Abstract
Polycomb protein histone methyltransferase, enhancer of Zeste homolog 2 (EZH2), is frequently overexpressed in human malignancy and is implicated in cancer cell proliferation and invasion. However, it is largely unknown whether EZH2 has a role in modulating the DNA damage response. Here, we show that polycomb repressive complex 2 (PRC2) is recruited to sites of DNA damage. This recruitment is independent of histone 2A variant X (H2AX) and the PI-3-related kinases ATM and DNA-PKcs. We establish that PARP activity is required for retaining PRC2 at sites of DNA damage. Furthermore, depletion of EZH2 in cells decreases the efficiency of DSB repair and increases sensitivity of cells to gamma-irradiation. These data unravel a crucial role of PRC2 in determining cancer cellular sensitivity following DNA damage and suggest that therapeutic targeting of EZH2 activity might serve as a strategy for improving conventional chemotherapy in a given malignancy.
Introduction
Polycomb group proteins (PcG) are essential epigenetic gene regulators involved in embryonic development and the maintenance of stem cells.Citation1 They exist in 2 main biochemical complexes, Polycomb repressive complex 1 and 2 (PRC1/PRC2).Citation1 These complexes are responsible for modifying chromatin with post-translational modifications associated with gene repression. PRC2 consists of an active methyltransferase, enhancer of Zeste homolog 2 (EZH2), and supporting proteins suppressor of Zeste 12 (SUZ12) and embryonic ectoderm development (EED). Together, these 3 proteins are responsible for the majority of trimethylation of histone 3 at lysine 27 (H3K27me3).Citation2 H3K27me3 is a marker of gene repression and serves as a recognition site for the chromodomain in the PRC1-associated paralogs CBX2, CBX4, CBX6, CBX7, and CBX8.Citation3-Citation5 PRC1 also consists of the mammalian polyhomeotic homolog (PH1, 2, or 3) and 2 ring domain-containing subunits, posterior sex combs homolog (PCGF 1–6), and the dRING homologs (RING1, RING2). PRC1 possesses E3-ubiquitin ligase activity that ubiquitylates lysine 119 on histone 2A (H2AK119).Citation6 Together, these 2 post-translational modifications repress gene transcription. Exactly how PcG proteins are targeted to specific genes is not entirely known, but it is known that PcG proteins are necessary during embryogenesis and for the maintenance of the stem cell phenotype. Knockout of PcG proteins causes embryonic lethality and hematopoietic, breast, and neuronal stem-cell depletion.Citation1,Citation3
Recent investigation of PcG proteins has revealed new roles outside of embryonic development and stem cell maintenance. It has recently been shown that PcG proteins play an important role in DNA double-strand break (DSB) repair. The PRC1 member, RING 2, has been shown to ubiquitylate histone 2A (H2A) and histone 2A variant X (H2AX) in response to DSBs.Citation7,Citation8 Post-translational modifications of H2AX, both phosphorylation and ubiquitylation, are essential for the recruitment and retention of several DSB repair proteins, such as MDC1 and RNF168.Citation9,Citation10 It was further shown that knockdown of BMI-1 decreases DSB repair efficiency and increases cellular sensitivity to ionizing radiation.Citation7 In addition to BMI-1 and RING 2, almost all PcG proteins have been shown to recruit to sites of laser-induced DNA damage, but their role in the DNA damage response remains elusive.Citation7,Citation11,Citation12
As mentioned previously, one current accepted mechanism of PcG gene repression requires PRC2 to first modify chromatin with H3K27me3, which is then recognized by PRC1.Citation4,Citation5 This raises an interesting hypothesis that PRC2 may in fact also play a role in the DSB repair pathway, and that PRC2 may be necessary for the recruitment of PRC1. There is already evidence supporting a role for PRC2 in the DSB repair pathway. It has been shown that PRC2 members do recruit to DNA damage sitesCitation13,Citation14; however, the recruitment mechanism and type of DNA damage PRC2 responds to are still unclear. One study showed PcG protein, MEL-18, recruitment at DNA damage to be PARP dependent,Citation11 while another report showed PHF1, a component in a small subset of PcG complexes, being Ku80-dependent.Citation15 The mechanism of PRC2 recruitment to DNA damage requires further evaluation. Furthermore, the methylation target of PRC2 at sites of DNA damage is also unclear. Some studies show increased H3K27me3 at DNA damage sites,Citation13,Citation14 but others report no increase in H3K27me3.Citation16 PRC2 has been implicated in the methylation of various targets besides H3K27,Citation17,Citation18 and thus it is possible there is a different methylation target at sites of DNA damage. EZH2 has been further implicated in cancer cells through regulation of the DNA damage checkpoint protein, Chk1, through unknown mechanisms.Citation19 It is evident that PRC2 plays an important role in the DNA damage response; however, this role needs to be further clarified and investigated.
