Abstract
Lapatinib, a dual EGFR/HER2 tyrosine kinase inhibitor, has been shown to have potent antitumor effects against human breast cancer. Recent studies have shown that lapatinib upregulates p27Kip1 (here after referred to as p27) expression and induces G1 cell cycle arrest in various types of cancer cells. However, the regulation of p27 in lapatinib-induced cell cycle arrest is not well studied. Here we demonstrate that lapatinib-induced cell growth inhibition and G1 cell cycle arrest in HER2-overexpressing human breast cancer cells were dependent on p27. We also show that lapatinib-induced upregulation of p27 expression was mediated through both transcriptional and post-translational mechanisms. On the one hand, lapatinib treatment led to increased FOXO3a expression and enhanced p27 transcription. On the other hand, lapatinib treatment resulted in increased DYRK1B expression, which correlated with increased p27 phosphorylation at Ser10 and decreased p27 degradation. Interestingly, we found that ERβ1 but not ERβ2 expression also upregulated p27 and enhanced lapatinib-induced cell proliferation inhibition and G1 cell cycle arrest in HER2-overexpressing breast cancer cells. Taken together, our results suggest that lapatinib induces p27 expression via both transcriptional and post-translational upregulations, leading to cell cycle arrest and cell proliferation inhibition, and that its effect on breast cancer cells may be modified by ER expression status.
Introduction
Breast cancer is the most frequently diagnosed cancer and the leading cause of cancer death in females worldwide, accounting for 23% (1.38 million) of the total new cancer cases and 14% (458 400) of the total cancer deaths in 2008.Citation1 In addition to early diagnosis, development of new therapies will be important to improve clinical outcome of breast cancer. In recent years, many efforts have been focused on the research of targeted therapy. Among the most studied molecular targets, the human epidermal growth factor receptor 2 (HER2) is a potent mediator of cellular growth and proliferation.Citation2,Citation3 HER2 gene overexpression occurs in approximately 20–30% of metastatic breast cancers and is associated with aggressive disease and decreased survival.Citation4,Citation5 Since HER2 plays important roles in breast cancer, several therapeutic approaches have been developed to target HER2, including monoclonal antibodies and small molecule TK inhibitors (TKIs). The humanized monoclonal antibody trastuzumab (Herceptin) has improved disease-free and overall survival in patients with both metastatic and early HER2-positive breast cancer.Citation6 Lapatinib (GW572016, GlaxoSmithKline) is a dual EGFR/HER2 tyrosine kinase inhibitor that prevents downstream cell proliferation and survival signaling via the PI3K/AKT and Erk/MAPK pathways.Citation7 Recent Phase III clinical studies have shown that lapatinib improves progression-free survival in patients who have progressed on trastuzumab and is approved for treatment of patients with advanced HER2-positive breast cancer in combination with capecitabine.Citation8
Previous studies have shown that lapatinib-mediated suppression of cancer cell proliferation correlates with G1 phase cell cycle arrest or apoptosis induction in vitro and in vivo.Citation9,Citation10 Many molecules are involved in cell cycle progression in eukaryotic cells, including a series of protein complexes composed of cyclins, cyclin-dependent kinases (CDKs), and their cyclin-dependent kinase inhibitors (CKIs). p27 is a member of the Cip/Kip family of CDK inhibitory proteins, which plays a pivotal role in the control of cell proliferation, differentiation, and apoptosis.Citation11 p27 is a negative regulator of the protein kinase CDK2/cyclin E and can block the cell cycle at G0/G1 phase.Citation12 In quiescent cells, p27 exhibits maximal translation and stability and contributes to blocked transition from G1 phase to S phase of the cell cycle via inhibition of cyclin-CDK complexes.Citation13,Citation14 Upon mitogenic stimulation, the level of p27 protein decreases rapidly, thus allowing the activation of cyclin E/CDK2 and cyclin A/CDK2 complexes to promote cell proliferation.Citation15 Forced expression of p27 results in cell cycle arrest in G1 phase; conversely, inhibition of p27 expression increases the number of cells in S phase. Thus, p27 is a major regulator of G1/S transition in the cell cycle. Numerous studies had indicated that p27 protein levels may be associated with the development and survival of human cancers and seem to be an important marker of cancer progression.Citation16,Citation17 In addition, several studies have demonstrated that regulation of p27 might be one of the mechanisms through which lapatinib blocks cell cycle progression in cancer cells.Citation9,Citation18,Citation19
Estrogen is essential for development and growth of mammary gland and has been associated with the proliferation and progression of breast cancer. The expression of estrogen receptors (ER) α and β plays an important role in cell cycle regulation. ERα is believed to regulate expression of genes involved in cell survival and proliferation, thus promoting tumor growth and differentiation, while the function of ERβ has been found to be anti-proliferative and pro-apoptotic.Citation20-Citation23 ERβ has several splicing variants, including ERβ1 (wild type), ERβ2 (identical to ERβcx), ERβ3, ERβ4, ERβ5, and ERΔ5.Citation24,Citation25 Interestingly, ERβ1 has been shown to act as a tumor suppressor,Citation26 and its expression declines during breast cancer progression.Citation27,Citation28 Furthermore, ERβ1 inhibits epithelial–mesenchymal transition (EMT) and invasion in basal-like breast cancer cells.Citation29 ERβ2 appears to be a powerful prognostic indicator in breast cancer, and nuclear and cytoplasmic expression differentially affect outcome. Nuclear ERβ2 immunoreactivity was shown to be significantly associated with better overall survival, while cytoplasmic ERβ2 was significantly associated with high-grade tumors.Citation28 Recently, it was shown that ERα blockade effectively arrested breast cancer cells at G1 cell cycle by stabilizing p27.Citation30,Citation31 However, the roles of ERβ1 and ERβ2 in this process have not yet been clearly established.
Several groups have demonstrated that lapatinib induces p27 in breast cancer cells.Citation32,Citation33 However, how p27 expression is regulated after lapatinib treatment is not fully understood. In this study, we found that p27 is required for the G1 cell cycle arrest effect of lapatinib in HER2-overexpressing breast cancer cells. We also established that lapatinib-induced p27 expression regulation is at both transcriptional level via promoter activation and post-translational level via Ser10 phosphorylation, which prevents protein degradation. Interestingly, we also found that ERβ1 and ERβ2 expression status may effect p27 expression and modify the effect of lapatinib.
