Abstract
Glucocorticoid therapy is an important treatment modality of hematological malignancies, especially T-cell acute lymphoblastic leukemia (T-ALL). Glucocorticoids are known to induce a cell cycle arrest and apoptosis in T-lymphoma cells. We could demonstrate that the cell cycle arrest induced by the synthetic glucocorticoid dexamethasone (Dex) clearly precedes apoptosis in human CEM T-ALL and murine S49.1 T-lymphoma cells. Cyclin D3 is strongly downregulated, whereas the CDK inhibitor p27Kip1 (p27) is strongly upregulated in response to dexamethasone in these cells. RNAi-mediated knockdown of p27 as well as overexpression of its negative regulator Skp2 revealed the critical function of p27 in the Dex-induced G1 arrest of CEM cells. Our studies indicate that several mechanisms contribute to the increase of p27 protein in our T-lymphoma cell lines. We found a significant upregulation of p27 mRNA in S49.1 and CEM cells. In addition, Dex treatment activated the mouse p27 promotor in reporter gene experiments, indicating a transcriptional regulation. However, the relatively moderate induction of p27 mRNA levels by Dex did not explain the strong increase of p27 protein in CEM and S49.1 cells. We found clear evidence for a posttranslational mechanism responsible for the robust increase in p27 protein. Dex treatment of S49.1 and CEM cells increases the half-life of p27 protein, which indicates that decreased protein degradation is the primary mechanism of p27 induction by glucocorticoids. Interestingly, we found that Dex treatment decreased the protein and mRNA levels of the negative regulator of p27 protein and E3 ubiquitin ligase subunit Skp2. We conclude that the cell cycle inhibitor p27 and its negative regulator Skp2 are key players in the glucocorticoid-induced growth suppression of T-lymphoma cells and should be considered as potential drug targets to improve therapies of T-cell malignancies.
Introduction
The effective depletion of malignant cells from a cancer patient is crucial in therapies against malignant diseases. Decreased proliferation and induction of cell death by apoptosis are two major cellular events to reduce the growth of and to eliminate cancer cells.
Glucocorticoids are widely used as immunosuppressive and anti-leukemic drugs. Especially sensitive to glucocorticoid therapy are tumor cells originated from T-cells. For example the synthetic glucocorticoid dexamethasone induces cell cycle arrest and apoptosis in many T-lymphoma cell lines and is therefore included in modern protocols of chemotherapies against lymphoblastic malignancies like T-ALL.Citation1-Citation3 The primary consequences of glucocorticoid action are changes in gene expression. Typically, glucocorticoids bind to the glucocorticoid receptor (GR), which translocates to the nucleus and, either directly, via specific DNA-sequences (glucocorticoid response elements, GREs), or indirectly, through interaction with other transcription factors, changes gene expression.Citation4-Citation6 During the last years there has been much success in identifying glucocorticoid-regulated genes partly explaining the immunosuppressive and anti-tumorigenic action of glucocorticoids.Citation7-Citation13 However, the importance of the regulated genes in the respective biological context is often not well understood. Glucocorticoid-induced cell cycle arrest and apoptosis seem to be two independent processes in T-lymphoma cells. In a human T-ALL model system, ectopically expressed Bcl-2 prevented glucocorticoid-induced apoptosis but not cell cycle arrest,Citation7 whereas conditional expression of cyclin D3 and/or c-myc prevented cell cycle arrest but did not affect apoptosis.Citation7 In parallel to the decreased expression of the anti-apoptotic protein Bcl2 and the pro-proliferative protein cyclin D3, expression of the cell cycle inhibitor p27 is induced by glucocorticoids. p27 upregulation by glucocorticoids is reported to occur in lymphoid and non-lymphoid cells.Citation7,Citation14-Citation17 p27 belongs to the CIP/KIP family of cell cycle inhibitors, which bind to and inhibit the activity of cyclin-CDK complexes. In contrast to cell cycle inhibitors of the INK4a/ARF family, the CIP/KIP proteins inhibit D-, E-, and A-type cyclin-CDK complexes and in vitro also B-type cyclin-CDK complexes. Consequently, when p27 levels are high, the cell cycle arrests in G1-phase, and cells stop to proliferate. p27 levels are regulated during the cell cycle. High amounts of p27 in early G1 decrease during progression toward S-phase and stay low until cells complete mitosis and enter G1-phase again.Citation18,Citation19 p27 is haploinsufficient for tumor suppression, and deregulated p27 expression is an important contributor to tumor formation.Citation19-Citation21 Transcriptional and posttranscriptional mechanisms are known to regulate p27 expression. The most powerful control mechanism is thought to be ubiquitin-dependent degradation accomplished by the E3 ubiquitin ligase complex SCFSkp2.Citation22,Citation23 The F-box protein Skp2 targets substrates like p27, p21, p57, or p130 for degradation.Citation24 Typically, Skp2 expression correlates inversely with p27 levels during the cell cycle. The importance of this “Skp2-p27 axis” is emphasized by results from mice, where the phenotypes found in Skp2−/− and p27−/− mice are partially reversed in Skp2−/− p27−/− double knockout mice.Citation25,Citation26 Skp2 plays a role in lymphomagenesis, and its expression correlates positively with tumor grade and negatively with p27 expression in human lymphomas.Citation27
Our study aimed to elucidate the importance of the cell cycle regulators p27 and cyclin D3 in glucocorticoid-induced cell cycle arrest. We found a strong upregulation of p27 at the protein level in mouse and human T-cell lymphomas. This was accompanied by a less pronounced induction of p27 mRNA in human and mouse T-lymphomas, indicating that post-transcriptional regulation is the primary mechanism leading to the increase of p27 protein by glucocorticoids. Induction of p27 was paralleled by a decrease of cyclin D3 after glucocorticoid treatment. Downregulation of p27 levels by RNAi completely abolished glucocorticoid-induced cell cycle arrest although cyclin D3 expression was still effectively reduced. This argues for a primary role of p27 in cell cycle arrest by glucocorticoids in T-lymphomas. Further we found a downregulation of the E3 ligase component Skp2 by dexamethasone. In addition stable expression of Skp2 in glucocorticoid-sensitive human T-lymphomas abolished p27 induction and, more importantly, also prevented the cell cycle arrest by dexamethasone, indicating the importance of the Skp2-p27 axis in glucocorticoid action on T-lymphomas.
Results
Cell cycle arrest precedes apoptosis in glucocorticoid-treated T-lymphomas
Glucocorticoids and its analogs are widely used in the treatment of inflammatory and malignant diseases. Glucocorticoids are an integral part of the therapy against acute lymphoblastic leukemia (ALL). The benefits of glucocorticoids are attributed to its effects on cell proliferation and viability by the induction of cell cycle arrest and apoptosis, respectively. In this study, we investigated the contribution of cell cycle proteins to the cell cycle arrest induced by glucocorticoids.
