progerin through a HIF family protein-independent pathway. We next examined the mechanism of pVHL-mediated progerin regulation. First, we examined progerin transcripts. However, progerin mRNA levels were not suppressed by pVHL transfection (Fig. S4A). This result is consistent with our previous results () and indicates that the regulation of progerin occurs at the post-translational level. Because pVHL is an E3 ligase,Citation24 we examined the effect of pVHL on the stability of progerin by pulse-chase analysis and found that pVHL levels were inversely correlated with progerin half-life (; Fig. S4B). However, pVHL also showed the reduction, coupling with PG (, lanes 6–8). About this, we speculated that pVHL is degraded with its target. In fact, we previously observed the similar feature when pVHL eliminates ER-α.Citation25 But, more detail working mechanism should be investigated. Anyway, the half-life of progerin (approximately 20 h) was clearly shortened to 3.5 h by pVHL transfection (Fig. S4B). In addition, a proteasome inhibitor was able to overcome the pVHL-induced progerin suppression (). However, consistent with our previous result (), the half-life of Lamin A was not reduced by pVHL (Fig. S4C). In addition, we observed an increase of Ub-conjugated progerin in pVHL-transfected cells (), indicating that progerin is a substrate for the E3 ligase activity of pVHL. To confirm the role of pVHL in the regulation of progerin, we tested the effect of mutant pVHL on progerin expression. In contrast to wild-type pVHL, E3 ligase-defective pVHL mutants did not suppress progerin expression (). Next, we examined the effect of pVHL on the nuclear deformation using progerin-transfected cells. Progerin-induced nuclear deformation was ameliorated by wild-type pVHL transfection (), while this deformation was not rescued by mutant pVHL (Fig. S4D). To confirm this rescue, we examined the effect of pVHL on the nuclear deformation in HGPS cells through IF staining of Lamin A/C. Similar to the results observed in RCC cells, pVHL overexpression ameliorated the defects in the nuclear morphology (Fig. S4E and F). These results suggest that pVHL can suppress progerin expression through its E3 ligase ability.
Direct interaction between pVHL and progerin
E3 ligases generally interact directly with their substrates. Therefore, we examined the interaction between pVHL and progerin. Immunoprecipitation of pVHL-Flag with a Flag antibody also co-precipitated progerin (Fig. S4G). Although Lamin A also co-precipitated with pVHL, the binding affinity between pVHL and progerin may have been stronger than that of pVHL and Lamin A, based upon the amount of input proteins. Through the GST pull-down assay, we confirmed the direct binding between progerin and pVHL (Fig. S4H). In contrast, Lamin C did not associate with GST-pVHL (Fig. S4H). These results indicate that the interaction between pVHL and progerin is stronger than that between pVHL and Lamin A/C. However, we did not exclude the possibility that farnesylation of Lamin A/C would involve in the binding between Lamin A and pVHL. To test this, we re-performed the IP with pVHL using farnesyl-transferase inhibitor (FTI)-treated cells. But, FTI did not alter the binding affinity between pVHL and Lamin A or PG (Fig. S4I).
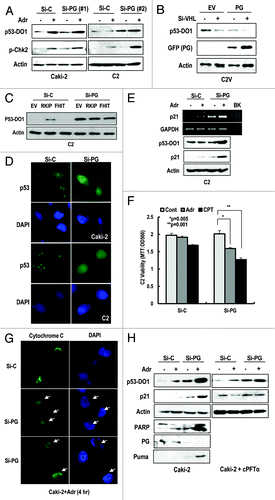
Progerin suppresses p53 function
As described above, p53 inactivation without genetic mutation is a well-known feature of RCCs.Citation6 To reveal the molecular mechanism leading to p53 inactivation, we examined the effect of well-known p53 inhibitors, including MDM2, COP1, and Parc-1.Citation26-Citation28 However, knockdown of these proteins did not induce the nuclear expression of p53 (Fig. S5A). Therefore, we investigated the link between p53 inactivation and progerin expression. Since p53 is mainly regulated at post-translation level, we monitored the p53 expression through WB analysis. First, we measured p53 expression in HGPS cells. Compared with normal fibroblasts, p53 expression was extremely low in HGPS cells (Fig. S5B). However, the elimination of progerin induced p53 expression (Fig. S5C). Thus, we examined the expression of p53 and responsiveness to DNA damage in si-progerin-transfected Caki-2 and C2 cells. p53 expression at protein level and sensitivity to adriamycin (Adr) were increased by si-progerin transfection (). In addition, we observed the phosphorylation of Chk2 in response to DNA damage by progerin elimination (). To test that progerin can suppress the p53, we measured the expression of p53 after transfection with progerin into C2V and found that progerin-mediated p53 suppression was strengthened by si-VHL (). Because FHIT and RKIP are known to be mutated in RCCs,Citation29,Citation30 we also examined their potential role in p53 inactivation. Compared with the partial induction of p53 by RKIP, si-progerin showed an apparent effect on p53 induction (). Immunofluorescence staining data also showed the enhancement of the p53 in response to DNA damage in si-progerin transfected cells (). Because it has been reported that HIF-2α, induced by pVHL defect, is related to p53 suppression,Citation21,Citation23 we compared the effect of si-HIF-2α and si-progerin on p53 expression in C2 cells. Consistent with previous reports, knockdown of HIF-2α induced p53 expression (Fig. S5D). However, the effect of si-progerin on p53 induction was greater than that of si-HIF-2α (Fig. S5D). In addition, we observed an increase of p21 expression at the transcriptional and translational level in response to Adr in progerin-knockdown cells (). However, p21 expression was not obviously induced by si-progerin, suggesting that full activation of p53 could be induced by additional stimulation, such as by DNA damage. Because p53 activation induces apoptosis, we measured cell viability after treatment with DNA-damaging agents (Adr and CPT). In progerin-knockdown cells, these agents reduced viability (). To confirm this reduction, we examined cytochrome C release. In progerin-knockdown cells, we observed an increase of cytochrome C release in response to Adr (; Fig. S5E). Consistent with our cell viability data, cytochrome C release was also increased approximately 2-fold in si-progerin-treated cells (Fig. S5F). To confirm this effect, we measured the expression of PUMA, an immediate target of p53-mediated apoptosis, and PARP cleavage and determined that si-progerin could promote apoptosis in response to Adr (). In addition, we tested the transcriptional activity of p53 using p53 specific transcription inhibitor, cPFTα, which blocked the induction of p21 in response to Adr as well as si-progerin (). These results strongly suggest that the elevated expression of progerin in RCCs could block p53-induced cell death and its tumor-suppressive function.