Results
Lapatinib inhibits cells proliferation and causes G1 phase cell cycle arrest in HER2-overexpressing breast cancer cells
To investigate the effects of lapatinib on cellular proliferation, we analyzed the inhibitory effects of increasing concentrations of lapatinib on SK-BR-3, MBA-MD-453, and MCF-7 cells by MTT assay. These human breast cancer cells were exposed to various concentrations (0.01, 0.03, 0.1, 0.3, 1.0, 3.0 µM) of lapatinib for different time durations (24, 48, or 72 h), and cell survival percentages were determined. Inhibition of the proliferation of SK-BR-3 and MBA-MD-453 cells by lapatinib treatment was readily detected, which was clearly time- and dose-dependent (). However, there was no significant effect of lapatinib on the growth of MCF-7 cells, which are HER2-negative ().
Figure 1. Lapatinib inhibits cell proliferation and causes G1 cell cycle arrest in EGFR+/HER2+ breast cancer cell lines. (A) SK-BR-3, MDA-MB-453, and MCF-7 cells were treated with various concentrations of lapatinib for indicated time, and MTT assay was performed to determine the cell viability. (B) SK-BR-3, MDA-MB-453, and MCF-7 cells were exposed to various concentrations of lapatinib for 24 h, or to 0.1 µM lapatinib for indicated time, and cell cycle distribution was determined by flow cytometry analysis, **P < 0.01.
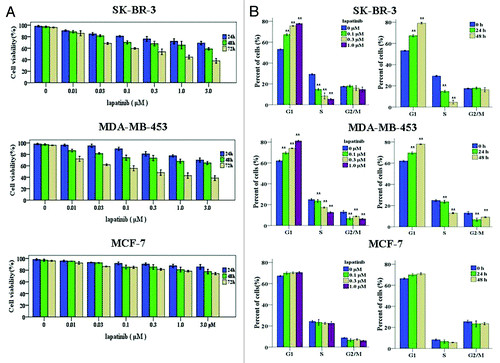
To determine the potential mechanisms by which lapatinib inhibits human breast cancer cell growth, the effect of lapatinib on cell cycle progression was evaluated by flow cytometry. When SK-BR-3 and MBA-MD-453 cells were treated with lapatinib at different doses (0.1, 0.3, 1 µM) for 24 or 48 h, G1 cell cycle arrest was induced in a time- and dose-dependent way (). In contrast, treatment of MCF-7 cells with lapatinib did not induce discernible changes in cell cycle distribution (). Taken together, these results showed that lapatinib could cause G1 phase cell cycle arrest and therefore inhibit cell proliferation in HER2-overexpressing breast cancer cells.
Lapatinib-induced G1 arrest in HER2-overexpressing breast cancer cell lines is dependent on p27
To understand how lapatinib induces G1 cell cycle arrest, we examined the expression and function of p27 in G1/S transition. To this end, SK-BR-3 and MBA-MD-453 breast cancer cells were treated with various concentrations of lapatinib for 48 h, and p27 expression was assessed. As shown in , lapatinib induced p27 expression in a dose-dependent manner. Similarly, when the cells were exposed to 0.1 µM lapatinib for different duration, a time-dependent p27 upregulation was observed ().
Figure 2. Lapatinib-induced G1 arrest in EGFR+/HER2+ breast cancer cell lines is dependent on p27. (A) SK-BR-3 and MDA-MB-453 cells were treated with various concentrations of lapatinib for 24 h, or 0.1 µM lapatinib for indicated time, and p27 protein levels were determined by western blot. (B) SK-BR-3 and MDA-MB-453 cells were transfected with control or p27 siRNA for 48 h, and treated with DMSO or 0.1 µM lapatinib for additional 24 h. Western blot was performed to determine the efficiency of p27 knockdown, and cell cycle distribution was quantified by flow cytometry analysis.
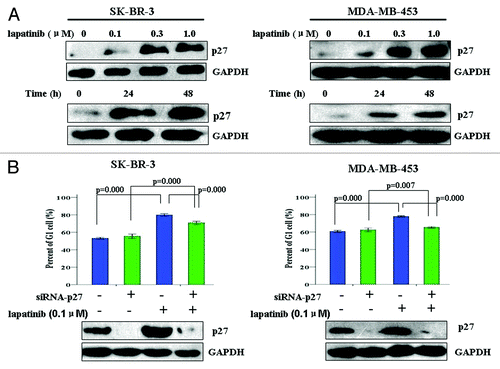
To determine whether lapatinib-induced upregulation of p27 expression is required for its G1 cell cycle arrest effect, we depleted p27 from SK-BR-3 and MBA-MD-453 cells and asked whether this would abolish lapatinib’s effect in cell cycle regulation. As shown in , when the cells were transfected with p27-targeting siRNA, p27 expression was efficiently suppressed, and lapatinib-induced G1 cell cycle arrest was also inhibited. Collectively, these results strongly suggest that lapatinib-induced G1 arrest in HER2-overexpressing breast cancer cell lines is dependent on its upregulation of p27.
Lapatinib increases promoter activity and mRNA levels of p27 in SK-BR-3 cells
Next we sought to determine how lapatinib regulates p27 expression. We first examined whether lapatinib treatment increased p27 transcription. To this end, we exposed SK-BR-3 cells to 0.1 µM lapatinib for 48 h, and assessed p27 mRNA levels by quantitative RT-PCR. As shown in , lapatinib treatment led to a 3.4-fold increase of p27 transcripts in SK-BR-3 cells, suggesting that lapatinib may enhance p27 transcription. To confirm whether this is the case, we used a luciferase reporter assay to assess the p27 promoted activity before and after lapatinib treatment. We transfected a p27 promoter driven luciferase reporter construct into SK-BR-3 cells and treated the cells with 0.1 µM lapatinib. As shown in , 48 h of lapatinib treatment resulted in significantly enhanced p27 promoter activity, as reflected by markedly increased luciferase expression. These data suggest that lapatinib potently upregulates the transcription of p27.
Figure 3. Lapatinb treatment upregulates p27 transcription in SK-BR-3 cells. (A) SK-BR-3 cells were treated with 0.1 µM lapatinib for 48 h, and p27 mRNA levels were determined by qRT-PCR. (B) SK-BR-3 cells were transfected with a p27 promoter driven firefly luciferase construct (p27PF) as well as a β-galactosidase (β-Gal) plasmid (as internal control). Twelve hours after transfection, cells were treated with 0.1 µM lapatinib for 24 or 48 h, and assayed for luciferase activity. (C) SK-BR-3 cells were exposed to 0.1 µM lapatinib for 48 h, and FOXO3a mRNA levels were determined by qRT-PCR. (D) SK-BR-3 cells were exposed to 0.1 µM lapatinib for 48 h, and FOXO3a protein levels were determined by western blot.