To analyze the temporal succession of cell cycle arrest and apoptosis induced by glucocorticoids, we treated mouse T-lymphoma cells S49.1 and a human ALL glucocorticoid-sensitive CEM subline with glucocorticoids. In the following, we looked into cell cycle distribution, apoptosis, and regulation of the key cell cycle proteins cyclin D3 and p27. We found a clear accumulation of cells in G1-phase of S49.1 and CEM cells after 24 h of dexamethasone (Dex) treatment () in the complete absence of apoptosis as indicated by the absence of subG1 cells. (). However, after 48 h Dex, treatment apoptosis was induced, as shown by increased subG1 peaks in S49.1 and CEM cells () and PARP cleavage in both cell lines (). In addition, we also found increased annexin V staining of S49.1 cells after 48 h of Dex treatment (Fig. S1A). The onset of apoptosis in S49.1 cells seemed to be delayed compared with CEM cells, as indicated by the lower increase in subG1 cell counts and the less dramatic PARP cleavage after 48 h of exposure to Dex. Indeed, after 72 h of Dex treatment, S49.1 cells showed a strong increase in subG1 cell count (Fig. S1B) and PARP cleavage (). In general, after 5 d of treatment, no more viable cells were detected (data not shown). These results indicate that glucocorticoid-induced cell cycle arrest and apoptosis are sequentially occurring processes in T-lymphomas. Apoptosis might depend on a preceding cell cycle arrest or both processes might be triggered by independent mechanisms. There is already good experimental evidence for the latter from studies in CEM cells where cyclin D3 or c-myc were conditionally overexpressed. Whereas overexpression of both proteins inhibited G1 arrest induced by glucocorticoids, apoptosis was not affected.Citation7
Figure 1. Glucocorticoids induce an accumulation in G1-phase of the cell cycle and apoptosis of T-lymphoma cells. (A) Cell cycle arrest precedes apoptosis in glucocorticoid-treated T-lymphomas. Mouse (S49.1) and human T-lymphomas (CCRF-CEM-C7H2, “CEM”) were cultured in the presence of Dex (100 nM) for the indicated time points. Cell cycle distribution (G1-, S+G2M-phase) and apoptosis (subG1) were determined by DNA-staining using propidium iodide (PI) as desribed in Materials and Methods. The diagrams to the right indicate rise in apoptosis and changes in cell cycle distribution upon Dex treatment (cell cycle distribution was calculated excluding subG1 cells). (B and C) Induction of PARP cleavage in S49.1 and CEM cells. Cells were treated for the indicated time points with 100 nM Dex, and 50 µg of protein extracts were subjected to immunoblot analysis. PARP and PARP cleavage product (PARP [cl.]) were detected by a specific antibody. (D and E) Regulation of cyclin D3 and p27 by glucocorticoids in T-lymphomas. S49.1 and CEM cells were treated for the indicated periods of time with Dex (100 nM) and 50 µg of protein extracts analyzed by western blotting for cyclin D3, p27, and α-tubulin expression using specific antibodies. (F) Inhibition of cyclin E-associated kinase activity by Dex. S49.1 cells were treated with Dex for the indicated time points. Cyclin E was immunoprecipitated from 400 µg of protein extracts and co-purified kinase activity estimated by an in vitro kinase assay using histone H1 as a substrate in the presence of γ-32P-ATP. Immunoprecipitated cyclin E, Cdk2, and p27 were detected by western blotting. Cyclin E and p27 signals were quantified from films using ImageJ software. Increased binding of p27 to cyclin E is expressed as the ratio of quantified p27 to cyclin E signal, the ratio of the control-treated sample is set to 1 (below p27 panel from cyclin E IP). Ten % of the amount of extract used for IP were analyzed by SDS-PAGE and indicate p27 levels during IP (WCE, lowest panel).
![Figure 1. Glucocorticoids induce an accumulation in G1-phase of the cell cycle and apoptosis of T-lymphoma cells. (A) Cell cycle arrest precedes apoptosis in glucocorticoid-treated T-lymphomas. Mouse (S49.1) and human T-lymphomas (CCRF-CEM-C7H2, “CEM”) were cultured in the presence of Dex (100 nM) for the indicated time points. Cell cycle distribution (G1-, S+G2M-phase) and apoptosis (subG1) were determined by DNA-staining using propidium iodide (PI) as desribed in Materials and Methods. The diagrams to the right indicate rise in apoptosis and changes in cell cycle distribution upon Dex treatment (cell cycle distribution was calculated excluding subG1 cells). (B and C) Induction of PARP cleavage in S49.1 and CEM cells. Cells were treated for the indicated time points with 100 nM Dex, and 50 µg of protein extracts were subjected to immunoblot analysis. PARP and PARP cleavage product (PARP [cl.]) were detected by a specific antibody. (D and E) Regulation of cyclin D3 and p27 by glucocorticoids in T-lymphomas. S49.1 and CEM cells were treated for the indicated periods of time with Dex (100 nM) and 50 µg of protein extracts analyzed by western blotting for cyclin D3, p27, and α-tubulin expression using specific antibodies. (F) Inhibition of cyclin E-associated kinase activity by Dex. S49.1 cells were treated with Dex for the indicated time points. Cyclin E was immunoprecipitated from 400 µg of protein extracts and co-purified kinase activity estimated by an in vitro kinase assay using histone H1 as a substrate in the presence of γ-32P-ATP. Immunoprecipitated cyclin E, Cdk2, and p27 were detected by western blotting. Cyclin E and p27 signals were quantified from films using ImageJ software. Increased binding of p27 to cyclin E is expressed as the ratio of quantified p27 to cyclin E signal, the ratio of the control-treated sample is set to 1 (below p27 panel from cyclin E IP). Ten % of the amount of extract used for IP were analyzed by SDS-PAGE and indicate p27 levels during IP (WCE, lowest panel).](/cms/asset/78f57ca4-c1c7-4c36-86ac-59b1a5351630/kccy_a_10925622_f0001.gif)
Biochemical analysis revealed a clear reduction of cyclin D3 and an induction of p27 expression already 24 h after treatment in both cell lines. This regulation became even more prominent after 48 h (). Cyclin E immunoprecipitates from control or Dex-treated S49.1 cells were examined for associated Cdk2 activity. Dex treatment abolished phosphorylation of the kinase substrate histone H1 already after 24 h (, upper panel). The kinase inhibition was accompanied by increased binding of p27 to cyclin E (, lower panel), indicating that p27 may be responsible for cyclin E/Cdk2 inhibition. We conclude that the described inverse regulation of cyclin D3 and p27 arrests the cell cycle and this represents a pathway conserved between mouse and human T-lymphomas.
Mechanisms of p27 regulation by glucocorticoids
Next we aimed to elucidate the mechanism leading to p27 protein induction. We treated S49.1 and CEM cells with Dex for different periods of time and determined the amount of p27 mRNA by reverse transcription quantitative PCR. In both cell lines, we found an approximately 2-fold significant increase in p27 mRNA after 24 h Dex treatment (). In S49.1 cells, p27 mRNA levels are elevated 2-fold after 6 h, and in CEM cells, 1.3-fold after 12 h Dex treatment. In northern blot analysis, p27 mRNA levels in S49.1 cells were constantly elevated until 48 h Dex treatment, indicating no further increase of p27 transcription rate by Dex over time (Fig. S2A). To analyze the potential activation of the p27 promoter by Dex, we transfected luciferase reporter constructs driven by different consecutive 5′ deletions of the mouse p27 promoter Citation52(). Our results indicate that the glucocorticoid responsiveness does not require the sequence −1609 to −615, since the promoter region from −615 to +178 is sufficient to mediate full activation by Dex (). The shortest −42-luc p27 promoter construct shows almost no Dex-inducibility (), suggesting that the glucocorticoid response element(s) lie between position −615 and −42 of the mouse p27 promoter. However, by microarray analysis, we were not able to detect a significant transcriptional regulation of p27 mRNA levels in Dex-treated childhood T-ALL patients (ref. Citation12 and data not shown). We consistently observed a stronger induction of protein levels relative to the mRNA levels in Dex-treated CEM and S49.1 cells ( and ). Therefore we asked whether other mechanisms might contribute to the robust increase in p27 protein levels. It is well documented that p27 protein half-life (stability) changes under different cellular conditions and cell cycle stages. We analyzed p27 protein stability in glucocorticoid-treated S49.1 and CEM cells by CHX chase experiments. In untreated cells, p27 levels decay with a half-life between 1 and 3 h (), which is in the range of half-lives reported previously for cycling cells.Citation22,Citation28 However, when S49.1 or CEM cells were pretreated with Dex for 24 h, p27 protein decreased more slowly in the presence of CHX (). The increase in stability did not depend on the presence of Dex during the CHX chase, since the same result was obtained with fresh Dex-free medium. (Fig. S2B). This clearly indicates that Dex induces cellular conditions where degradation of the p27 protein is strongly impaired.