Figure 3. Progerin suppresses p53. (A) Si-progerin (Si-PG) induces p53 expression in Caki-2 and C2 cell line. These cells were transfected with si-C or si-PG for 24 h. After washing, cells were incubated with 1 μg/ml of adriamycin (Adr) for 2 h. (B) Progerin suppresses p53 expression in VHL-expressed C2V. C2V cells were transfected with progerin (PG) with/without si-VHL for 24 h. VHL knockdown could enhance the PG-induced p53 suppression. (C) FHIT or RKIP do not induce p53 in C2 cells. Because FHIT is located in the same chromosome as pVHL and frequently deleted in RCCs, we examined the effects of RKIP and FHIT on p53. However, these genes did not significantly induce p53 compared with si-PG. (D) IF staining of p53 in si-progerin (Si-PG)-transfected Caki-2 and C2 cells. Increased and nuclear localized p53 could be detected by IF with p53 (DO-1) antibody. (E) Si-progerin (Si-PG) induced p21 at the transcription and translation level. To address the functionality of p53, we measured the expression of p21 using RT-PCR and WB analysis. (F) Si-progerin (Si-PG) enhances the sensitivity to DNA damaging agents in C2. Cells were transfected with the indicated si-RNA for 24 h and incubated with adriamycin (Adr;1 μg/ml) or camptothecin (CPT: 1 μM) for 6 h. After washing, cell viability was determined by MTT assay. Data were analyzed with student’s t-test. (G) Si-progerin (Si-PG) can promote apoptosis. Caki-2 cells were transfected with si-PG or si-C for 24 h and incubated with adriamycin (Adr 2 μg/ml) for 6 h. Cells were fixed and stained with cytochrome C antibody. Compared with si-C, in which cytochrome C was stained as a cytoplasmic punctured pattern, diffused cytochrome C was detected in si-PG-transfected cells (arrows). (H) Si-progerin (Si-PG) can induce PUMA and PARP cleavage. To confirm that si-PG could induce apoptosis, we examined the expression of PUMA and PARP cleavage. cPTFα blocked the induction of p21 by si-PG or Adr, indicated that the effect of si-PG was dependent on p53 transcription activity.
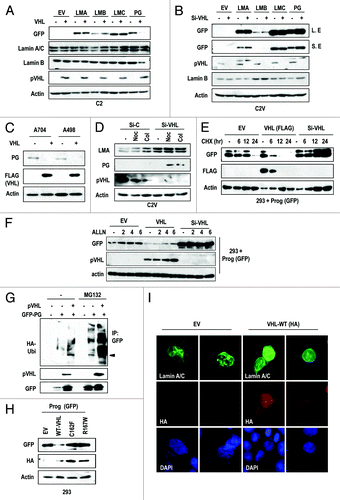
pVHL activates p53
Because pVHL inhibits progerin expression at post-translation level (), we examined the effect of pVHL on p53 expression in RCCs. In addition, the p53 expression level and its impact on DNA damage in RCC cell lines seem to be tightly related with progerin expression and VHL status (Fig. S6A). In fact, ACHN and C2V showed higher expression and normal response of p53 to Adr (Fig. S6A). Forced expression of pVHL could induce basal levels of p53 as well as DNA damage-mediated p53 induction in VHL-defective C2 and Caki-2 cells (; Fig. S6B). In addition, we observed p53 induction in response to the MDM2 inhibitor nutlin-3 in these cell lines (; Fig. S6B). In fact, basal p53 expression in C2V cells was higher than that in the C2 cell line (data not shown). Because pVHL can activate p53 through the direct interaction,Citation31 we also measured the effect of pVHL in the progerin-negative cell lines HCT116 and A549. However, in these cell lines, we did not observe a significant induction of p53 or a synergistic effect with DNA-damaging agents after pVHL overexpression (; Fig. S6C). Moreover, co-transfection of si-progerin and pVHL showed a synergistic effect on p53 expression as well as its target gene expression in Caki-2 and C2 cells (), whereas C2V cells did not exhibit an enhancement of p53 expression or influence DNA damage response by si-progerin (Fig. S6D). These results indicate that pVHL-mediated p53 induction is achieved by the suppression of progerin, and p53 inactivation in RCCs is a result of elevated progerin levels derived from VHL mutation. To confirm this result, we measured the p53 expression in si-progerin- or pVHL-transfected HGPS cells and revealed that p53 was increased by si-progerin as well as by pVHL overexpression (). Given these findings, we propose that pVHL-mediated p53 activation is achieved by progerin suppression.
Figure 4. pVHL induces p53 through the inhibition of progerin-mediated p14 suppression. (A) pVHL transfection can induce p53 expression in C2 cells. C2 cells were transfected with pVHL for 24 h and incubated with 1 μg/ml of adriamycin (Adr) or 2 μM of Nutlin-3 (Nut) for 2 h. (B) pVHL does not induce p53 in A549. In the progerin-negative cell line, overexpression of pVHL did not induce p53. A549 cells were transfected with pVHL and incubated with Adr and CPT for 2 h. (C) pVHL and si-progerin (si-PG) show a synergistic effect on p53 induction in Caki-2 cells. Cells were transfected with pVHL alone or co-transfected with pVHL and si-PG for 24 h and incubated with Etoposide (Etop; 5 μM) or Nutlin-3 (Nut; 2 μM) for 2 h. The expression of p21 was used for marker of p53 activity. (D) Si-progerin (Si-PG) induces p53 in HGPS (HS) cells. HGPS cells were transfected with si-PG (5 μg/ml [+] or 10 μg/ml [++]) for 48 h and the expression of progerin (PG) and p53 was measured. (E) pVHL induces p53 in HGPS (HS) cells. HGPS cells were transfected with pVHL (1.5 μg/ml [+] and 3 μg/ml [++]) for 48 h. (F) Nutlin-3 (Nut) partially induces p53 in HGPS (HS) cells. Cells were incubated with 2 μM of Nut for the indicated time. Expression of p53 was calculated by densitometry analysis and presented as graph (right panel). (G) Si-Progerin (Si-PG) enhances the DNA damage response in C2. Si-PG could induce p53 as well as p-CHK2 in response to camptothecin (CPT). In addition, p14/ARF (p14) expression was increased by si-PG transfection. (H) Si-p14 eliminates p53 induction by si-progerin (Si-PG) Caki-2 cells were transfected with the indicated si-RNA for 24 h and incubated with drugs for 2 h. Si-PG-induced p53 activation was completely blocked by si-p14 co-transfection.