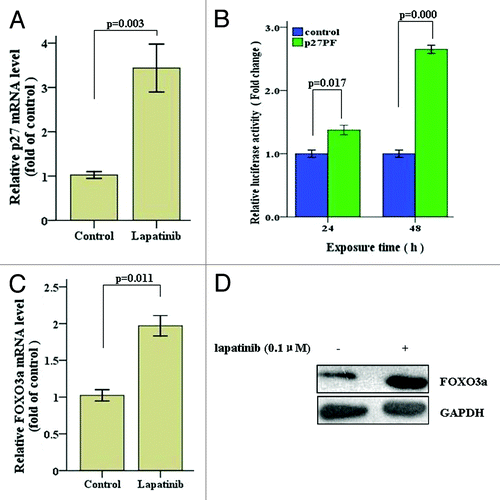
Previous studies have shown that the expression of FOXO3a (forkhead transcription factor in rhabdomyosarcoma-like1), which is one of the principal regulators of p27 gene transcription, correlated with that of p27.Citation34,Citation35 To investigate whether lapatinib enhances transcription of p27 by upregulating FOXO3a expression, we examined FOXO3a mRNA and protein levels upon lapatinib treatment. As shown in , lapatinib significantly upregulates FOXO3a expression, as reflected by increased mRNA level detected by quantitative RT-PCR and protein level detected by western blot. Collectively, these results suggest that lapatinib enhances p27 transcription by upregulating FOXO3a, one of the principle transcription factors that control p27 expression.
Lapatinib induces p27 phosphorylation at Serine10 and inhibits its degradation
To understand whether lapatinib also confers p27 expression regulation at post-translational level, we first measured the protein half-life of p27 in the absence and presence of lapatinib. SK-BR-3 cells were treated with the protein synthesis inhibitor cycloheximide (CHX) together with DMSO or lapatinib and harvested at different time points, and the dynamics of p27 protein levels were measured by western blot. As shown in , SK-BR-3 cells treated with lapatinib showed delayed p27 degradation compared with those treated with DMSO. When p27 expression was normalized against GAPDH (internal control), the half-life of p27 in lapatinib-treated cells was calculated to be ~7.0 h, whereas the half-life of p27 in DMSO-treated cells was ~2.5 h (). These data suggest that lapatinib significantly prolongs the half-life of p27 protein by inhibiting its degradation.
Figure 4. Lapatinib treatment promotes p27 phosphorylation at Serine10 and inhibits p27 degradation in SK-BR-3 cells. (A) SK-BR-3 cells were treated with 0.1 µM lapatinib or DMSO for 48 h, washed, and treated with 5 μg/mL of cycloheximide (CHX) for the indicated time, and p27 protein levels were determined by western blot. Representative blots from multiple experiments are shown. (B) Quantification of p27 protein levels as normalized to the levels of GAPDH. (C) SK-BR-3 cells were treated with 0.1 µM lapatinib for 48 h, and p27 phosphorylation at Serine 10 and Threonine 187 was determined by western blot.
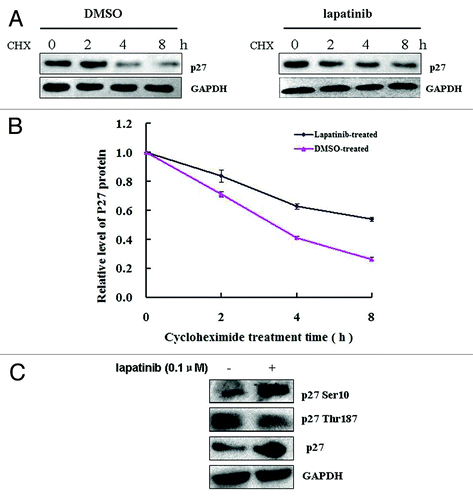
Phosphorylation of p27 protein has been widely recognized to be one of the major post-translational mechanisms that regulate the abundance of this protein.Citation36 To determine whether lapatinib regulates p27 phosphorylation, we exposed SK-BR-3 cells to 0.1 µM lapatinib, and found that lapatinib treatment increased p27 phosphorylation at Ser10 but decreased its phosphorylation at Thr187. Phosphorylation of p27 at Thr187 has been shown to enhance p27 degradation.Citation37,Citation38 Therefore, decreased Thr187 phosphorylation is associated with increased p27 stability. The effect of Ser10 phosphorylation on p27 stability is complicated by different kinases, including DYRK1B and KIST, which inhibit and enhance p27 degradation, respectively.Citation39,Citation40 We found that upon lapatinib treatment, DYRK1B mRNA and protein expression was significantly upregulated (). In contrast, the mRNA and protein levels of KIST were not significantly reduced (). These data suggest that lapatinib may regulate DYRK1B, but not KIST, expression and p27 phosphorylation at Ser10, thereby affecting its stability.
Figure 5. Lapatinib treatment increases DYRK1B expression in SK-BR-3 cells. (A) SK-BR-3 cells were treated with 0.1 µM lapatinib for 48 h, and mRNA expression levels of DYRK1B and KIST were determined by qRT-PCR. (B) SK-BR-3 cells were treated with 0.1 µM lapatinib for 48 h, and protein expression levels of DYRK1B and KIST were determined by western blot. (C) SK-BR-3 cells were transfected with a DYRK1B expressing plasmid or a vector-only plasmid, protein levels of DYRK1B were determined by western blot 48 h posttransfection. (D) SK-BR-3 cells were transfected with a DYRK1B expressing plasmid for 24 h, and then treated with 0.1 µM lapatinib for 48 h. Cell cycle distribution was quantified by flow cytometry analysis.
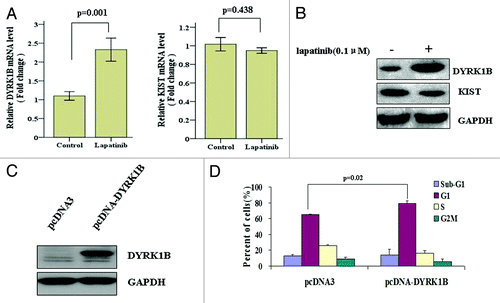
To further determine whether DYRK1B expression and p27 phosphorylation at Ser10 are involved in lapatinib-induced G1 cell cycle arrest, we transfected SK-BR-3 cells with a DYRK1B-expressing plasmid, treated the cells with 0.1 µM lapatinib for 48 h, and examined the cell cycle distribution. We found that overexpression of DYRK1B () led to a further increase of cell population at G1 phase (), suggesting that p27 phosphorylation at Ser10 by DYRK1B enhances its stability and leads to more efficient cell cycle arrest at G1 phase.