EZH2 overexpression is prevalent in cancer, and the poor prognosis associated with EZH2 overexpression could reflect an increased capacity to repair DNA damage induced by radiation or chemotherapy treatments.Citation20 EZH2 is overexpressed in breast, prostate, colon, and other cancers and correlates with a very aggressive, metastatic phenotype (reviewed in ref. Citation20). In addition, it has been shown that overexpression of EZH2 can induce a more stem-like phenotype in cancers (reviewed in ref. Citation21), and that less differentiated cell types are typically more resistant to radiation and chemotherapy.Citation22
To directly test the role of EZH2 in the DNA damage response, we used laser-microirradiation to introduce localized DNA damage, and we then monitored the recruitment of GFP-tagged PRC2 proteins to the damage. We found that PRC2 proteins recruit with kinetics similar to other early DSB repair proteins. This recruitment is independent of H2AX, DNA-PKcs, and ATM. We also found that depletion of EZH2 decreased DSB repair and increased cellular sensitivity to ionizing radiation, demonstrating the importance of PRC2 in DNA damage repair. Surprisingly, we found that EZH2 does not recruit to ionizing radiation-induced foci, but chromatin IP experiments demonstrate that it does recruit to restriction enzyme-mediated double-strand breaks. Our data collectively implicates PRC2 in the DSB repair pathway and supports other findings that PcG proteins are important in DSB repair and cellular survival following ionizing radiation exposure.
Results
PRC2 proteins recruit to laser microirradiation-induced DNA damage tracks
We have previously shown that PRC1 recruits to DSBs and participates in DNA repair.Citation23 Although PRC1 recruitment was not inhibited by EZH2 knockdown, we wished to determine whether or not PRC2 also participates in the DNA damage response. We used laser micro-irradiation to introduce damage and monitored the recruitment of GFP-tagged PRC2 components to DNA damage sites in real-time. This approach is commonly employed to study the recruitment of DSB repair proteins,Citation13,Citation15,Citation23,Citation24 although it introduces other types of DNA damage as well. Laser micro-irradiation involves first sensitizing cells with a DNA intercalator, such as Hoechst. The DNA intercalator is then excited using a laser, and this leads to damage to the DNA in a region that the experimentalist predetermines. DNA damage is introduced through the generation of free radicals upon excitation of the Hoechst. We used Hoechst 33258 excited with 2-photon excitation at 750 nm. We transfected U2OS cells with GFP-tagged EZH2 and then used laser micro-irradiation to determine whether or not GFP-EZH2 recruits to DNA damage. We found that GFP-EZH2 recruits to laser-induced DNA damage within 2 seconds and continues to recruit until a maximum fluorescence is reached at around 1 min ().
Figure 1. Ezh2, Suz12, and Eed recruit to laser-induced DNA damage. U2OS cells expressing GFP-Ezh2, GFP-Suz12, or GFP-Eed were laser micro-irradiated and monitored using time-lapse microscopy. Representative images at 0, 2, and 100 s are shown after laser micro-irradiation. Accumulation of GFP-Ezh2, GFP-Suz12, and GFP-Eed on the laser damage tracks over time were quantified and plotted (n = 20). Increased fluorescence on the damage tracks is plotted over time. Ezh2 does not recruit to UV damage. (B) U2OS cells were grown on a coverslip for 24 h prior to the experiment. The coverslip was then placed below a polycarbonate membrane and exposed to UV radiation (120 J/m2). Cells were then left to recover for 20 min and then fixed with 4% PFA. Cells were immuno-stained with Ezh2 and γ-H2AX antibodies. (C) U2OS cells were transfected with GFP-Ezh2 24 h before the experiment. Cells were either treated with UV (120 J/m2), MNNG (100μM) or left untreated and fluorescent recovery after photobleaching was then performed to determine the kinetics of GFP-Ezh2. Relative intensity of the bleached region was plotted over time. (D) Ezh2 recruitment is not dependent on H2AX, ATM, or DNA PK. GFP-Ezh2 was transfected in H2AX KO, EBS, or MO59J cells 24 h before the experiment. Cells were then laser micro-irradiated and imaged over time. Representative images at three different time points are shown and quantifications (an average of 12–15 cells was plotted) of the micro-irradiated regions were shown on the right.