Figure 2. Transcriptional and post-transcriptional regulation of p27 expression. (A) Regulation of p27 mRNA by Dex in S49.1 and CEM cells. S49.1 and CEM cells were treated with Dex for the indicated time periods and p27 mRNA quantified by RT-qPCR. p27 mRNA levels are expressed relative to the 0 h time point, which was set to 1. All samples were normalized to the amount of TBP control mRNA. Data are expressed as fold increase relative to the normalized control; error bars indicate SD derived from 2–3 independent experiments, each analyzed in triplicate; statistical analysis was performed by unpaired Student t test (*P < 0.05; **P < 0.01). (B) Mouse p27 promoter activation by glucocorticoids. S49.1 cells were transfected with the indicated luciferase reporter constructs as described in “Materials and Methods”. Cells were treated with Dex (100 nM) or ethanol (0.07%) 24 h after transfection and analyzed for luciferase activity after another 24 h. Luciferase activity is expressed as relative light units (RLU) per µg protein. (C and D) Increased p27 protein stability by Dex in S49.1 and CEM cells. Twenty-four hours Dex (100 nM) or control (0.07% ethanol) treated S49.1 and CEM cells were incubated in the presence of cycloheximide (CHX, 20 µM) as indicated, and p27 expression was monitored during the indicated period of time. Upper graphs show western blot analyses, lower panels quantification of p27 levels obtained from 3 independent experiments. p27 amounts (y-axis, log scale) are plotted against time (x-axis), p27 levels at time point zero are set to 100%. Images and quantitative data were obtained by using the ImageQuant LAS 4000 digital imaging system. Statistical analysis was performed by unpaired Student t test (*P < 0.05; 0P = 0.08).
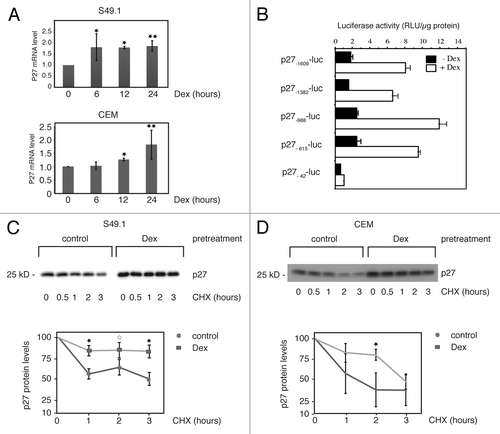
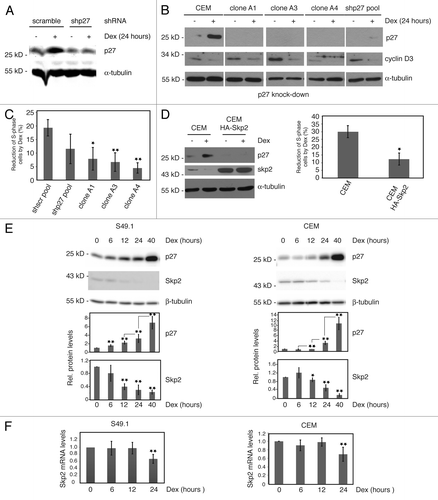
Figure 3. Cell cycle arrest by glucocorticoids requires p27 induction. (A) p27 knockdown in CEM cells. CEM cells were transduced with control (scramble) or p27 shRNA (p27–30) expressing constructs by lentiviral infection and analyzed for p27 expression 5 d after infection. Additionally, cells were treated with Dex (100 nM) for 24 h. Fifty µg of protein extracts were resolved by SDS-PAGE and p27 levels determined by immunoblotting using the Odyssey infrared imaging system. (B) Parental CEM cells and single clones derived from p27 shRNA (p27–30) pools were tested for p27 and cyclin D3 expression in the absence and presence of Dex (100 nM, 24 h). Protein extracts from cells were subjected to SDS-PAGE and immunoblotting using specific antibodies. α-tubulin was used as a loading control. A representative immunoblot is shown. (C) Reduced cell cycle arrest in p27-knockdown cells. Proliferation and cell cycle distribution of cells used in (B) were determined by BrdU incorporation and PI staining. BrdU-positive S-phase cells were determined by anti BrdU labeling (see “Materials and Methods”; Fig. S4). Changes in the number of S-phase (BrdU-positive) cells by Dex after 24 h were calculated and expressed as % reduction of S-phase cells compared with control (0.07% ethanol) treated cells. Data show mean values and SD from at least 3 independent experiments; statistical analysis was performed by unpaired Student t test (*P < 0.05; **P < 0.01). (D) Endogenous Skp2 is downregulated by Dex and Skp2 overexpression reduces p27 levels and glucocorticoid-induced cell cycle arrest. Parental control CEM cells and lentivirally HA-Skp2 transduced cells were analyzed for glucocorticoid response (24 h Dex) by immunoblotting and FACS analyses as described in (C). Skp2 and p27 expression was determined by using specific antibodies. α-tubulin levels indicate equal loading. Diagram shows mean values and SD from 3 independent experiments; statistical analysis was performed by unpaired Student t test (*P < 0.05). (E) Inverse regulation of Skp2 and p27 protein expression by Dex. S49.1 and CEM cells were treated for the indicated time periods with Dex (100 nM) and 50 µg of protein extracts subjected to immunoblot analysis. Images and quantitative data were obtained by using the ImageQuant LAS 4000 digital imaging system. Data are expressed as fold-induction relative to the control; error bars indicate SD and are derived from n > 3 independent experiments analyzed in triplicates; statistical analysis was performed by unpaired Student t test from consecutive time points (p27) or relative to control (Skp2) (*P < 0.05; **P < 0.01). (F) Regulation of Skp2 mRNA by Dex in S49.1 and CEM cells. Procedure is identical to the one described in . Data are expressed as fold-induction relative to the control, error bars indicate SD and are derived from n = 3 independent experiments analyzed in triplicates; statistical analysis was performed by unpaired Student t test (**P < 0.01).