ERβ1and ERβ2 affect p27 expression and lapatinib-induced G1 cell cycle arrest
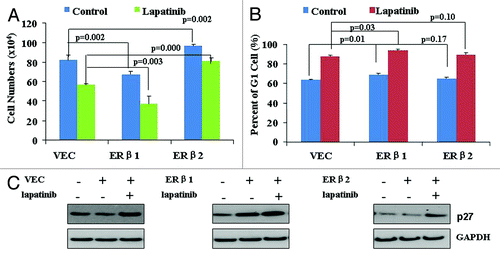
ERβ1 and ERβ2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell lineCitation41 and seem to have different effects on breast cancer cell proliferation.Citation42,Citation43 We asked whether this is also the case in our system, and investigated whether this affects cellular response to lapatinib. To this end, we transiently transfected SK-BR-3 cells with ERβ1- or ERβ2-expressing vectors, and treated them with 0.1 µM lapatinib for 48 h. We found that ERβ1 expression inhibited cell proliferation and enhanced G1 cell cycle arrest, both in the presence and absence of lapatinib (). However, ERβ2 expression increased cell proliferation, although it did not seem to alter cell cycle distribution (). In addition, we found that ERβ1 expression increased p27 expression, while ERβ2 did not affect p27 protein levels (). These results suggest that ERβ1 but not ERβ2 expression could regulate p27 expression, and ER expression status may affect lapatinib-induced p27 expression and cell cycle arrest.
Figure 6. The effects of ERβ1 and ERβ2 on cell proliferation and G1 cell phase arrest and p27 expression. (A) SK-BR-3 cells were transiently transfected with ERβ1- or ERβ2-expressing vectors, and treated 6 h later with 0.1 µM lapatinib for 48 h. Cell proliferation was assayed by cell counting. (B) SK-BR-3 cells were transiently transfected with ERβ1- or ERβ2-expressing vectors and treated 6 h later with 0.1 µM lapatinib for 48 h. Cell cycle distribution was quantified by flow cytometry analysis. (C) SK-BR-3 cells were transiently transfected with ERβ1- or ERβ2-expressing vectors, and treated 6 h later with 0.1 µM lapatinib for 48 h. Protein levels of p27 were determined by western blot.
Discussion
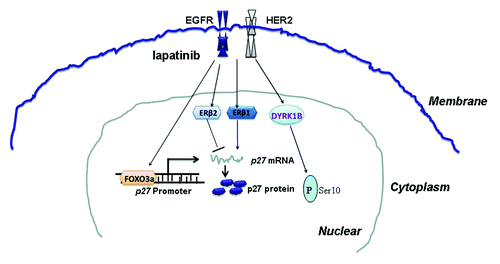
Lapatinib has been shown to induce cell cycle arrest in many cancer cells. It also induces p27 expression, but mechanisms underlying p27 expression regulation remain unclear. In the present study, we confirmed that lapatinib could induce G1 cell cycle arrest and upregulation of p27 expression in HER2-overexpressing breast cancer cells. Importantly, we demonstrated that the lapatinib-induced G1 arrest was dependent on p27, suggesting that regulation of p27 expression is a key mechanism in lapatinib causing cell cycle arrest and cell growth inhibition. In addition, we found that lapatinib regulates p27 expression at both transcriptional and post-translational levels. Moreover, we demonstrated that expression of ERβ1 also regulates p27 expression and affects cell cycle progression. Collectively, our study provides insights into regulation of p27 expression in breast cancer cells by targeted therapy as well as hormone receptors ().
Figure 7. A schematic model for the roles of lapatinib in p27 expression regulation.
Both promoter activity and mRNA level of p27 were increased upon lapatinib treatment of SK-BR-3 cells. Lapatinib may exert its effects on p27 transcription through multiple transcription factors, including forkhead transcription factor, c-myc, E2F, SP1, and NF-Y,Citation44-Citation46 which are all known to bind directly to the p27 promoter. In our study, we found that lapatinib treatment increased FOXO3a expression, indicating that FOXO3a may mediate the activation of p27 promoter in this setting. However, whether other transcription factors are also involved in the lapatinib-induced transcriptional regulation of p27 needs to be elucidated. A possible approach to identify the transcription factors responsible for lapatinib induced p27 transcription activation would be through microarray analysis. Understanding which transcription factors are altered by lapatinib treatment not only could help to further elucidate the detailed transcription regulation of p27 by this drug, but also to explore other mechanisms through which lapatinib exerts its cell cycle and cell proliferation regulation.
We demonstrated that lapatinib increased the half-life of p27 protein by regulating its phosphorylation, thereby inhibiting its degradation. Thr187 and Ser10 are 2 major sites of p27 phosphorylation that regulate p27 subcellular localization, stability, and function in the cells.Citation47 Lapatinib is able to decrease p27 phosphorylation at Thr187 and increase p27 phosphorylation at Ser10. The process of p27 protein degradation partially depends on phosphorylation at Thr187 by cyclin E/CDK2 complex, which binds to ubiquitin ligase SCFSkp2, leading to proteasome degradation.Citation48,Citation49 Therefore, by reducing phosphorylation of p27 protein at Thr187, lapatinib could stabilize p27 in SK-BR-3 cells. Phosphorylation of Ser10 also contributes to regulation of the stability of this protein. Ser10 phosphorylation is catalyzed by DYRK1B as well as KIST but is associated with different outcomes. p27 phosphorylation at Ser10 by KIST increases p27 binding through its nuclear export signal (NES) to CRM1 (or exportin 1), thereby promoting relocation of p27 from nucleus to cytoplasm, where it is degraded.Citation50,Citation51 In contrast, dual-specificity tyrosine-phosphorylationregulated kinase 1B (DYRK1B, also called Mirk), a member of the conserved Dyrk/minibrain family of tyrosine regulated kinases, was shown to phosphorylate p27 Ser10 and stabilize p27 during the G0 phase of the cell cycle by sequestrating it within the nucleus, where it can bind to CDK2 and induce cell cycle arrest.Citation40 Although how cells can recognize and differentiate phosphorylation of the same site catalyzed by different kinases, leading to different outcomes, remains an interesting question, our data clearly show that lapatinib treatment increases DYRK1B, but not KIST expression. Thus, lapatinib inhibits nuclear export of p27 and stabilizes the protein in the nucleus, preventing its degradation.
Estrogen receptor (ER) β was discovered over a decade ago. Recent studies indicate that ERβ has an anti-proliferative effect in breast cancer cell lines and in vivo.Citation22,Citation23,Citation52 However, ERβ has multiple isoforms, and their functions are not quite clear. We found that ERβ1 but not ERβ2 expression could upregulate p27 and enhance lapatinib-induced G1 cell cycle arrest and cell proliferation inhibition in SK-BR-3 cells. These results suggest that ER expression status may affect lapatinib-induced p27 expression and cell cycle arrest. Therefore, more detailed analysis of ER status in breast cancer patients may be warranted, since expression of different ERβ isoforms may predict different responses to lapatinib. Moreover, further functional studies of ERα and different ERβ isoforms are needed, so that anti-estrogen therapies and other targeted therapies could be better formulated in a personalized manner.