![Figure 4. pVHL induces p53 through the inhibition of progerin-mediated p14 suppression. (A) pVHL transfection can induce p53 expression in C2 cells. C2 cells were transfected with pVHL for 24 h and incubated with 1 μg/ml of adriamycin (Adr) or 2 μM of Nutlin-3 (Nut) for 2 h. (B) pVHL does not induce p53 in A549. In the progerin-negative cell line, overexpression of pVHL did not induce p53. A549 cells were transfected with pVHL and incubated with Adr and CPT for 2 h. (C) pVHL and si-progerin (si-PG) show a synergistic effect on p53 induction in Caki-2 cells. Cells were transfected with pVHL alone or co-transfected with pVHL and si-PG for 24 h and incubated with Etoposide (Etop; 5 μM) or Nutlin-3 (Nut; 2 μM) for 2 h. The expression of p21 was used for marker of p53 activity. (D) Si-progerin (Si-PG) induces p53 in HGPS (HS) cells. HGPS cells were transfected with si-PG (5 μg/ml [+] or 10 μg/ml [++]) for 48 h and the expression of progerin (PG) and p53 was measured. (E) pVHL induces p53 in HGPS (HS) cells. HGPS cells were transfected with pVHL (1.5 μg/ml [+] and 3 μg/ml [++]) for 48 h. (F) Nutlin-3 (Nut) partially induces p53 in HGPS (HS) cells. Cells were incubated with 2 μM of Nut for the indicated time. Expression of p53 was calculated by densitometry analysis and presented as graph (right panel). (G) Si-Progerin (Si-PG) enhances the DNA damage response in C2. Si-PG could induce p53 as well as p-CHK2 in response to camptothecin (CPT). In addition, p14/ARF (p14) expression was increased by si-PG transfection. (H) Si-p14 eliminates p53 induction by si-progerin (Si-PG) Caki-2 cells were transfected with the indicated si-RNA for 24 h and incubated with drugs for 2 h. Si-PG-induced p53 activation was completely blocked by si-p14 co-transfection.](/cms/asset/ee4be71b-21e8-4851-8550-7bc460934c37/kccy_a_10925371_f0004.gif)
Progerin inhibits p14/ARF
To address the molecular mechanism that underlies progerin-mediated p53 suppression, we examined the expression of p53 in human progeria syndrome cells (HGPS and WS). Compared with the induction of p53 in normal cells in response to Adr, HGPS, and Werner syndrome (WS) cells did not exhibit the induction of p53 (Fig. S6E). In fact, it has been reported that WS cells are insensitive to DNA damage-induced p53 activation,Citation32 and we have previously observed that WS cells also express progerin (data not shown). This result implies that progerin can block DNA damage-induced p53 activation. Because p53 is tightly regulated by the MDM2 pathway, we tested the involvement of progerin in MDM2-mediated p53 suppression. Treatment with Nutlin-3 (Nut-3), which induces p53 via the blocking of the interaction between MDM2 and p53,Citation33 could induce p53 in HGPS cells (). However, compared with normal cells, in which Nut-3 clearly induced p53 and p21, the response of p53 and p21 to Nut-3 in HGPS was less effective (). Moreover, we have previously observed that Nut-3 did not obviously induce p53 in the C2 and Caki2 cell lines (; Fig. S6B). Taken together, we propose that the elevated expression of progerin impairs DNA damage-induced p53 activation and is involved in MDM2-mediated p53 suppression.
Because p14/ARF is an inhibitor of MDM2 and is activated by ATM/ATR-dependent DNA damage signaling,Citation34 we assumed that p14/ARF is a strong candidate for the missing link in progerin-mediated p53 suppression. To examine the involvement of p14/ARF in progerin-mediated p53 suppression, we examined p53 expression in p14/ARF-transfected cells. Consistent with a previous result, si-progerin treatment increased the sensitivity of p53 to CPT and also led to elevated p14/ARF levels (). To confirm this result, we measured the expression of p53 in HGPS cells after transfection with si-progerin and p14/ARF and found that the elimination of progerin and the overexpression of p14/ARF exhibited a synergistic effect on p53 expression (Fig. S6F). We further explored the effect of p14/ARF on progerin-induced p53 inactivation, and found that the overexpression of p14/ARF could overcome the suppressive effects of progerin on p53 expression (Fig. S6G). In addition, p14/ARF knockdown blocked the si-progerin-induced p53 activation in Caki-2 (; Fig. S6H). These results indicate that progerin blocks p53 activation through the suppression of p14/ARF. Because progerin can induce the nuclear deformation, we next examined the effect of p14/ARF on the nuclear deformation of HGPS. However, differently from the effect of si-progerin or pVHL ( and ), p14/ARF alone did not demonstrate an ameliorating effect on the nuclear deformation of HGPS (Fig. S7A and B). This result implies that progerin-p14 linkage is restricted to p53 regulation and the nuclear deformation occurs through a separate function of progerin.
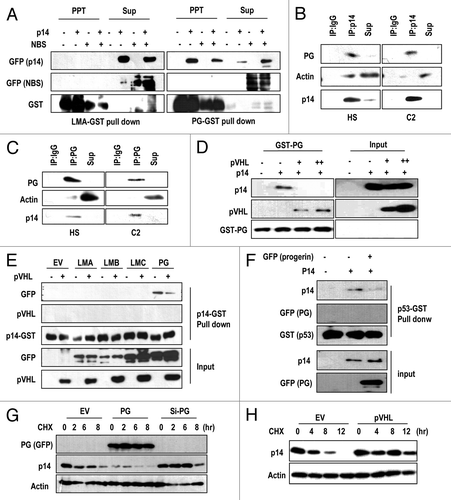
pVHL blocks the interaction between progerin and p14/ARF
To investigate in more detail molecular relevance between p14/ARF and progerin, we performed a GST pull-down assay using recombinant Lamin A, and progerin and found that p14 interacted with progerin exclusively (). The progerin-p14/ARF interaction was specific, because progerin failed to interact with a non-specific control, NBS (Nijmegen breakage syndrome protein, ). To confirm this interaction, we tested the interaction between endogenous p14/ARF and progerin in C2 and HGPS cells through IP. From reciprocal IP analysis, we could confirm the binding between progerin and p14/ARF (). Because we previously showed the interaction between progerin and pVHL (Fig. S4G and H), we checked the effect of pVHL on the p14-progerin interaction using a competition assay. Addition of pVHL blocked the interaction between p14/ARF-progerin, whereas progerin-pVHL binding was increased (). Similarly, we found that pVHL interfered with p14/ARF-progerin binding using a GST-p14 pull-down assay (). To verify the mechanism of progerin-induced p53 inactivation, we examined the effect of progerin on p14/ARF-p53 association,Citation35 and found that progerin had a disruptive effect on p53-p14 binding (). Considering these results, we propose that in a pVHL-deficient context, progerin inhibits p14/ARF, leading to p53 inactivation. Next, we monitored the effect of progerin on p14/ARF stability because the elimination of progerin induced p14/ARF expression () and p14/ARF has a very short half-life.Citation36 Through a pulse-chase analysis, we found that progerin promoted p14/ARF degradation, whereas si-progerin suppressed this degradation (). We also observed an increase in the p14/ARF half-life by pVHL transfection (). These results strongly suggest that pVHL can affect p14/ARF expression through the suppression of progerin.