Reducing p27 expression by RNAi and forced degradation by Skp2 overexpression reveals an essential role of p27 in glucocorticoid-induced G1 arrest
Cyclin D3 and p27 are oppositely regulated in T-lymphoma cells ( and ref. Citation7) and are the most likely candidate cell cycle regulators important for glucocorticoid-induced G1 arrest. D-type cyclins are critical for the activation of Cdk4 and Cdk6, whose kinase activities are responsible for the early steps in G1-phase progression, mostly by phosphorylating proteins of the Rb-family. A previous report showed that cell cycle arrest of CEM cells by glucocorticoids could be reversed by ectopically expressed cyclin D3, indicating an important role of this cyclin in T-lymphoma proliferation.Citation7 However, the relevance of the observed upregulation of p27 by glucocorticoids for cell cycle arrest has not been determined. In order to characterize the role of p27 in glucocorticoid-induced cell cycle arrest, we knocked down p27 in CEM cells by shRNA-mediated RNA interference (RNAi). The knockdown efficiency of various targeting constructs was determined by the successful depletion of transiently overexpressed HA-tagged human p27 protein in 293T cells (Fig. S3A and B). The most efficient construct was selected to generate lentiviruses to infect CEM cells.Citation13 p27 shRNA (p27–30)-expressing cells were first tested 5 d post infection for expression of p27 and glucocorticoid response. Single clones were additionally isolated and analyzed. Compared with wild-type CEM or control (scramble)-infected cells (), we found a clear reduction of p27 expression in the p27–30 pool (), which was even more pronounced in single clones (). Strikingly, treatment with Dex for 24 h did not or only marginally induced p27 levels in p27 knockdown cells (). Moreover, this was associated with reduced cell cycle arrest, as indicated by a strongly diminished Dex effect on actively proliferating (S-phase) cells compared with wild-type CEM cells or control (scramble) pools (). Interestingly, proliferation during p27 knockdown was unaffected in the presence of Dex despite reduced cyclin D3 levels (). This suggests that the upregulation of p27 is an essential event in glucocorticoid-induced cell cycle arrest of CEM cells. In general but in particular in the single knockdown clones, p27 depletion in CEM cells increased the amount of S-phase and reduced the amount of G1-phase cells without significantly changing the G2M proportion (Fig. S4). This is in accordance with the cell cycle-inhibitory function of p27. Obviously, p27 had no antiapoptotic function in CEM cells, since p27 knockdown did not increase the amount of subG1 cells and did not sensitize CEM cells to Dex treatment at 24 h (Fig. S4).
To obtain further experimental support for the role of p27 in the G1 arrest of T-ALL cells, we asked whether forced degradation of p27 might prevent cell cycle arrest by glucocorticoids. We enhanced the activity of the E3 ubiquitin ligase complex SCFSkp2 by overexpressing Skp2 in CEM cells (, “CEM HA-Skp2”). As expected, p27 levels were lower in Skp2-overexpressing cells, and also induction of p27 protein by glucocorticoids was strongly reduced (). In agreement with an essential role of p27 in the glucocorticoid-induced cell cycle arrest, Skp2 overexpression in CEM cells reduced the response of actively proliferating (S-phase) cells to Dex treatment (, right panel).
Interestingly, we observed a downregulation of endogenous Skp2 protein by Dex in CEM and S49.1 cells (). When we analyzed the expression of Skp2 and p27 in a time-course experiment, we found a significant induction of p27 protein after 6 h of Dex treatment in S49.1 cells and between 12 and 24 h in CEM cells (). Skp2 levels were clearly downregulated between 6 and 12 h in S49.1 and after 12 h in CEM cells (). Although p27 and Skp2 levels perfectly inversely correlate, downregulation of Skp2 did not precede upregulation of p27, but rather occurred simultaneously (). Skp2 downregulation by Dex might therefore represent a positive feedback loop leading to the robust induction of p27 levels to keep p27 protein high enough to arrest the cell cycle of T-lymphomas. Skp2 transcription was previously reported to be regulated during the cell cycle in HeLa cells. Skp2 protein and mRNA levels are generally low in G1, but both rise as cells approach and enter S-phase.Citation29 We analyzed Skp2 mRNA abundance in S49.1 and CEM cells, which were treated for increasing periods of time with Dex. We found significantly reduced Skp2 mRNA levels after 24 h Dex in S49.1 and CEM cells (). However, the reduction of Skp2 protein occurs earlier and seems to be more pronounced ().
Discussion
The aim of our work was to investigate the role of p27 in glucocorticoid-induced cell cycle arrest of T-lymphomas. We found that cell cycle arrest and apoptosis induced by glucocorticoids are two temporally seperate processes (). This finding supports earlier studies, where conditional overexpression of cyclin D3 in human T-ALL CEM cells prevented glucocorticoid-induced cell cycle arrest but not apoptosis.Citation7 As overexpression of cyclin D3 was able to rescue the cell cycle arrest by glucocorticoids, downregulation of cyclin D3 expression by glucocorticoids seemed to be the primary cause for the growth inhibitory actions of glucocorticoids. However, the contribution of p27 to the G1 arrest was not determined.
We now provide experimental evidence that the increase in p27 protein is pivotal for glucocorticoid-induced cell cycle arrest. Both reduction of p27 levels by RNAi and forced protein degradation by overexpression of the SCF E3 ubiquitin ligase complex component Skp2 abolished Dex-dependent cell cycle arrest. This occurred even under conditions of decreased cyclin D3 expression ().
The amount of p27 is tightly regulated in proliferating cells. Already a small increase in p27 might block the cell cycle by binding to and inhibiting CDKs.Citation30 Multiple mechanisms are responsible for the precise control of p27 expression.Citation21 Transcriptional and post-transcriptional mechanisms regulate p27 levels in cells. The most significant is reported to be ubiquitin-dependent degradation by the E3 ubiquitin ligase complex SCFSkp2. In the present study, we found clear evidence for diverse regulatory mechanisms leading to p27 upregulation by Dex. We detected a weak induction of p27 mRNA levels in the human T-ALL (CEM) cell line as well as in the murine T-lymphoma (S49.1) cell line (). Upregulation of p27 mRNA seems to be a consequence of increased p27 transcription, since we observed activation of p27 promotor reporter constructs by Dex in transient transfection experiments in S49.1 cells (). The precise mechanism of transcriptional activation by glucocorticoids in S49.1 cells is not known at present. The “classical” mechanism of glucocorticoid action is by binding of the hormone-bound, activated GR to glucocorticoid-response elements (GREs) in the promoter region of the respective genes.Citation31-Citation33 Predicted from several databases, there are putative GREs in the promoters of the mouse and human p27 gene. We identified a putative GRE in the mouse promoter at position −653 relative to the translational start site (5′-TGTCCT-3′). The presence of this hexanucleotide sequence was shown to be essential for GR binding.Citation34 Whether this or any other predicted GRE are functional needs to be investigated in further studies. Beside the direct activation of p27 transcription through binding of the GR to promoter elements, the GR could regulate p27 transcription indirectly by changing the expression of transcription factors binding to the p27 promoter. Although p27 transcript levels are not changed significantly during the cell cycle, a growing number of transcription factors that regulate p27 transcription are reported.Citation21,Citation35 The activated dioxin receptor and AFX-like Forkhead transcription factors activate p27 transcription and induce cell cycle arrest,Citation36,Citation37 and the level of the forkhead transcription factor FOXO3A is induced by glucocorticoids in mouse lymphocytes.Citation38 Regarding the relatively weak contribution of p27 transcriptional regulation to the strong induction of p27 protein level, ( and compared with ), posttranscriptional and/or posttranslational mechanisms seem to be dominant in the upregulation of p27. Based on our results and published data, we conclude that p27 plays a central role in Dex-induced cell cycle arrest in the investigated mouse and human T-ALL cell systems. According to our model, glucocorticoids activate the transcription of the p27 gene, which moderately increases levels of p27 protein (). p27 mRNA was also induced by Dex in human CEM cells. By using qPCR we were able to detect a 1.3–2-fold increase in p27 mRNA after 12 and 24 h Dex (). The induction was weak and might have been missed previously by less sensitive methods.Citation7 In parallel, cyclin D3 protein levels rapidly decline after Dex treatment, most likely through decreased mRNA stability.Citation7 The reduction of cyclin D3 is not sufficient to arrest T-ALL cells (). This is in accordance with a central role of p27 in the glucocorticoid-induced cell cycle arrest. Based on earlier studies,Citation19,Citation20 where cyclin D/Cdk complexes were characterized as a sink for non-inhibitory p27, we propose that downregulation of cyclin D3 protein leads to a release of p27, which is then available for binding and inhibiting cyclin E/Cdk2 (), leading to a G1 arrest. The downregulation of Skp2 protein occurs simultaneously with p27 induction and might not contribute to the early increase in p27 levels. However, the more drastic increase in p27 protein upon longer Dex treatment is most likely a consequence of decreased ubiquitin-dependent proteolysis of p27 through repression of Skp2 (). This notion is supported by our results showing increased p27 protein stability in Dex-treated S49.1 and CEM cells (). The regulation of Skp2 levels by glucocorticoids is a novel finding. By RT-qPCR we detected a downregulation of Skp2 mRNA levels by glucocorticoids between 12 and 24 h of treatment (). Transcriptional repression of Skp2 by glucocorticoids is an attractive model that is supported by previous reports. Particularly, known transcriptional activators of Skp2 like c-mycCitation40 and E2FCitation41,Citation42 are strongly downregulated by glucocorticoids in T-lymphomas.Citation7
Figure 4. Central role of p27 regulation in the glucocorticoid-induced cell cycle arrest of T-lymphoma cells. The model depicts mechanisms leading to the strong increase of p27 by glucocorticoids and, as a consequence, to G1 arrest. Previous results exploring the role of cyclin D3 in glucocorticoid-induced cell cycle arrest are integrated in the model. The modes of action are described in the figure and explained in the “Discussion” section.