Materials and Methods
Reagents
Lapatinib with a purity of 99% was purchased from Selleck Chemicals. 3-[4, 5-dimethylthiazol-2-yl]-2, 5-diphenyl tetrazolium bromide (MTT), dimethyl sulfoxide (DMSO), and cycloheximide (CHX) were from Sigma Chemicals. Cell Cycle Detection Kit was a product of Nanjing Kaiji Co. The antibodies used included the following: anti-p27 Ser10 (Abcam), anti-p27 Thr187 (Santa Cruz), anti-p27 (610241, BD Biosciences), anti-DYRK1B (AP7538b, Abgent), and anti-KIST (AP8067a, Abgent). All culture media and serum were obtained from GIBCO/BRL Life Technologies, Inc.
Cell lines and cell culture
SK-BR-3, MDA-MB-453, and MCF-7 cell lines were purchased from the American Type Culture Collection. The cells were cultured in RPMI 1640 medium (GIBCO) supplemented with 10% fetal bovine serum (FBS), 100 U/ml penicillin, and 100 μg/ml streptomycin, and maintained at 37°C with 5% CO2 in a humidified atmosphere.
Cell viability
The effect of lapatinib on the viability of cells was determined by MTT assay. Human breast cancer cells were plated at a density of 3 × 104 cells per well in flat-bottomed 96-well plates. The next day, cells were provided with fresh medium including lapatinib at doses of 0.01, 0.03, 0.1, 0.3, 1.0, 3.0 µM for another 24, 48, 72 h. After that the cells were incubated with 5 mg/L MTT solution at 37 °C for 4 h. The resulting crystal was dissolved in 200 μl DMSO per well, and the plates were read on microplate reader at 570 nm. The effect of lapatinib on growth inhibition was assessed as percent cell viability, where DMSO-treated cells were taken as 100% viable. DMSO at the concentrations used was without any effect on cell viability. Assays were performed 3 times independently.
Flow cytometry analysis
The cell cycle distribution was examined by flow cytometry. Briefly, the cells cultured with or without lapatinib, were trypsinized, washed with PBS, and fixed with 70% ethanol overnight at 4 °C. The fixed cells were washed with PBS and resuspended at the concentration of 1 × 106 cells/ml in a staining solution containing PI (20 μg/ml) and DNase-free RNase (100 μg/ml) and incubated for 30 min at 37°C in the dark. Measurements and analyses were performed on a FACS Calibur flow cytometer with CELLQUEST software (BD Biosciences, San Diego). For each experiment, 30 000 events per sample were recorded.
RNA interference
p27-siRNA and nonspecific (control) siRNA were from Santa Cruz Biotechnology. Cells were seeded in 6-well culture plates at 2.5 × 105 cells per well (30–50% confluence) in RPMI-1640 medium supplemented with 10% fetal bovine serum. The next day, cells were transfected with p27 siRNA or a control siRNA at a final concentration of 100 nM by Lipofectamine2000 (Invitrogen). The next day, cells were fed with fresh medium with DMSO or lapatinib (0.1µM) for 48 h, and harvested for cell cycle analysis or western blotting.
Transient transfections
SK-BR-3 cells at 60–70% confluency were seeded in a 12-well plate with 10% FBS. After 24 h, the cells were transiently transfected with Lipofectamine (Invitrogen, Inc) according to the manufacturer's protocol. Two hours before the transfection, the cells were fed with serum-free medium. Six hours after the transfection, the cells were fed with complete RPMI1640 medium, containing 10% FBS. Transient transfection of SK-BR-3 cells with ERβ1 or ERβ2 expression plasmid has previously been described.Citation41
Cell proliferation assay
Three × 105 SK-BR-3 cells were seeded in a 12-well plate with 10% FBS. After 24 h, the cells were transiently transfected with ERβ1- and ERβ2-expressing vectors or the control vectors. Six hours after the transfection, the cells were fed with complete RPMI1640 medium containing 10% FBS. Forty-eight hours after transfection, the cells were counted with a Neubauer chamber for cell proliferation assay. In addition, cell proliferation was quantified by an MTS [3-(4,5-dimethylthiazol-2-yl)-5-(3-carboxymethoxyphenyl)-2-(4-sulfophenyl)- 2H-tetrazolium] assay on a 96-well plate, as described in the manufacturer’s protocol (Promega). The experiments were performed in triplicates and were repeated 3 times.
Western blot analysis
Cells were lysed in RIPA buffer supplemented with protease and phosphatase inhibitors. The cell suspension was kept on ice for 20 min and then centrifuged at 15 000 × g for 15 min at 4 °C. Protein concentration was quantified with the BCA Protein Assay Kit. Protein samples were separated on a sodium dodecylsulfate-polyacrylamide gel and blotted onto nitrocellulose. Blots were blocked with 5% dry milk in tris-buffered saline/0.1% tween-20 and incubated overnight with a diluted solution of primary antibody, and then with the horseradish peroxidase-conjugated secondary antibody (1:50 000) for 2 h. The specific antibodies used were mouse anti-p27 (1:100 dilution), rabbit anti-phospho-p27 (1:1000 dilution), rabbit anti-MIRK/DYRK1B (1:100 dilution), rabbit anti-KIST (1:100 dilution), and mouse anti-GAPDH (1:5000 dilution). The blot was detected with the enhanced chemiluminescence detection system (Santa Cruz Biotechnology).