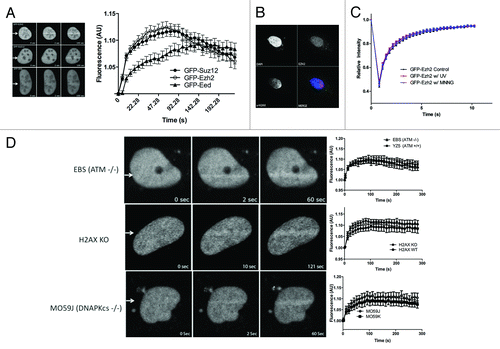
PRC2 also consists of 2 other essential subunits, EED and SUZ12. Therefore, we added a GFP tag to each of these proteins and proceeded with laser micro-irradiation experiments. Both GFP-EED and GFP-SUZ12 also recruit to laser-induced DNA damage (). GFP-EZH2 and GFP-SUZ12 recruitment profiles are almost identical, supporting that these proteins may recruit as a complex. GFP-EED recruitment is slightly slower and lesser than the other PRC2 members, indicating it may recruit individually or as a part of a complex, or both. The recruitment kinetics of PRC2 closely resembles the recruitment kinetics of other well-established DSB repair proteins, such as NBS1, MRE11, and RNF8, supporting the hypothesis that PRC2 acts early in the DNA damage response.Citation23
Because laser micro-irradiation can induce several types of DNA damage, we tested the potential of the other major types of damage, UV and single-strand breaks, to recruit PRC2. We placed a 10 μm polycarbonate membrane over cells and exposed the cells to UV (1 J/s) for 2 min. Cells were allowed to recover for 20 min and then fixed and stained for EZH2. We were unable to detect any EZH2 enrichment at sites of UV damage (). We also performed fluorescence recovery after photobleaching (FRAP) to determine if treating cells with UV or an alkylating agent, MNNG, altered the mobility of EZH2, reflecting its retention at sites of DNA damage. We found that neither UV (60 J) nor MNNG (0.1 mM) affected the kinetics of EZH2 (). We were therefore able to conclude that EZH2 is unlikely to be recruited directly by single-strand breaks or UV damage.
Determination of the requirement for the recruitment of PRC2 to sites of laser-induced DNA damage
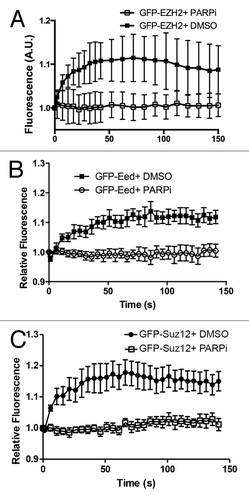
ATM and DNA-PKcs are 2 structurally related PI-3 kinases that coordinate and mediate the cellular response to DSBs. To determine if ATM and/or DNA-PKcs are required for the recruitment of EZH2, we microirradiated cells that lack each kinase and examined the recruitment of EZH2 in these cells using the GFP-tagged EZH2 construct. We found that neither of these kinases are responsible for the initial recruitment or retention of PRC2 at laser-induced DNA damage (). The histone 2A variant, H2AX is a central platform for the accumulation of several DNA repair proteins at the sites of DNA damage. We therefore wanted to investigate the requirement for H2AX in the recruitment of PRC2 to laser-induced DNA damage. We found EZH2 efficiently recruited in both H2AX WT and H2AX KO MEFs, suggesting that H2AX is dispensable for the recruitment of PRC2 to the sites of DNA damage (). Similar results have been reported by ourselves and others for the recruitment of PRC1 to sites of laser micro-irradiation.Citation13,Citation23 In both cases, the recruitment of PRC1 proteins to laser micro-irradiation-induced DNA damage was found to be PARP-dependent.Citation13,Citation25 To test the potential role of PARP in recruitment, we transfected U2OS cells with GFP-EZH2 and treated the cells with 5 μM PARP1/2 inhibitor AG14361 for 1 h prior to laser-microirradation. This treatment resulted in rapid inhibition of PARP1/2 activity in cells (data not shown). GFP-EZH2 recruitment was completely abrogated in the presence of the PARP inhibitor (). We obtained identical results with GFP-SUZ12 and GFP-EED, further establishing the PARP dependency of PRC2 recruitment (). Similar results were obtained using PARP1 knockout cells (data not shown). Taken together, these data indicate that PRC2 recruitment to laser-induced DNA damage is dependent on PARP1/2 activity.