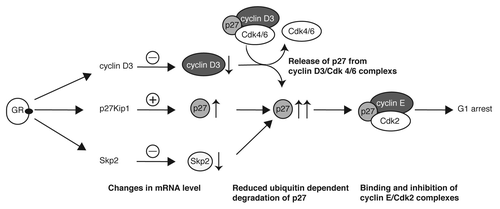
Our studies could not address the necessity of Skp2 downregulation for the p27-induced cell cycle arrest. At this stage, we can only speculate whether Skp2 repression is the cause or the consequence of p27 upregulation and G1 arrest. Both, p27 and Skp2 are useful prognostic and therapeutic markers in a variety of cancers. p27 function is deregulated by altered synthesis, enhanced degradation, and mislocalization. Low levels of the cdk inhibitor often correlate with poor prognosis in many cancers.Citation21 Conversely, Skp2 is often overexpressed in cancers and serves as an independent marker for the prognosis of malignant diseases.Citation43 Skp2 exerts its oncogenic function predominantly by the ubiqutin-dependent degradation of p27.Citation25,Citation26 Recent achievements in developing drugs inhibiting Skp2 emphasize the potential importance of the “Skp2-p27” axis in cancer therapies.Citation43 Treatment of certain cancer cell lines with a molecule that inhibits the assembly of the E3 ubiquitin ligase complex SCFSkp2 resensitized these cancer cells to therapeutic agents like dexamethasone or doxorubicin.Citation44 Whether considering Skp2 could improve the design and success of glucocorticoid therapies has to be verified in future studies. Our data suggest that high Skp2 expression might be predictive for a weaker response to glucocorticoid therapy. A combinatorial therapy involving synthetic glucocorticoids together with Skp2 inhibitors might be more effective under such circumstances.
Materials and Methods
Cell culture, transfection, and cell lysis
The glucocorticoid-sensitive mouse T-lymphoma cell line S49.1Citation7,Citation45 was obtained from the American Type Culture Collection (ATCC) and cultured in DMEM (Sigma Aldrich, D5671) supplemented with 10% FBS plus 100 U/ml penicillin, 100 µg/ml streptomycin according to ATCC guidelines. The human T-ALL cell line CCRF-CEM-C7H2 (CEM)Citation46 and their sublines were cultured as described.Citation7 Cells were treated with 100 nM Dexamethasone (Dex). Stably transfected cells were selected with 1 µg/mL of puromycine. Cyclohexamide (CHX) was used at a concentration of 20 µg/mL.
S49.1 cells were transfected with p27 reporter gene constructs by DEAE-Dextran methodCitation47 at a density of 3 × 105 per ml. Five µg DNA was used for the transfection of 2 × 106 cells. Before transfection, cells were washed with TBS (137 mM NaCl; 25 mM TRIS-HCl pH 7.4; 5 mM KCl; 0.7 mM CaCl2; 0.5 mM MgCl2; 0.6 mM Na2HPO4) and resuspended in 1× transfection mix (5 µg DNA, 0.5 µg/µl DEAE-Dextran in 200 µl TBS). After 20 min of incubation, cells were DMSO shocked by adjusting the cell suspension to 1% DMSO and further incubated for 3 min. A large volume of TBS was added, and cells were washed with TBS and resuspended in fresh growth media. Cells were treated (0.07% ethanol as a vehicle control, 100 nM Dex) 16 h post-transfection and harvested for reporter gene assay 24 h after treatment.
Plasmids, RNA interference, and lentiviral infection
The lentiviral GATEWAY destination vector pHR-SFFV-DEST-IRES-Puro (U416) was generated by exchanging the eGFP sequence with PCR amplified IRES-PURO cassette (pLIB-rtTAM2-IRES-Puro)Citation48 in the lentiviral plasmid pHR-SIN-CSGW-delta Not (kindly provided by Mary Collins, UCL, London). To render the plasmid GATEWAY-compatible, a DEST cassette (rfa, Invitrogen) was cloned into the blunt-ended BamHI site.
The lentiviral HA-Skp2 expression construct was obtained by recombining Skp2-cDNA into the U416 vector using the Gateway® Cloning technology (Life Technologies). The resulting bicistronic plasmid allowed the puromycin selection for stable integrants that expressed Skp2. Control (scramble) and pLKO1.puro (Addgene) were describedCitation49 earlier. p27 shRNA plasmids TRCN0000009856, TRCN0000009857, TRCN0000039928, TRCN0000039930, TRCN0000039931 were obtained from Sigma (Sigma Aldrich).
The plasmid pcDNA3.1 HA-p27Kip1 contains one hemagglutinin-tag fused to the N-terminus of human p27.Citation50
293T cells were transfected by the calcium phosphate method.Citation51 For a 6 cm dish, 0.5 µg HA-p27 pcDNA3.1 plasmid together with 4 µg of single p27 shRNA plasmid or 5 µg of single p27 shRNA plasmid alone were transfected into 293T cells. Typically cells were harvested 40 to 48 h post-transfection and processed for immunoblot analysis.