Quantitative real-time polymerase chain reaction (qRT-PCR)
Total RNA was isolated using the TRIzol reagent (Invitrogen) and was reverse transcribed using a PrimeScript First Strand cDNA Synthesis Kit (TaKaRa) according to the manufacturer’s instructions. qRT-PCR was performed with SYBR®PrimeScript® RT-PCR Kit II. qPCR reactions for indicated genes were performed using SYBR green qPCR kit by a fluorescent temperature cycler (Mx3000P Real Time PCR System; Stratagene). Sequences of primers (forward and reverse, respectively) were as follows: p27: 5′-GTCAAACGTAAACAGCTCGAAT-3′ and 5′-TGCATAATGCTACATCCAACG-3′; DYRK1B: 5′-GAGCTGATGAACCAGCATGA-3′ and 5′-CCTCAGGTGAGCGGTAGAAG-3′; KIST: 5′-CATTCCTTTTGCCCCTCATA-3′ and 5′-GCTTTGGAATCACCAGCATT-3′; FOXO3a: 5′-AGGGAAGTTTGGTCAATCAGAA-3′ and 5′-TGGAGATGAGGGAATCAAAGTT-3′; GAPDH:5′-CCATGGAGAAGGCTGGGG-3′ and 5′-CAAAGTTGTCATGGATGACC-3′. Transcript quantification was done in triplicate for every sample and reported relative to GAPDH. The Relative gene expression was quantified according to the comparative Ct method using GAPDH as an internal standard.Citation53,Citation54
Luciferase reporter assays
The reporter plasmids p27PF-Luc and control plasmid, pGVB2, were the generous gifts of Dr Toshiyuki Sakai (Kyoto Prefectural University of Medicine). SK-BR-3 cells (3 × 105 cells per well) were seeded into 24-well plates and transfected the second day using Lipofectamine 2000 (Invitrogen). For transfection experiments, 0.8 μg of reporter plasmid, or pGVB2 plasmid and 0.2 μg of pCMV-β Gal expression plasmid were used. After 24 h of transfection, lapatinib, or DMSO (vehicle) were added and incubated for another 24 or 48 h. Cells were washed twice with cold PBS and lysed with 150 μl of cell culture lysis reagent (Promega). Thirty microliters of cell extract was used for luciferase activity assays, and 10 μl of cell extract was used for β-Gal assays. LumiCount was used to quantitate luciferase activity (Luciferase Assay System, Promega), and the β-Gal assay kit (Invitrogen, cat. #45-0449) was used for β-Gal activities following the kit protocol. All transfections were normalized by measuring β-galactosidase activity of the samples. The luciferase activity was normalized to β-Gal activity. Data are the average of at least 3 independent experiments, and error bars indicate standard deviation.
Statistical analysis
The results were expressed as mean ± SE calculated from the specified numbers of determination. A Student t test was used to compare individual data with control value. All analyses were performed at a significance level of P < 0.05 using SPSS version 16.0.
| Abbreviations: | ||
| HER2 | = | human epidermal growth factor receptor 2 |
| TKIs | = | tyrosine kinase inhibitors |
| CDKs | = | cyclin-dependent kinase |
| CDKI | = | cyclin-dependent kinase inhibitors |
| Ser | = | serine |
| Thr | = | threonine |
| PI3K | = | phosphatidylinositol 3-kinase |
| MAPK | = | mitogen-activated protein |
| DYRK1B | = | dual-specificity tyrosine-phosphorylation regulated kinase 1B |
| qPCR | = | quantitative real-time PCR |
| MTT | = | 3-(4,5-Dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| FOXO3a | = | Forkhead box O3a |
| CHX | = | cycloheximide |
Acknowlegements
This work was supported by The Cancer Prevention and Research Institute of Texas Grants HIRP100680 and RP110444, the Texas Emerging Technology Fund under Agreement 300-9-1958, the Robert A. Welch Foundation Grant E-0004, and the Swedish Cancer Fund (to JAG), and by grants from National Natural Science Foundation of China (no.81272252), Natural Science Foundation of Jiangsu Province (no. BK2011656) (to XG).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Jemal A, Bray F, Center MM, Ferlay J, Ward E, Forman D. Global cancer statistics. CA Cancer J Clin 2011; 61:69 - 90; http://dx.doi.org/10.3322/caac.20107; PMID: 21296855
- Cufí S, Vazquez-Martin A, Oliveras-Ferraros C, Corominas-Faja B, Urruticoechea A, Martin-Castillo B, et al. Autophagy-related gene 12 (ATG12) is a novel determinant of primary resistance to HER2-targeted therapies: utility of transcriptome analysis of the autophagy interactome to guide breast cancer treatment. Oncotarget 2012; 3:1600 - 14; PMID: 23307622
- Waterhouse BR, Gijsen M, Barber PR, Tullis ID, Vojnovic B, Kong A. Assessment of EGFR/HER2 dimerization by FRET-FLIM utilizing Alexa-conjugated secondary antibodies in relation to targeted therapies in cancers. Oncotarget 2011; 2:728 - 36; PMID: 21908901
- Slamon DJ, Godolphin W, Jones LA, Holt JA, Wong SG, Keith DE, et al. Studies of the HER-2/neu proto-oncogene in human breast and ovarian cancer. Science 1989; 244:707 - 12; http://dx.doi.org/10.1126/science.2470152; PMID: 2470152
- Esteva FJ, Cheli CD, Fritsche H, Fornier M, Slamon D, Thiel RP, et al. Clinical utility of serum HER2/neu in monitoring and prediction of progression-free survival in metastatic breast cancer patients treated with trastuzumab-based therapies. Breast Cancer Res 2005; 7:R436 - 43; http://dx.doi.org/10.1186/bcr1020; PMID: 15987448
- Piccart-Gebhart MJ, Procter M, Leyland-Jones B, Goldhirsch A, Untch M, Smith I, et al, Herceptin Adjuvant (HERA) Trial Study Team. Trastuzumab after adjuvant chemotherapy in HER2-positive breast cancer. N Engl J Med 2005; 353:1659 - 72; http://dx.doi.org/10.1056/NEJMoa052306; PMID: 16236737
- Xia W, Mullin RJ, Keith BR, Liu LH, Ma H, Rusnak DW, et al. Anti-tumor activity of GW572016: a dual tyrosine kinase inhibitor blocks EGF activation of EGFR/erbB2 and downstream Erk1/2 and AKT pathways. Oncogene 2002; 21:6255 - 63; http://dx.doi.org/10.1038/sj.onc.1205794; PMID: 12214266