Figure 5. Direct interaction between p14/ARF-progerin can be blocked by pVHL. (A) p14/ARF (p14) directly and specifically interacts with progerin. GST-progerin (bead; PG) and GST-Lamin A (LMA) were incubated with p14- or p14-/NBS-transfected 293 lysates for 2 h at RT. Precipitated proteins with GST-beads were subjected to SDS-PAGE and WB analysis. (B and C) Interaction of p14/ARF and progerin in C2 and HGPS (HS) cells. Each cell lysate was incubated with p14 Ab or Progerin Ab. After IP, associated progerin, or p14, was detected by SDS-PAGE and WB. Actin was used as a control. (D) pVHL blocks the interaction between p14-progerin. Using GST-PG, a pull-down assay was performed. Associated p14 was diminished by the addition of pVHL-lysate. Instead, pVHL was associated with progerin (GST-PG). (E) GST pull-down assay using GST-p14. The association of p14-progerin was confirmed by GST-p14 and GFP-PG lysate. In addition, other types of Lamin proteins did not show an interaction with GST-p14. Moreover, pVHL could block the interaction between GST-p14 and GFP-PG. (F) Progerin (PG) blocked the interaction between p53-p14. The interaction between GST-p53 and p14 was reduced by the addition of PG-lysate. (G) Progerin (PG) regulates the half-life of p14. To address the effect of progerin on p14 half-life, we performed a pulse-chase analysis using CHX. The half-life of p14 (approximately 2 h) was obviously reduced by GFP-progerin (PG) transfection; whereas si-PG extended the half-life to 6 h. (G) pVHL extends the half-life of p14. Using a similar procedure, we also measured the effect of pVHL on the half-life of p14.
Progerin is elevated in human leukemia
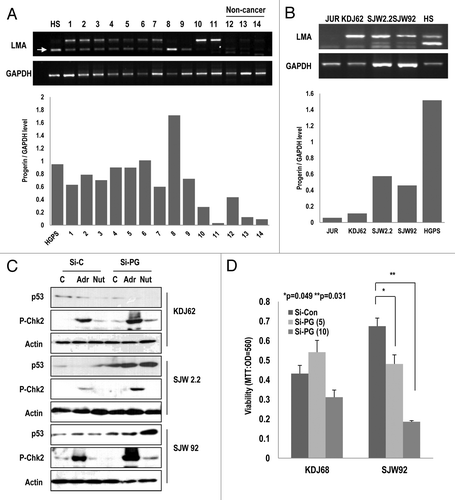
Because progerin can suppress p53 through p14/ARF inactivation, we speculated that other types of cancers also exhibited the elevated expression of the Lamin A splice variant. To test this effect, we examined the expression of progerin in 16 leukemia and 3 healthy blood samples. Leukemia cells also show the resistance to chemotherapy and radiation therapy and exhibit a dysmorphic nucleus.Citation37,Citation38 Among 19 samples, half of them expressed higher levels of progerin relative to normal cells (; Table S1). In , we showed only 14 samples. Instead, we showed the expression of progerin in Table S1 with clinicopathological information). We did not observe a difference between acute myeloid leukemia and acute lymphatic leukemia (Table S1). From these samples, we established 3 types of leukemia cell lines (KDJ62, SJW2.2, and SJW 92) and evaluated the expression of progerin. Among these cell lines, 2 cell lines (SJW 2.2 and 92) showed high levels of progerin expression (). We also observed the expression of progerin at the translational level through WB analysis (Fig. S8A). Therefore, we measured the activation of p53 and Chk2 in these cell lines after the elimination of progerin using siRNA (Fig. S8A). In KDJ 62, si-progerin did not induce p53 and Chk2 activation in response to Adr and Nut-3. In contrast, SJW2.2 and 92 showed the induction of p53 and p-Chk2 in response to Adr in the elimination of progerin (). We also examined cell viability after the elimination of progerin, and found that it reduced the viability of SJW 92 but not that of KDJ62 (). These results suggest that p53 inactivation of SJW 92 and 2.2 is dependent on progerin expression. Moreover, it has been reported that progerin is elevated in human prostate cancer.Citation39 Considering our results and those of others, several types of cancers, including RCCs, leukemia, and prostate cancer, could overcome the p53-induced tumor suppression through progerin overexpression, which blocks p14/ARF-induced p53 activation and DNA damage signaling (Fig. S8B).
Figure 6. Elevated expression of progerin in human leukemia samples. (A) A large portion of leukemia samples express progerin. Using RT-PCR, we explored the expression of progerin and found that among 11 leukemia samples (1–11), 9 samples expressed progerin (arrow). In contrast, normal samples did not show progerin expression (12–14). HGPS (HS) cells were used as a positive control of progerin. Among them, 5 samples (#1, 3, 5, 7, and 8) exhibited a smaller band that was confirmed to correspond to progerin using the cloning and sequencing method. The relative expression of progerin/GAPDH was provided as a graph (below). (B) Progerin expression in 3 established cell lines. Using the patient’s white blood cells, 3 types of cell lines were established (KDJ62 from #11, SJW2.2 from #8, and SJW92 from #2). Among them, two cell lines (SJW2.2 and SJW92) expressed progerin. Jurkat (JUR) and HGPS (HS) were used for control cell lines. The relative expression of progerin/GAPDH was provided as a graph (below). (C) Si-progerin (Si-PG) enhances p53 expression and the DNA damage response. In progerin-positive cell lines (SJW2.2 and SJW92), si-PG enhanced the responsibility of p53 and p-Chk2 to adriamycin (Adr) and Nutlin-3 (Nut). In contrast, KDJ62 did not show a significant difference. (D) Si-progerin (Si-PG) suppressed the cell viability in a progerin-dependent manner. Si-PG suppressed the cell viability in SJW92 cell lines. In contrast, KDJ62 did not show a reduction of viability. Cells were transfected with 5 or 10 μg/ml of si-progerin for 24 h.