CEM cells were transduced with lentiviral particles containing p27 shRNAs or HA-tagged Skp2 according to the procedure previousely described.Citation13
Antibodies and reagents
Rabbit polyclonal anti-Actin (A2066), mouse monoclonal anti-α-Tubulin (T9026) were obtained from Sigma (Sigma Aldrich); rabbit polyclonal anti-cyclin D3 (C-16, sc-182), rabbit polyclonal anti-cyclin E (M-20, sc-481), mouse monoclonal anti-Skp2 (A-2, sc-74477) from Santa Cruz (Santa Cruz Biotechnology), mouse monoclonal anti-p27 (610241) from BD Transduction Laboratories (BD Bioscience), anti-GAPDH (6C5, CB1001) from Millipore, mouse monoclonal anti-HA tag antibody (clone 12CA5, ab16918) from Abcam. Mouse monoclonal anti-PARP antibody was a kind gift from Jose Yelamos.
Dexamethasone and cycloheximide were purchased from Sigma Aldrich.
Western blotting, reporter gene assay, and assay for cyclin E-Cdk2 activity
For immunoblot analysis, cells were washed in ice-cold phosphate-buffered saline and lysed in NP-40 lysis buffer (50 mM Tris pH 7.5, 150 mM NaCl, 0.5% NP-40) and protease inhibitor cocktail (Sigma Aldrich). The lysate was sonicated, centrifugated at 20 000 g for 20 min and the supernatant stored at −80°C. For immunoblot analysis typically 50 µg lysates were separated by SDS-PAGE. The subsequent immunoblot analyses were performed by using ECL (Pierce, Thermo Fisher Scientific) either by exposing to sensitive films or by using an ImageQuant LAS 4000 digital imaging system (GE Healthcare) or Odyssey infrared fluorescence detection (Li-Cor) technology.
The luciferase reporter constructs p27-1609-luc, p27-1382-luc, p27-988-luc, p27-615-luc, and p27-42-luc were previousely described.Citation52 Transfected cells were lysed in Reporter Lysis Buffer, and firefly luciferase expression was determined using the Luciferase Assay System (Promega). Luciferase activity was expressed as relative light units per µg extract used in the assay. Extracts from 1.5 × 107 S49.1 cells were used to determine endogenous Cyclin E-associated kinase activity according to the literature.Citation36
RNA isolation, northern blotting, and quantification by reverse transcription quantitative PCR (RT-qPCR)
Total RNA was isolated from cells using RNeasy Mini Kit (Qiagen) according to the manual. Five µg PolyA+ mRNA was isolated and processed for northern blot analysis,Citation53 and probes for p27 and GAPDH were obtained and used for hybridization as described.Citation36
For mouse S49.1 cells, 1 µg of total RNA was reverse transcribed with the QuantiTect Reverse Transcription Kit (Qiagen). Eighty ng of cDNA was used in quantitative PCR using QuantiFast Probe PCR Kit (Qiagen) by using gene-specific primer pairs for p27 (forward: 5′-CAAACTCTGAGGACCGGCAT-3′, reverse: 5′-TCTTAATTCGGAGCTGTTTACGTC-3′), Skp2 (forward: 5′-ACCTGATACTCTACTTGAACTTGGA-3′, reverse: 5′-ATGGTGGTGAAATAGGCG-CA-3′) and TBP (forward: 5′-ACTTCGTGCAAGAAATG-CTGAA-3′, reverse: 5′-TGTCCGTGGCT-CTCTTATTCTCA-3′) on an Mx3005P cycler (Stratagene) instrument. For human CCRF-CEM-C7H2, total RNA from cells cultured in the presence or absence of 10−7 M Dex for the times indicated in the “Results” section was reversely transcribed into cDNA using Superscript II as recommended by the manufacturer (Invitrogen). Twenty ng cDNA was amplified in TaqMan Universal MasterMix (Promega) in 96-well plates (94 °C for 10 min, 40 cycles of 94 °C for 15 s alternating with 60 °C for 1 min) using commercially available assays for detection of p27 and SKP2 (CDKN1B/p27: Hs01597588_m1 SKP2: Hs00394998_m1). For normalization of mRNA content, TATA box-binding protein (TBP) mRNA was used (forward primer: GCCCGAAACGCCGAATA; reverse primer: CGTGGCTCTCTTATCCTCATGAT; probe: ATCCCAAGCGGTTTGCTGCGGT). Measurements of 3 technical replicates from 1–3 independent experiments were averaged and normalized against the TBP housekeeping gene. The difference between TBP-normalized CT values (ΔCT) of Dex-treated and control samples was reported as M values (log2-fold change values, ΔΔCT). Statistical analyses were performed using an unpaired Student t test.
To control the specificity of the amplicons, melting curves were performed for each analysis, the size of PCR products was determined and the gene specificity was verified by sequencing.
Cell cycle and apoptosis analyses
Analyses of cell cycle distribution and subG1 cells were performed by staining ethanol-fixed cells with propidium iodide (PI) and flow cytometry as previousely reported.Citation54 To determine the amount of apoptotic cells, the quantity of PI signal less than 2N or 4N in case of the tetraploid CEM cells, DNA amount was calculated and expressed as percent of all cells. For cell cycle analyses, subG1-cells were excluded. Poly-(ADP-Ribose)-Polymerase (PARP)-cleavage was detected by SDS-PAGE followed by immunoblot analysis using PARP antibodies. Apoptotic cells were quantified by annexin V staining and analyzed by flow cytometry as described.Citation55
For anti-BrdU and propidium iodide (Pl) staining, cells were labeled with 20 μM bromodeoxyuridine (BrdU), washed with PBS, and fixed in 70% ethanol at –20 °C for several hours. Fixed cells were processed as described,Citation50 labeled with anti-BrdU-FITC antibody (clone BU20A, eBioscience), stained with PI, and analyzed with a FACScan flow cytometer (BD Bioscience) and FlowJo software (Tree Star).