- Gomez HL, Doval DC, Chavez MA, Ang PC, Aziz Z, Nag S, et al. Efficacy and safety of lapatinib as first-line therapy for ErbB2-amplified locally advanced or metastatic breast cancer. J Clin Oncol 2008; 26:2999 - 3005; http://dx.doi.org/10.1200/JCO.2007.14.0590; PMID: 18458039
- Kim JW, Kim HP, Im SA, Kang S, Hur HS, Yoon YK, et al. The growth inhibitory effect of lapatinib, a dual inhibitor of EGFR and HER2 tyrosine kinase, in gastric cancer cell lines. Cancer Lett 2008; 272:296 - 306; http://dx.doi.org/10.1016/j.canlet.2008.07.018; PMID: 18774637
- Diaz R, Nguewa PA, Parrondo R, Perez-Stable C, Manrique I, Redrado M, et al. Antitumor and antiangiogenic effect of the dual EGFR and HER-2 tyrosine kinase inhibitor lapatinib in a lung cancer model. BMC Cancer 2010; 10:188; http://dx.doi.org/10.1186/1471-2407-10-188; PMID: 20459769
- Belletti B, Baldassarre G. New light on p27(kip1) in breast cancer. Cell Cycle 2012; 11:3701 - 2; http://dx.doi.org/10.4161/cc.21573; PMID: 22895010
- Akli S, Zhang XQ, Bondaruk J, Tucker SL, Czerniak PB, Benedict WF, et al. Low molecular weight cyclin E is associated with p27-resistant, high-grade, high-stage and invasive bladder cancer. Cell Cycle 2012; 11:1468 - 76; http://dx.doi.org/10.4161/cc.19882; PMID: 22441703
- Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501 - 12; http://dx.doi.org/10.1101/gad.13.12.1501; PMID: 10385618
- Hengst L, Reed SI. Inhibitors of the Cip/Kip family. Curr Top Microbiol Immunol 1998; 227:25 - 41; http://dx.doi.org/10.1007/978-3-642-71941-7_2; PMID: 9479824
- Nigg EA. Targets of cyclin-dependent protein kinases. Curr Opin Cell Biol 1993; 5:187 - 93; http://dx.doi.org/10.1016/0955-0674(93)90101-U; PMID: 8507490
- Guan X, Wang Y, Xie R, Chen L, Bai J, Lu J, et al. p27(Kip1) as a prognostic factor in breast cancer: a systematic review and meta-analysis. J Cell Mol Med 2010; 14:944 - 53; http://dx.doi.org/10.1111/j.1582-4934.2009.00730.x; PMID: 19298520
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253 - 67; http://dx.doi.org/10.1038/nrc2347; PMID: 18354415
- Li J, Cho YY, Langfald A, Carper A, Lubet RA, Grubbs CJ, et al. Lapatinib, a preventive/therapeutic agent against mammary cancer, suppresses RTK-mediated signaling through multiple signaling pathways. Cancer Prev Res (Phila) 2011; 4:1190 - 7; http://dx.doi.org/10.1158/1940-6207.CAPR-10-0330; PMID: 21791570
- Chu I, Blackwell K, Chen S, Slingerland J. The dual ErbB1/ErbB2 inhibitor, lapatinib (GW572016), cooperates with tamoxifen to inhibit both cell proliferation- and estrogen-dependent gene expression in antiestrogen-resistant breast cancer. Cancer Res 2005; 65:18 - 25; PMID: 15665275
- Ikeda K, Inoue S. Estrogen receptors and their downstream targets in cancer. Arch Histol Cytol 2004; 67:435 - 42; http://dx.doi.org/10.1679/aohc.67.435; PMID: 15781984
- Al-Dhaheri M, Wu J, Skliris GP, Li J, Higashimato K, Wang Y, et al. CARM1 is an important determinant of ERα-dependent breast cancer cell differentiation and proliferation in breast cancer cells. Cancer Res 2011; 71:2118 - 28; http://dx.doi.org/10.1158/0008-5472.CAN-10-2426; PMID: 21282336
- Lazennec G, Bresson D, Lucas A, Chauveau C, Vignon F. ER beta inhibits proliferation and invasion of breast cancer cells. Endocrinology 2001; 142:4120 - 30; http://dx.doi.org/10.1210/en.142.9.4120; PMID: 11517191
- Paruthiyil S, Parmar H, Kerekatte V, Cunha GR, Firestone GL, Leitman DC. Estrogen receptor beta inhibits human breast cancer cell proliferation and tumor formation by causing a G2 cell cycle arrest. Cancer Res 2004; 64:423 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-03-2446; PMID: 14729654
- Ogawa S, Inoue S, Watanabe T, Orimo A, Hosoi T, Ouchi Y, et al. Molecular cloning and characterization of human estrogen receptor betacx: a potential inhibitor ofestrogen action in human. Nucleic Acids Res 1998; 26:3505 - 12; http://dx.doi.org/10.1093/nar/26.15.3505; PMID: 9671811
- Ström A, Hartman J, Foster JS, Kietz S, Wimalasena J, Gustafsson JA. Estrogen receptor beta inhibits 17beta-estradiol-stimulated proliferation of the breast cancer cell line T47D. Proc Natl Acad Sci U S A 2004; 101:1566 - 71; http://dx.doi.org/10.1073/pnas.0308319100; PMID: 14745018
- Mak P, Leav I, Pursell B, Bae D, Yang X, Taglienti CA, et al. ERbeta impedes prostate cancer EMT by destabilizing HIF-1alpha and inhibiting VEGF-mediated snail nuclear localization: implications for Gleason grading. Cancer Cell 2010; 17:319 - 32; http://dx.doi.org/10.1016/j.ccr.2010.02.030; PMID: 20385358
- Yan M, Rayoo M, Takano EA, Fox SB, kConFab Investigators. Nuclear and cytoplasmic expressions of ERβ1 and ERβ2 are predictive of response to therapy and alters prognosis in familial breast cancers. Breast Cancer Res Treat 2011; 126:395 - 405; http://dx.doi.org/10.1007/s10549-010-0941-9; PMID: 20490651
- Shaaban AM, Green AR, Karthik S, Alizadeh Y, Hughes TA, Harkins L, et al. Nuclear and cytoplasmic expression of ERbeta1, ERbeta2, and ERbeta5 identifies distinct prognostic outcome for breast cancer patients. Clin Cancer Res 2008; 14:5228 - 35; http://dx.doi.org/10.1158/1078-0432.CCR-07-4528; PMID: 18698041
- Thomas C, Rajapaksa G, Nikolos F, Hao R, Katchy A, McCollum CW, et al. ERβ1 represses basal-like breast cancer epithelial to mesenchymal transition by destabilizing EGFR. Breast Cancer Res 2012; 14:R148; http://dx.doi.org/10.1186/bcr3358; PMID: 23158001
- Simpkins F, Hevia-Paez P, Sun J, Ullmer W, Gilbert CA, da Silva T, et al. Src Inhibition with saracatinib reverses fulvestrant resistance in ER-positive ovarian cancer models in vitro and in vivo. Clin Cancer Res 2012; 18:5911 - 23; http://dx.doi.org/10.1158/1078-0432.CCR-12-1257; PMID: 22896656