Discussion
Although cancer is a well-known aging-related disease, the specific concordance between the disease and aging depends on the cancer type. One well-known example of an aging-related cancer is RCCs, the incidence of which is logarithmically increased from 50–60 y. Although the aging process may contribute to the increased incidence by telomere shortening, many types of cancers can overcome telomere shortening by the re-expression of telomeraseCitation40 or the alternative lengthening of telomeres.Citation41 Therefore, additional reasons are likely to exist for the significantly increased incidence of cancer in the aging population. Here, we considered the possibility that nuclear shape defects, which are used for the grading index of RCCs, might also be a clue to mechanistic aspects of RCCs progression.Citation5 As described previously, these shape defects resemble the nuclear deformation observed in HGPS cells, which are mediated by the aberrant splicing of LMNA that leads to the expression of a variant termed progerin. According to recent findings, progerin is expressed at the basal level in normal cells and accumulates during the aging process.Citation17 Indeed, normal fibroblasts isolated from aged individuals show the nuclear deformation,Citation17 raising the possibility that alternative LMNA splicing might contribute to normal aging.Citation6 In this study, we revealed that RCC cell lines also expressed progerin, which is responsible for irregularities in the nuclear shape. In fact, progerin was strongly expressed in RCC cell lines (), and its elimination restored the normal nuclear shape and p53 activation (; Fig. S2A–D). In addition, we observed the elevated expression of progerin in human leukemia samples and cell lines (; Fig. S8A). These results indicate that the elevated expression of progerin may contribute to cancer occurrence or progression.
Interestingly, the LMNA locus has been linked to cancer progression in many other studies, in which the expression of Lamin A and/or Lamin C were found to be reduced in tumors relative to normal tissues. In many of these cases, progerin expression was not assessed, and our findings call for this issue to be revisited. Taken together, these findings demand a specific assessment of the relative expression levels of Lamin A and C and progerin across a range of tumor samples and cell lines. A better understanding of the complex regulatory pathways involved in these events may shed an important light on links between nuclear intermediate filaments, nuclear organization, and cancer progression.
In addition, RCCs exhibit resistance to chemotherapy and IR, which evokes many problems in the treatment of RCCs after metastasis.Citation42 However, a detailed molecular mechanism for the chemo-resistance of RCCs has not been demonstrated until now. Because p53 can induce apoptosis or senescence in response to DNA damage, metabolic stress, hypoxic stress, or oncogenic stress, the genetic mutation of p53 has been proposed as an explanation. Indeed, approximately 50% of human cancers possess the mutation or deletion of p53.Citation43 However, the mutation rate of p53 is very low in RCCs.Citation6 Thus, there is an additional mechanism that can inactivate p53 in RCCs. A confirmed well-known p53 inactivation mechanism is the overexpression of MDM2, which promotes p53 degradation through its E3 ligase activity.Citation43 In addition, the inactivation of p14/ARF by hypermethylation or deletion has been proposed as the p53 inactivation mechanism. Indeed, p14/ARF stabilizes p53 by sequestering MDM2. Thus, the inactivation of p14/ARF induces MDM2 activation and p53 inactivation. In this study, we found that the elevated expression of progerin could suppress p53 activation through p14/ARF degradation () and sequestration (). However, we did not observe the induction or restoration of p53 activity by knockdown of MDM2 and other p53 E3 ligases (Fig. S5). Considering this result, there would be a novel function of p14/ARF, which activates p53 in a MDM2-independent mechanism. Until now, the mechanism has not been identified.
In RCCs, it has been proposed that the elevated expression of HIF-2α suppresses p53.Citation21,Citation23 Indeed, the elimination of HIF-2α could induce p53 in VHL-deficient C2 cells (Fig. S5D). Thus, HIF-2α can contribute to p53 inactivation of RCCs (Fig. S8B). However, HIF-2α did not alter the expression of progerin (Fig. S3J), indicating that it is not responsible for the nuclear deformation of pVHL-defective RCCs. Through further investigation, we will verify the molecular functions and the relevance between HIF-2α and our system.
Concerning the expression of p53 in HGPS, it has been reported that a chronic activation of p53 would be a cause of senescence.Citation17,Citation44,Citation45 This result is not consistent with our finding that p53 expression and function are suppressed by progerin. However, inconsistent reports have also been published that state the suppression of p53 function or basic expression in progeria syndrome cells.Citation32,Citation46 In addition, the reduction of p53 function in aging has also been suggested.Citation4 In our system, we consistently observe the reduction or impairment of p53 in progeria syndrome cells and RCCs (Fig. S5B and C). Although we did not provide a clear explanation of this inconsistency, the differentiation of cell lines or culture conditions would contribute to it. Another possible mechanism is the differential dependency of p53 expression on p14/ARF. In fact, DNA damage-induced p53 stabilization is not related to p14/ARF but to phosphorylation. Thus, if cells are maintained in a state of sustained DNA damage, which can be induced by various stimulations, including telomere shortening by long-term culture, p53 expression would be elevated in HGPS cells. To ascertain this hypothesis, further investigation and collaboration are required.
Concerning the molecular mechanism for the elevated expression of progerin in RCCs, we propose the loss of pVHL through genetic mutation. In fact, pVHL can suppress progerin expression, and si-pVHL enhances it (). Moreover, pVHL suppresses progerin expression in a HIF-1α-independent mechanism (Fig. S3I), indicating a novel tumor-suppressive function of pVHL. In fact, it remains to be determined why the loss of VHL is required for RCC development. The kidney possesses a well-organized blood vessel system, and tumor angiogenesis is required at the late stage, whereas VHL mutation and deletion are required for the early stage. In addition, using a knockout mouse model, it has been reported that although pVHL is essential for HIF-1α regulation, the loss of pVHL does not promote tumor growth.Citation47 This result suggests that pVHL would exert a novel tumor-suppressive function, which is not related to angiogenesis or HIF-1α degradation. Here, we suggested that pVHL activates p53-p14/ARF function through the blocking of progerin (Fig. S8B).
Considering the fact that nuclear deformation is frequently detected in other types of cancer,Citation48-Citation50 and progerin expression has been reported in human prostate cancers,Citation39 progerin-mediated p53 inactivation may be a frequent event in human cancer, particularly in those lacking p53 mutation or in age-related cancers. Thus, a progerin-specific inhibitor would be useful as an anticancer drug for RCC and other types of cancer. One candidate would be farnesyltransferase inhibitors, which block progerin farnesylation and are shown to improve survival in a mouse model of HGPS.Citation51 Moreover, a more specific inhibitor might be the one that blocks the progerin-p14/ARF association, and we have obtained novel chemical entities that have this activity (data not shown) through chemical screening. These agents promote cell death in RCC cell lines. These results suggest that the suppression of progerin may be a rational treatment for RCC and other cancers.