| Abbreviations: | ||
| ALL | = | acute lymphoblastic leukemia |
| CDK | = | cyclin dependent kinase |
| Cdk2 | = | cyclin-dependent kinase 2 |
| Dex | = | Dexamethasone |
| GR | = | glucocorticoid receptor |
| GRE | = | glucocorticoid response element |
| SD | = | standard deviation |
| shRNA | = | small hairpin RNA |
| Skp2 | = | S-phase kinase-associated protein 2 |
Additional material
Download Zip (3.7 MB)Acknowledgments
We thank Peter Herrlich and Martin Göttlicher for critical and helpful comments during the preparation of the manuscript, Jonathan Vosper and all members of the Hengst lab for support, stimulating discussions and critical reading of the manuscript, Johanna Gostner for help with the RT-qPCR and statistical analysis of the data, and Barbara Gschirr for TaqMan real-time RT-PCR. We appreciate Albert Nordin providing us the p27 reporter constructs. Part of the work was funded by the FWF (Grant P24031-B20 and SFB F21-B12). AT was supported by ONCOTYROL, a COMET Center funded by the Austrian Research Promotion Agency (FFG), the Tiroler Zukunftsstiftung and the Styrian Business Promotion Agency (SFG). The Tyrolean Cancer Research Institute is supported by the Tiroler Krankenanstalten Ges.m.b.H (TILAK), the Tyrolean Cancer Aid Society, various businesses, financial institutions, and the people of Tyrol.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: http://www.landesbioscience.com/journals/cc/article/25622
Related Research Data
References
- Claman HN. Corticosteroids and lymphoid cells. N Engl J Med 1972; 287:388 - 97; http://dx.doi.org/10.1056/NEJM197208242870806; PMID: 5043524
- Norman MR, Thompson EB. Characterization of a glucocorticoid-sensitive human lymphoid cell line. Cancer Res 1977; 37:3785 - 91; PMID: 269011
- Inaba H, Pui C-H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol 2010; 11:1096 - 106; http://dx.doi.org/10.1016/S1470-2045(10)70114-5; PMID: 20947430
- Wright AP, Zilliacus J, McEwan IJ, Dahlman-Wright K, Almlöf T, Carlstedt-Duke J, et al. Structure and function of the glucocorticoid receptor. J Steroid Biochem Mol Biol 1993; 47:11 - 9; http://dx.doi.org/10.1016/0960-0760(93)90052-X; PMID: 8274424
- Reichardt HM, Schütz G. Glucocorticoid signalling–multiple variations of a common theme. Mol Cel Endocrine 1998; 146:1 - 6; http://dx.doi.org/10.1016/S0303-7207(98)00208-1
- Kassel O, Herrlich P. Crosstalk between the glucocorticoid receptor and other transcription factors: molecular aspects. Mol Cell Endocrinol 2007; 275:13 - 29; http://dx.doi.org/10.1016/j.mce.2007.07.003; PMID: 17689856
- Ausserlechner MJ, Obexer P, Böck G, Geley S, Kofler R. Cyclin D3 and c-MYC control glucocorticoid-induced cell cycle arrest but not apoptosis in lymphoblastic leukemia cells. Cell Death Differ 2004; 11:165 - 74; http://dx.doi.org/10.1038/sj.cdd.4401328; PMID: 14576768
- Harrison RW 3rd, Lippman SS, Hendry WJ 3rd, Chien MC. Isolation of a genomic sublibrary enriched for glucocorticoid-regulated genes. DNA Cell Biol 1990; 9:95 - 102; http://dx.doi.org/10.1089/dna.1990.9.95; PMID: 2344394
- Smith JB, Herschman HR. Targeted identification of glucocorticoid-attenuated response genes: in vitro and in vivo models. Proc Am Thorac Soc 2004; 1:275 - 81; http://dx.doi.org/10.1513/pats.200402-017MS; PMID: 16113446
- Wang J-C, Derynck MK, Nonaka DF, Khodabakhsh DB, Haqq C, Yamamoto KR. Chromatin immunoprecipitation (ChIP) scanning identifies primary glucocorticoid receptor target genes. Proc Natl Acad Sci USA 2004; 101:15603 - 8; http://dx.doi.org/10.1073/pnas.0407008101; PMID: 15501915
- Obexer P, Certa U, Kofler R, Helmberg A. Expression profiling of glucocorticoid-treated T-ALL cell lines: rapid repression of multiple genes involved in RNA-, protein- and nucleotide synthesis. Oncogene 2001; 20:4324 - 36; http://dx.doi.org/10.1038/sj.onc.1204573; PMID: 11466613
- Schmidt S, Rainer J, Riml S, Ploner C, Jesacher S, Achmüller C, et al. Identification of glucocorticoid-response genes in children with acute lymphoblastic leukemia. Blood 2006; 107:2061 - 9; http://dx.doi.org/10.1182/blood-2005-07-2853; PMID: 16293608
- Ploner C, Rainer J, Niederegger H, Eduardoff M, Villunger A, Geley S, et al. The BCL2 rheostat in glucocorticoid-induced apoptosis of acute lymphoblastic leukemia. Leukemia 2008; 22:370 - 7; http://dx.doi.org/10.1038/sj.leu.2405039; PMID: 18046449
- Baghdassarian N, Catallo R, Mahly MA, Ffrench P, Chizat F, Bryon PA, et al. Glucocorticoids induce G1 as well as S-phase lengthening in normal human stimulated lymphocytes: differential effects on cell cycle regulatory proteins. Exp Cell Res 1998; 240:263 - 73; http://dx.doi.org/10.1006/excr.1998.3942; PMID: 9596999
- Baghdassarian N, Peiretti A, Devaux E, Bryon PA, Ffrench M. Involvement of p27Kip1 in the G1- and S/G2-phase lengthening mediated by glucocorticoids in normal human lymphocytes. Cell Growth Differ 1999; 10:405 - 12; PMID: 10392902
- Peiretti A, Baghdassarian N, Gerland LM, Ffrench P, Bryon PA, Magaud JP, et al. CDK2 is involved in the S-phase lengthening induced by glucocorticoids in normal human lymphocytes. Eur J Cell Biol 2003; 82:253 - 61; http://dx.doi.org/10.1078/0171-9335-00298; PMID: 12800980
- Rogatsky I, Trowbridge JM, Garabedian MJ. Glucocorticoid receptor-mediated cell cycle arrest is achieved through distinct cell-specific transcriptional regulatory mechanisms. Mol Cell Biol 1997; 17:3181 - 93; PMID: 9154817
- Hengst L, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol 1998; 227:25 - 41; http://dx.doi.org/10.1007/978-3-642-71941-7_2; PMID: 9479824
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501 - 12; http://dx.doi.org/10.1101/gad.13.12.1501; PMID: 10385618
- Sherr CJ, Roberts JM. Inhibitors of mammalian G1 cyclin-dependent kinases. Genes Dev 1995; 9:1149 - 63; http://dx.doi.org/10.1101/gad.9.10.1149; PMID: 7758941
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253 - 67; http://dx.doi.org/10.1038/nrc2347; PMID: 18354415
- Pagano M, Tam SW, Theodoras AM, Beer-Romero P, Del Sal G, Chau V, et al. Role of the ubiquitin-proteasome pathway in regulating abundance of the cyclin-dependent kinase inhibitor p27. Science 1995; 269:682 - 5; http://dx.doi.org/10.1126/science.7624798; PMID: 7624798
- Sutterlüty H, Chatelain E, Marti A, Wirbelauer C, Senften M, Müller U, et al. p45SKP2 promotes p27Kip1 degradation and induces S phase in quiescent cells. Nat Cell Biol 1999; 1:207 - 14; http://dx.doi.org/10.1038/12027; PMID: 10559918
- Kitagawa K, Kotake Y, Kitagawa M. Ubiquitin-mediated control of oncogene and tumor suppressor gene products. Cancer Sci 2009; 100:1374 - 81; http://dx.doi.org/10.1111/j.1349-7006.2009.01196.x; PMID: 19459846