- Cariou S, Donovan JC, Flanagan WM, Milic A, Bhattacharya N, Slingerland JM. Down-regulation of p21WAF1/CIP1 or p27Kip1 abrogates antiestrogen-mediated cell cycle arrest in human breast cancer cells. Proc Natl Acad Sci U S A 2000; 97:9042 - 6; http://dx.doi.org/10.1073/pnas.160016897; PMID: 10908655
- D’Alessio A, De Luca A, Maiello MR, Lamura L, Rachiglio AM, Napolitano M, et al. Effects of the combined blockade of EGFR and ErbB-2 on signal transduction and regulation of cell cycle regulatory proteins in breast cancer cells. Breast Cancer Res Treat 2010; 123:387 - 96; http://dx.doi.org/10.1007/s10549-009-0649-x; PMID: 19946741
- Hegde PS, Rusnak D, Bertiaux M, Alligood K, Strum J, Gagnon R, et al. Delineation of molecular mechanisms of sensitivity to lapatinib in breast cancer cell lines using global gene expression profiles. Mol Cancer Ther 2007; 6:1629 - 40; http://dx.doi.org/10.1158/1535-7163.MCT-05-0399; PMID: 17513611
- Dijkers PF, Medema RH, Pals C, Banerji L, Thomas NS, Lam EW, et al. Forkhead transcription factor FKHR-L1 modulates cytokine-dependent transcriptional regulation of p27(KIP1). Mol Cell Biol 2000; 20:9138 - 48; http://dx.doi.org/10.1128/MCB.20.24.9138-9148.2000; PMID: 11094066
- Lynch RL, Konicek BW, McNulty AM, Hanna KR, Lewis JE, Neubauer BL, et al. The progression of LNCaP human prostate cancer cells to androgen independence involves decreased FOXO3a expression and reduced p27KIP1 promoter transactivation. Mol Cancer Res 2005; 3:163 - 9; http://dx.doi.org/10.1158/1541-7786.MCR-04-0163; PMID: 15798096
- Slingerland J, Pagano M. Regulation of the cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10 - 7; http://dx.doi.org/10.1002/(SICI)1097-4652(200004)183:1<10::AID-JCP2>3.0.CO;2-I; PMID: 10699961
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997; 16:5334 - 44; http://dx.doi.org/10.1093/emboj/16.17.5334; PMID: 9311993
- DeSalle LM, Pagano M. Regulation of the G1 to S transition by the ubiquitin pathway. FEBS Lett 2001; 490:179 - 89; http://dx.doi.org/10.1016/S0014-5793(01)02121-4; PMID: 11223033
- Deng X, Ewton DZ, Mercer SE, Friedman E. Mirk/dyrk1B decreases the nuclear accumulation of class II histone deacetylases during skeletal muscle differentiation. J Biol Chem 2005; 280:4894 - 905; http://dx.doi.org/10.1074/jbc.M411894200; PMID: 15546868
- Deng X, Mercer SE, Shah S, Ewton DZ, Friedman E. The cyclin-dependent kinase inhibitor p27Kip1 is stabilized in G(0) by Mirk/dyrk1B kinase. J Biol Chem 2004; 279:22498 - 504; http://dx.doi.org/10.1074/jbc.M400479200; PMID: 15010468
- Dey P, Jonsson P, Hartman J, Williams C, Ström A, Gustafsson JA. Estrogen receptors β1 and β2 have opposing roles in regulating proliferation and bone metastasis genes in the prostate cancer cell line PC3. Mol Endocrinol 2012; 26:1991 - 2003; http://dx.doi.org/10.1210/me.2012.1227; PMID: 23028063
- Murphy LC, Peng B, Lewis A, Davie JR, Leygue E, Kemp A, et al. Inducible upregulation of oestrogen receptor-beta1 affects oestrogen and tamoxifen responsiveness in MCF7 human breast cancer cells. J Mol Endocrinol 2005; 34:553 - 66; http://dx.doi.org/10.1677/jme.1.01688; PMID: 15821116
- Fox EM, Davis RJ, Shupnik MA. ERbeta in breast cancer--onlooker, passive player, or active protector?. Steroids 2008; 73:1039 - 51; http://dx.doi.org/10.1016/j.steroids.2008.04.006; PMID: 18501937
- Yang W, Shen J, Wu M, Arsura M, FitzGerald M, Suldan Z, et al. Repression of transcription of the p27(Kip1) cyclin-dependent kinase inhibitor gene by c-Myc. Oncogene 2001; 20:1688 - 702; http://dx.doi.org/10.1038/sj.onc.1204245; PMID: 11313917
- Wang C, Hou X, Mohapatra S, Ma Y, Cress WD, Pledger WJ, et al. Activation of p27Kip1 Expression by E2F1. A negative feedback mechanism. J Biol Chem 2005; 280:12339 - 43; http://dx.doi.org/10.1074/jbc.C400536200; PMID: 15713665
- Inoue T, Kamiyama J, Sakai T. Sp1 and NF-Y synergistically mediate the effect of vitamin D(3) in the p27(Kip1) gene promoter that lacks vitamin D response elements. J Biol Chem 1999; 274:32309 - 17; http://dx.doi.org/10.1074/jbc.274.45.32309; PMID: 10542271
- De Vita F, Riccardi M, Malanga D, Scrima M, De Marco C, Viglietto G. PKC-dependent phosphorylation of p27 at T198 contributes to p27 stabilization and cell cycle arrest. Cell Cycle 2012; 11:1583 - 92; http://dx.doi.org/10.4161/cc.20003; PMID: 22441823
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997; 11:1464 - 78; http://dx.doi.org/10.1101/gad.11.11.1464; PMID: 9192873
- Tsvetkov LM, Yeh KH, Lee SJ, Sun H, Zhang H. p27(Kip1) ubiquitination and degradation is regulated by the SCF(Skp2) complex through phosphorylated Thr187 in p27. Curr Biol 1999; 9:661 - 4; http://dx.doi.org/10.1016/S0960-9822(99)80290-5; PMID: 10375532
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J 2001; 20:6672 - 82; http://dx.doi.org/10.1093/emboj/20.23.6672; PMID: 11726503
- Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem 2002; 277:14355 - 8; http://dx.doi.org/10.1074/jbc.C100762200; PMID: 11889117
- Wada-Hiraike O, Hiraike H, Okinaga H, Imamov O, Barros RP, Morani A, et al. Role of estrogen receptor beta in uterine stroma and epithelium: Insights from estrogen receptor beta-/- mice. Proc Natl Acad Sci U S A 2006; 103:18350 - 5; http://dx.doi.org/10.1073/pnas.0608861103; PMID: 17110437
- Chen W, Wei F, Xu J, Wang Y, Chen L, Wang J, et al. Trastuzumab enhances the anti-tumor effects of the histone deacetylase inhibitor sodium butyrate on a HER2-overexpressing breast cancer cell line. Int J Mol Med 2011; 28:985 - 91; PMID: 21887460
- Le XF, Pruefer F, Bast RC Jr.. HER2-targeting antibodies modulate the cyclin-dependent kinase inhibitor p27Kip1 via multiple signaling pathways. Cell Cycle 2005; 4:87 - 95; http://dx.doi.org/10.4161/cc.4.1.1360; PMID: 15611642