Materials and Methods
Cell cultures and reagents
Cell lines (293; DMEM, A549, HCT116; RPMI-1640) used in this study were obtained from the ATCC, and other cell lines (UMRC2; C2, UMRC2V; C2V, Caki-2; DMEM) were kindly provided by Dr Jung YJ (Pusan National University). Other types of RCC cell lines (A498, A704, and ACHN) were obtained from the Korea Cell Line Bank. All cell lines were maintained in liquid medium containing 10% FBS and 1% antibiotics in a 37 °C growth chamber. Human fibroblast cells from a patient with Hutchinson Gilford progeria syndrome (HGPS) (AG01972; 14-y-old female) and a healthy person (AG06013; 29-y-old male, AG09603; 81-y-old female) were obtained from the Coriell Cell Repositories and maintained in EMEM containing 15% FBS and 2 mM Glu without antibiotics. General chemical inhibitors, including nocodazole and colcemide, were purchased from Calbiochem. p14/ARF (MS-850-P0) Emerin was obtained from Novocastra, antibodies against progerin (sc-81611), Lamin A (sc-20680), GST (sc-138), GFP (sc-9996), VHL (sc-17780), p53 (DO-1) (sc-126), Actin (sc-1616), MDM2 (sc-965), and His (sc-8036) were purchased from Santa Cruz; α-FLAG (F3165) was obtained from Sigma; p21 (#2946) and p-chk2 (#2661) were obtained from Cell Signaling. The farnensyltransferase inhibitor (FTI) FTI-277 was obtained from Sigma.
Vector and transfection
GFP-fused progerin and GFP-fused Lamin A expression vectors were kindly provided by Scaffidi and Misteli (NCI). pVHL mammalian expression vectors were obtained from Dr Jung YJ (Pusan National University). Myc-ARF, pVHL mutant vectors, and p21-luciferase vector were obtained from Addgene. si-progerin and si-p14 were designed as described previously.Citation18,Citation19 For transfection, Jetpei (polyplus) was used according to the manufacturer’s protocol. In brief, 1.5 μg of the vector was mixed with 1.5 μl of Jetpei reagent in 150 mM NaCl solution. The mixture was incubated for 15 min at RT. After incubation, the mixture was added to the cell. After 3 h, the serum-free medium was replaced with 10% FBS-containing medium.
Recombinant proteins and GST-Pull down
For the investigation of protein-protein interactions, recombinant proteins were generated. The recombinant Lamin A-C-terminal region (LMA; 564-664 AA) and progerin-C-terminal region (PG; 564-614 AA) were produced by cloning 100 AAs upstream from the termination codon through PCR. Full-length p14 and VHL were produced by PCR and cloned into the pGEX vector. Each clone was confirmed by DNA sequencing. These recombinant proteins were purified using a nickel column. For the binding assay, GST-bead-fused Lamin A-C or progerin-C was incubated with 293 cell lysate obtained from p14/ARF-transfected cells for 2 h at RT. In addition, GST-p14 or pVHL was incubated with GFP-Lamin A or progerin-transfected 293 lysate for 2 h at RT. After washing twice with PBS and once with RIPA buffer, precipitated materials were collected and subjected to SDS-PAGE and western blot analysis with anti-p14 and GST.
Immunoprecipitation and western blot analysis
Whole-cell lysates were prepared in RIPA buffer, and the lysates were centrifuged at 14 000 rpm for 30 min. Twenty micrograms of cell extracts was separated by SDS-PAGE and transferred onto a PVDF membrane. The membrane was incubated for 1 h overnight at 4 °C with an appropriate primary antibody, followed by reaction with a secondary antibody at room temperature for 1 h. Peroxidase activity was detected by chemiluminescence with an ECL kit (Intron) as recommended by the manufacturer. To examine the interaction between pVHL and Lamin A, the protein extracts were added to the antibody against pVHL or Lamin A/C (2 μg/sample). After incubation for 2 h at 4 °C with agitation, protein A, and protein G were added. After washing twice with PBS, the precipitates were dissolved in RIPA buffer and SDS sample buffer.
Immunofluorescence staining
To detect the nuclear morphology, immunofluorescence (IF) staining was performed. The cells that were transfected with the indicated vectors were fixed with 100% Me-OH for 10 min at 4 °C. After washing with PBS, the cells were incubated with blocking buffer (PBST with 1% BSA) for 1 h. After washing with PBS twice, the cells were incubated with anti-Lamin A/C antibody, p53 antibody or cytochrome C in blocking buffer (1:200) for 2 h and, sequentially, with anti-Rabbit Ab-FITC or anti-Rabbit Ab-Rhodamin in blocking buffer (1: 1000) for 2 h; finally, the cells were mounted. Nuclei were stained by DAPI, and the immunofluorescence signal was detected through fluorescence microscopy (Zeiss).
RNA isolation and RT-PCR
For RT-PCR, total cellular RNA was extracted using the Qiagen RNA extraction kit. After measuring the RNA concentration, 1 μg of total RNA was reverse transcribed into cDNA using the MMLV RT (Invitrogen) and a random hexamer. RT-PCR was performed with the primers specific to Lamin A/C (5′-AAGGAGATGACCTGCTCCATC-3′ and 5′ - TTTCTTTGGCTTCAAG CCCCC-3′) or GAPDH (5′-ATCTTCCAGGAGCGAGATCCC-3′ and 5′ -AGTGAGCTTCCC GTTCAGCTC- 3′) cDNA.
MTT assay
To measure the cell viability, cells were transfected with the indicated vectors or si-RNA for 24 h. After washing, the cells were treated with adriamycin (Adr) or camptothecin (CPT) for 2 h. For the MTT assay, the cells were incubated with 0.5 mg/ml of MTT solution for 4 h at 37 °C. After removing excess solution, the precipitated materials were dissolved in 200 μl DMSO and quantified by measuring absorbance at 540 nm.
Luciferase assay
To measure the p21 activity, the p21-luc vector was co-transfected with siRNA to 293 cells for 24 h. After washing, the cells were treated by adriamycin (Adr) for 2 h. After washing with wash buffer (Promega), the cells were lysed with lysis buffer. Luciferase activity was determined using a luminometer.
Human leukemia samples
Blood samples from leukemia patients and healthy subjects were provided by the Medical Center of Pusan National University. After the collection of white blood cells (WBC), cells were stored at −70 °C before use. Diagnosis was performed via the general procedure. To establish the cell line, we cultured the WBC in DMEM (15% FBS) and obtained 3 types of stably growing cells.