- Nakayama K, Nagahama H, Minamishima YA, Miyake S, Ishida N, Hatakeyama S, et al. Skp2-mediated degradation of p27 regulates progression into mitosis. Dev Cell 2004; 6:661 - 72; http://dx.doi.org/10.1016/S1534-5807(04)00131-5; PMID: 15130491
- Kossatz U, Dietrich N, Zender L, Buer J, Manns MP, Malek NP. Skp2-dependent degradation of p27kip1 is essential for cell cycle progression. Genes Dev 2004; 18:2602 - 7; http://dx.doi.org/10.1101/gad.321004; PMID: 15520280
- Latres E, Chiarle R, Schulman BA, Pavletich NP, Pellicer A, Inghirami G, et al. Role of the F-box protein Skp2 in lymphomagenesis. Proc Natl Acad Sci USA 2001; 98:2515 - 20; http://dx.doi.org/10.1073/pnas.041475098; PMID: 11226270
- Hengst L, Reed SI. Translational control of p27Kip1 accumulation during the cell cycle. Science 1996; 271:1861 - 4; http://dx.doi.org/10.1126/science.271.5257.1861; PMID: 8596954
- Zhang H, Kobayashi R, Galaktionov K, Beach D. p19Skp1 and p45Skp2 are essential elements of the cyclin A-CDK2 S phase kinase. Cell 1995; 82:915 - 25; http://dx.doi.org/10.1016/0092-8674(95)90271-6; PMID: 7553852
- Hengst L, Göpfert U, Lashuel HA, Reed SI. Complete inhibition of Cdk/cyclin by one molecule of p21(Cip1). Genes Dev 1998; 12:3882 - 8; http://dx.doi.org/10.1101/gad.12.24.3882; PMID: 9869641
- Evans RM. Molecular characterization of the glucocorticoid receptor. Recent Prog Horm Res 1989; 45:discussion 22–7 1 - 22, discussion 22-7; PMID: 2682841
- Beato M, Herrlich P, Schütz G. Steroid hormone receptors: many actors in search of a plot. Cell 1995; 83:851 - 7; http://dx.doi.org/10.1016/0092-8674(95)90201-5; PMID: 8521509
- Mangelsdorf DJ, Thummel C, Beato M, Herrlich P, Schütz G, Umesono K, et al. The nuclear receptor superfamily: the second decade. Cell 1995; 83:835 - 9; http://dx.doi.org/10.1016/0092-8674(95)90199-X; PMID: 8521507
- Scheidereit C, Westphal HM, Carlson C, Bosshard H, Beato M. Molecular model of the interaction between the glucocorticoid receptor and the regulatory elements of inducible genes. DNA 1986; 5:383 - 91; http://dx.doi.org/10.1089/dna.1986.5.383; PMID: 3096677
- Borriello A, Cucciolla V, Oliva A, Zappia V, Della Ragione F. p27Kip1 metabolism: a fascinating labyrinth. Cell Cycle 2007; 6:1053 - 61; http://dx.doi.org/10.4161/cc.6.9.4142; PMID: 17426451
- Kolluri SK, Weiss C, Koff A, Göttlicher M. p27(Kip1) induction and inhibition of proliferation by the intracellular Ah receptor in developing thymus and hepatoma cells. Genes Dev 1999; 13:1742 - 53; http://dx.doi.org/10.1101/gad.13.13.1742; PMID: 10398686
- Medema RH, Kops GJ, Bos JL, Burgering BM. AFX-like Forkhead transcription factors mediate cell-cycle regulation by Ras and PKB through p27kip1. Nature 2000; 404:782 - 7; http://dx.doi.org/10.1038/35008115; PMID: 10783894
- Ma J, Xie Y, Shi Y, Qin W, Zhao B, Jin Y. Glucocorticoid-induced apoptosis requires FOXO3A activity. Biochem Biophys Res Commun 2008; 377:894 - 8; http://dx.doi.org/10.1016/j.bbrc.2008.10.097; PMID: 18957280
- Bretones G, Acosta JC, Caraballo JM, Ferrándiz N, Gómez-Casares MT, Albajar M, et al. SKP2 oncogene is a direct MYC target gene and MYC down-regulates p27(KIP1) through SKP2 in human leukemia cells. J Biol Chem 2011; 286:9815 - 25; http://dx.doi.org/10.1074/jbc.M110.165977; PMID: 21245140
- Yung Y, Walker JL, Roberts JM, Assoian RKA. A Skp2 autoinduction loop and restriction point control. J Cell Biol 2007; 178:741 - 7; http://dx.doi.org/10.1083/jcb.200703034; PMID: 17724117
- Muth D, Ghazaryan S, Eckerle I, Beckett E, Pöhler C, Batzler J, et al. Transcriptional repression of SKP2 is impaired in MYCN-amplified neuroblastoma. Cancer Res 2010; 70:3791 - 802; http://dx.doi.org/10.1158/0008-5472.CAN-09-1245; PMID: 20424123
- Hershko DD. Oncogenic properties and prognostic implications of the ubiquitin ligase Skp2 in cancer. Cancer 2008; 112:1415 - 24; http://dx.doi.org/10.1002/cncr.23317; PMID: 18260093
- Chen Q, Xie W, Kuhn DJ, Voorhees PM, Lopez-Girona A, Mendy D, et al. Targeting the p27 E3 ligase SCF(Skp2) results in p27- and Skp2-mediated cell-cycle arrest and activation of autophagy. Blood 2008; 111:4690 - 9; http://dx.doi.org/10.1182/blood-2007-09-112904; PMID: 18305219
- Daniel V, Litwack G, Tomkins GM. Induction of cytolysis of cultured lymphoma cells by adenosine 3′:5′-cyclic monophosphate and the isolation of resistant variants. Proc Natl Acad Sci USA 1973; 70:76 - 9; http://dx.doi.org/10.1073/pnas.70.1.76; PMID: 4346041
- Strasser-Wozak EM, Hattmannstorfer R, Hála M, Hartmann BL, Fiegl M, Geley S, et al. Splice site mutation in the glucocorticoid receptor gene causes resistance to glucocorticoid-induced apoptosis in a human acute leukemic cell line. Cancer Res 1995; 55:348 - 53; PMID: 7812967
- Schwartz O, Virelizier JL, Montagnier L, Hazan U. A microtransfection method using the luciferase-encoding reporter gene for the assay of human immunodeficiency virus LTR promoter activity. Gene 1990; 88:197 - 205; http://dx.doi.org/10.1016/0378-1119(90)90032-M; PMID: 2189784
- Ausserlechner MJ, Obexer P, Deutschmann A, Geiger K, Kofler R. A retroviral expression system based on tetracycline-regulated tricistronic transactivator/repressor vectors for functional analyses of antiproliferative and toxic genes. Mol Cancer Ther 2006; 5:1927 - 34; http://dx.doi.org/10.1158/1535-7163.MCT-05-0500; PMID: 16928812
- Jäkel H, Weinl C, Hengst L. Phosphorylation of p27Kip1 by JAK2 directly links cytokine receptor signaling to cell cycle control. Oncogene 2011; 30:3502 - 12; http://dx.doi.org/10.1038/onc.2011.68; PMID: 21423214
- Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007; 128:269 - 80; http://dx.doi.org/10.1016/j.cell.2006.11.047; PMID: 17254966
- Graham FL, van der Eb AJ. A new technique for the assay of infectivity of human adenovirus 5 DNA. Virology 1973; 52:456 - 67; http://dx.doi.org/10.1016/0042-6822(73)90341-3; PMID: 4705382
- Kwon TK, Nagel JE, Buchholz MA, Nordin AA. Characterization of the murine cyclin-dependent kinase inhibitor gene p27Kip1. Gene 1996; 180:113 - 20; http://dx.doi.org/10.1016/S0378-1119(96)00416-7; PMID: 8973354
- Aviv H, Leder P. Purification of biologically active globin messenger RNA by chromatography on oligothymidylic acid-cellulose. Proc Natl Acad Sci USA 1972; 69:1408 - 12; http://dx.doi.org/10.1073/pnas.69.6.1408; PMID: 4504350
- Kalejta RF, Brideau AD, Banfield BW, Beavis AJ. An integral membrane green fluorescent protein marker, Us9-GFP, is quantitatively retained in cells during propidium iodide-based cell cycle analysis by flow cytometry. Exp Cell Res 1999; 248:322 - 8; http://dx.doi.org/10.1006/excr.1999.4427; PMID: 10094838
- Kullmann M, Schneikert J, Moll J, Heck S, Zeiner M, Gehring U, et al. RAP46 is a negative regulator of glucocorticoid receptor action and hormone-induced apoptosis. J Biol Chem 1998; 273:14620 - 5; http://dx.doi.org/10.1074/jbc.273.23.14620; PMID: 9603979