Additional material
Download Zip (1.8 MB)Acknowledgments
This study was supported by a grant of the Korea Health technology R&D Project, Ministry of Health & Welfare, Republic of Korea. (A100287) and the Research Fund Program of Research Institute for Basic Science, Pusan National University, Korea, 2013, Project No. RIBS-PNU-2013-202.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/cc/article/25371
Related Research Data
References
- Vogelstein B, Kinzler KW. The multistep nature of cancer. Trends Genet 1993; 9:138 - 41; http://dx.doi.org/10.1016/0168-9525(93)90209-Z; PMID: 8516849
- Latif F, Tory K, Gnarra J, Yao M, Duh FM, Orcutt ML, et al. Identification of the von Hippel-Lindau disease tumor suppressor gene. Science 1993; 260:1317 - 20; http://dx.doi.org/10.1126/science.8493574; PMID: 8493574
- Varley JM, Evans DG, Birch JM. Li-Fraumeni syndrome--a molecular and clinical review. Br J Cancer 1997; 76:1 - 14; http://dx.doi.org/10.1038/bjc.1997.328; PMID: 9218725
- Feng Z, Hu W, Teresky AK, Hernando E, Cordon-Cardo C, Levine AJ. Declining p53 function in the aging process: a possible mechanism for the increased tumor incidence in older populations. Proc Natl Acad Sci USA 2007; 104:16633 - 8; http://dx.doi.org/10.1073/pnas.0708043104; PMID: 17921246
- Delahunt B. Advances and controversies in grading and staging of renal cell carcinoma. Mod Pathol 2009; 22:Suppl 2 S24 - 36; http://dx.doi.org/10.1038/modpathol.2008.183; PMID: 19494851
- Gurova KV, Hill JE, Razorenova OV, Chumakov PM, Gudkov AV. p53 pathway in renal cell carcinoma is repressed by a dominant mechanism. Cancer Res 2004; 64:1951 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-03-1541; PMID: 15026329
- Gnarra JR, Tory K, Weng Y, Schmidt L, Wei MH, Li H, et al. Mutations of the VHL tumour suppressor gene in renal carcinoma. Nat Genet 1994; 7:85 - 90; http://dx.doi.org/10.1038/ng0594-85; PMID: 7915601
- Maxwell PH, Wiesener MS, Chang GW, Clifford SC, Vaux EC, Cockman ME, et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 1999; 399:271 - 5; http://dx.doi.org/10.1038/20459; PMID: 10353251
- Ema M, Taya S, Yokotani N, Sogawa K, Matsuda Y, Fujii-Kuriyama Y. A novel bHLH-PAS factor with close sequence similarity to hypoxia-inducible factor 1α regulates the VEGF expression and is potentially involved in lung and vascular development. Proc Natl Acad Sci USA 1997; 94:4273 - 8; http://dx.doi.org/10.1073/pnas.94.9.4273; PMID: 9113979
- Majmundar AJ, Wong WJ, Simon MC. Hypoxia-inducible factors and the response to hypoxic stress. Mol Cell 2010; 40:294 - 309; http://dx.doi.org/10.1016/j.molcel.2010.09.022; PMID: 20965423
- Hanahan D, Folkman J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996; 86:353 - 64; http://dx.doi.org/10.1016/S0092-8674(00)80108-7; PMID: 8756718
- Seagroves TN, Peacock DL, Liao D, Schwab LP, Krueger R, Handorf CR, et al. VHL deletion impairs mammary alveologenesis but is not sufficient for mammary tumorigenesis. Am J Pathol 2010; 176:2269 - 82; http://dx.doi.org/10.2353/ajpath.2010.090310; PMID: 20382704
- Burtner CR, Murakami CJ, Kennedy BK, Kaeberlein M. A molecular mechanism of chronological aging in yeast. Cell Cycle 2009; 8:1256 - 70; http://dx.doi.org/10.4161/cc.8.8.8287; PMID: 19305133
- Worman HJ, Fong LG, Muchir A, Young SG. Laminopathies and the long strange trip from basic cell biology to therapy. J Clin Invest 2009; 119:1825 - 36; http://dx.doi.org/10.1172/JCI37679; PMID: 19587457
- De Sandre-Giovannoli A, Bernard R, Cau P, Navarro C, Amiel J, Boccaccio I, et al. Lamin a truncation in Hutchinson-Gilford progeria. Science 2003; 300:2055; http://dx.doi.org/10.1126/science.1084125; PMID: 12702809
- Eriksson M, Brown WT, Gordon LB, Glynn MW, Singer J, Scott L, et al. Recurrent de novo point mutations in lamin A cause Hutchinson-Gilford progeria syndrome. Nature 2003; 423:293 - 8; http://dx.doi.org/10.1038/nature01629; PMID: 12714972
- Scaffidi P, Misteli T. Lamin A-dependent nuclear defects in human aging. Science 2006; 312:1059 - 63; http://dx.doi.org/10.1126/science.1127168; PMID: 16645051
- Scaffidi P, Misteli T. Reversal of the cellular phenotype in the premature aging disease Hutchinson-Gilford progeria syndrome. Nat Med 2005; 11:440 - 5; http://dx.doi.org/10.1038/nm1204; PMID: 15750600
- Voorhoeve PM, Agami R. The tumor-suppressive functions of the human INK4A locus. Cancer Cell 2003; 4:311 - 9; http://dx.doi.org/10.1016/S1535-6108(03)00223-X; PMID: 14585358
- Burtner CR, Kennedy BK. Progeria syndromes and ageing: what is the connection?. Nat Rev Mol Cell Biol 2010; 11:567 - 78; http://dx.doi.org/10.1038/nrm2944; PMID: 20651707
- Bertout JA, Majmundar AJ, Gordan JD, Lam JC, Ditsworth D, Keith B, et al. HIF2α inhibition promotes p53 pathway activity, tumor cell death, and radiation responses. Proc Natl Acad Sci USA 2009; 106:14391 - 6; http://dx.doi.org/10.1073/pnas.0907357106; PMID: 19706526
- Frew IJ, Krek W. Multitasking by pVHL in tumour suppression. Curr Opin Cell Biol 2007; 19:685 - 90; http://dx.doi.org/10.1016/j.ceb.2007.10.001; PMID: 18006292
- Roberts AM, Watson IR, Evans AJ, Foster DA, Irwin MS, Ohh M. Suppression of hypoxia-inducible factor 2α restores p53 activity via Hdm2 and reverses chemoresistance of renal carcinoma cells. Cancer Res 2009; 69:9056 - 64; http://dx.doi.org/10.1158/0008-5472.CAN-09-1770; PMID: 19920202
- Lisztwan J, Imbert G, Wirbelauer C, Gstaiger M, Krek W. The von Hippel-Lindau tumor suppressor protein is a component of an E3 ubiquitin-protein ligase activity. Genes Dev 1999; 13:1822 - 33; http://dx.doi.org/10.1101/gad.13.14.1822; PMID: 10421634
- Jung YS, Lee SJ, Yoon MH, Ha NC, Park BJ. Estrogen receptor α is a novel target of the Von Hippel-Lindau protein and is responsible for the proliferation of VHL-deficient cells under hypoxic conditions. Cell Cycle 2012; 11:4462 - 73; http://dx.doi.org/10.4161/cc.22794; PMID: 23159849
- Dornan D, Wertz I, Shimizu H, Arnott D, Frantz GD, Dowd P, et al. The ubiquitin ligase COP1 is a critical negative regulator of p53. Nature 2004; 429:86 - 92; http://dx.doi.org/10.1038/nature02514; PMID: 15103385
- Momand J, Zambetti GP, Olson DC, George D, Levine AJ. The mdm-2 oncogene product forms a complex with the p53 protein and inhibits p53-mediated transactivation. Cell 1992; 69:1237 - 45; http://dx.doi.org/10.1016/0092-8674(92)90644-R; PMID: 1535557
- Nikolaev AY, Li M, Puskas N, Qin J, Gu W. Parc: a cytoplasmic anchor for p53. Cell 2003; 112:29 - 40; http://dx.doi.org/10.1016/S0092-8674(02)01255-2; PMID: 12526791
- Al-Mulla F, Hagan S, Behbehani AI, Bitar MS, George SS, Going JJ, et al. Raf kinase inhibitor protein expression in a survival analysis of colorectal cancer patients. J Clin Oncol 2006; 24:5672 - 9; http://dx.doi.org/10.1200/JCO.2006.07.5499; PMID: 17179102
- Sükösd F, Kuroda N, Beothe T, Kaur AP, Kovacs G. Deletion of chromosome 3p14.2-p25 involving the VHL and FHIT genes in conventional renal cell carcinoma. Cancer Res 2003; 63:455 - 7; PMID: 12543802
- Roe JS, Kim H, Lee SM, Kim ST, Cho EJ, Youn HD. p53 stabilization and transactivation by a von Hippel-Lindau protein. Mol Cell 2006; 22:395 - 405; http://dx.doi.org/10.1016/j.molcel.2006.04.006; PMID: 16678111
- Blander G, Zalle N, Leal JF, Bar-Or RL, Yu CE, Oren M. The Werner syndrome protein contributes to induction of p53 by DNA damage. FASEB J 2000; 14:2138 - 40; PMID: 11023999
- Vassilev LT, Vu BT, Graves B, Carvajal D, Podlaski F, Filipovic Z, et al. In vivo activation of the p53 pathway by small-molecule antagonists of MDM2. Science 2004; 303:844 - 8; http://dx.doi.org/10.1126/science.1092472; PMID: 14704432
- Zhang Y, Xiong Y, Yarbrough WG. ARF promotes MDM2 degradation and stabilizes p53: ARF-INK4a locus deletion impairs both the Rb and p53 tumor suppression pathways. Cell 1998; 92:725 - 34; http://dx.doi.org/10.1016/S0092-8674(00)81401-4; PMID: 9529249
- Kamijo T, Weber JD, Zambetti G, Zindy F, Roussel MF, Sherr CJ. Functional and physical interactions of the ARF tumor suppressor with p53 and Mdm2. Proc Natl Acad Sci USA 1998; 95:8292 - 7; http://dx.doi.org/10.1073/pnas.95.14.8292; PMID: 9653180
- Rodway H, Llanos S, Rowe J, Peters G. Stability of nucleolar versus non-nucleolar forms of human p14(ARF). Oncogene 2004; 23:6186 - 92; http://dx.doi.org/10.1038/sj.onc.1207854; PMID: 15286709
- Brynes RK, Golomb HM, Desser RK, Recant W, Reese C, Rowley J. Acute monocytic leukemia. Cytologic, histologic, cytochemical, ultrastructural, and cytogenetic observations. Am J Clin Pathol 1976; 65:471 - 82; PMID: 57715
- Gorre ME, Mohammed M, Ellwood K, Hsu N, Paquette R, Rao PN, et al. Clinical resistance to STI-571 cancer therapy caused by BCR-ABL gene mutation or amplification. Science 2001; 293:876 - 80; http://dx.doi.org/10.1126/science.1062538; PMID: 11423618
- Tang Y, Chen Y, Jiang H, Nie D. Promotion of tumor development in prostate cancer by progerin. Cancer Cell Int 2010; 10:47; http://dx.doi.org/10.1186/1475-2867-10-47; PMID: 21106101
- Hahn WC, Stewart SA, Brooks MW, York SG, Eaton E, Kurachi A, et al. Inhibition of telomerase limits the growth of human cancer cells. Nat Med 1999; 5:1164 - 70; http://dx.doi.org/10.1038/13495; PMID: 10502820
- Dunham MA, Neumann AA, Fasching CL, Reddel RR. Telomere maintenance by recombination in human cells. Nat Genet 2000; 26:447 - 50; http://dx.doi.org/10.1038/82586; PMID: 11101843
- Rini BI, Atkins MB. Resistance to targeted therapy in renal-cell carcinoma. Lancet Oncol 2009; 10:992 - 1000; http://dx.doi.org/10.1016/S1470-2045(09)70240-2; PMID: 19796751
- Meek DW. Tumour suppression by p53: a role for the DNA damage response?. Nat Rev Cancer 2009; 9:714 - 23; PMID: 19730431
- Kudlow BA, Stanfel MN, Burtner CR, Johnston ED, Kennedy BK. Suppression of proliferative defects associated with processing-defective lamin A mutants by hTERT or inactivation of p53. Mol Biol Cell 2008; 19:5238 - 48; http://dx.doi.org/10.1091/mbc.E08-05-0492; PMID: 18843043
- Liu B, Wang J, Chan KM, Tjia WM, Deng W, Guan X, et al. Genomic instability in laminopathy-based premature aging. Nat Med 2005; 11:780 - 5; http://dx.doi.org/10.1038/nm1266; PMID: 15980864
- O’Neill M, Núñez F, Melton DW. p53 and a human premature ageing disorder. Mech Ageing Dev 2003; 124:599 - 603; http://dx.doi.org/10.1016/S0047-6374(03)00007-1; PMID: 12735900
- Mack FA, Rathmell WK, Arsham AM, Gnarra J, Keith B, Simon MC. Loss of pVHL is sufficient to cause HIF dysregulation in primary cells but does not promote tumor growth. Cancer Cell 2003; 3:75 - 88; http://dx.doi.org/10.1016/S1535-6108(02)00240-4; PMID: 12559177
- Artacho-Pérula E, Roldán-Villalobos R, Blanco-García F, Blanco-Rodríguez A. Objective differential classification of thyroid lesions by nuclear quantitative assessment. Histol Histopathol 1997; 12:425 - 31; PMID: 9151131
- Fenner A. Prostate cancer. Nuclear irregularity is a predictor of clinical outcome. Nat Rev Urol 2010; 7:475; http://dx.doi.org/10.1038/nrurol.2010.131; PMID: 20839382
- Swerdlow SH, Pelstring RJ, Collins RD. Morphometric analysis of follicular center cell lymphomas. Am J Pathol 1990; 137:953 - 63; PMID: 2221019
- Fong LG, Frost D, Meta M, Qiao X, Yang SH, Coffinier C, et al. A protein farnesyltransferase inhibitor ameliorates disease in a mouse model of progeria. Science 2006; 311:1621 - 3; http://dx.doi.org/10.1126/science.1124875; PMID: 16484451