Figure 2. Ezh2 recruitment to laser induced DNA damage is PARP dependent. U2OS cells expressing (A) GFP-Ezh2, (B) GFP-Eed, or (C) GFP-Suz12 were treated with PARP inhibitor AG14361 (5 μM) or DMSO (control) for 1 h prior to damage induction. Cells were then laser micro-irradiated and imaged over time. Quantifications (an average of 12–15 cells was plotted) of the GFP signal at the micro-irradiated regions were plotted and shown as indicated. Error bars represent SEM.
H3K27me3 does not increase at laser micro-irradiation tracks
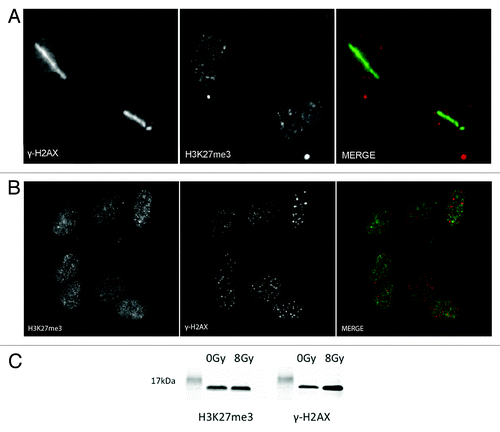
Post-translational modifications are essential in the signaling cascade and repair of DSBs. It has been reported that methylation of H4K20 and H3K36 are important in the DSB repair pathway.Citation26,Citation27 We next determined if the main substrate for PRC2 methylation, H3K27me3, is also methylated upon DNA damage. Using U2OS cells, we laser micro-irradiated and fixed cells 5 min after damage and then stained the cells using a specific antibody to H3K27me3. We also stained the cells using a specific antibody to γ-H2AX to mark the presence of DNA damage. Using indirect immunofluorescence, we were unable to detect an increase of H3K27me3 at the laser damage sites (). Similar results were obtained when cells were exposed to radiation (2 Gy) and examined for H3K27me3 colocalization with γ-H2AX ionizing radiation-induced foci (IRIF) (). Similar results were obtained when cells were allowed to recover for 30 min, and 1 h after DNA damage (data not shown). These results were further confirmed by immunoblot using H3K27me3 antibody. Immunoblotting of extracts prepared from cells exposed (8 Gy) or not exposed to radiation showed that H3K27me3 levels did not significantly increase after DNA damage (). An antibody specific to γ-H2AX was used to show the induction of DNA damage. Several separate commercial antibodies were additionally tested, and all were found not to stain laser micro-irradiation sites (data not shown).
Figure 3. H3K27 is not methylated upon laser induced DNA damage. (A) U2OS cells were laser micro-irradiated and then fixed with 4% paraformaldehyde 5 min after laser induced DNA damage. Cells were then co-immunostained with H3K27me3 and γ-H2AX antibodies. (B) U2OS cells were exposed to 2 Gy and then left to recover for 30 min. Cells were then fixed with 4% PFA and co-immunostained with H3K27me3 and γ-H2AX antibodies. (C) U2OS cells were either not treated or treated with 8 Gy and left to recover for 30 min before harvesting. Nuclei were extracted, sonicated, and run on an 18% polyacrilamide gel. The western blots were stained with H3K27me3 and γ- H2AX to show the induction upon ionizing radiation.
EZH2 does not significantly enrich at sites of ionizing radiation induced foci (IRIF)
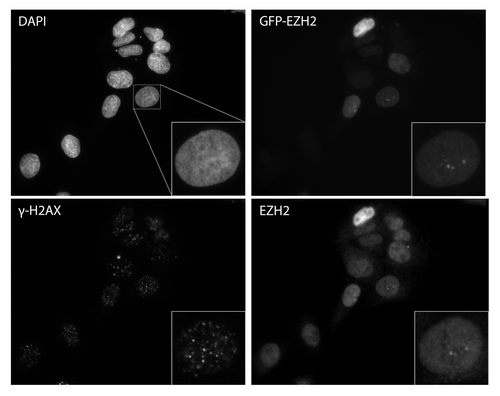
We further tested the ability of PRC1 to recruit to DSBs by testing whether or not EZH2 is recruited to ionizing radiation-induced foci. Upon treatment with ionizing radiation, DSB repair proteins localize to DSBs and are commonly seen to enrich in IRIF, although there are exceptions (e.g., Ku70/80,Citation28 data not shown). We treated U2OS cells with 2 Gy and examined EZH2 accumulation at IRIF using indirect immunofluorescence (). We were unable to detect any enrichment of EZH2 at IRIF marked with γ-H2AX. We were also unable to detect enrichment of SUZ12 or EED at IRIF (data not shown).
Figure 4. Ezh2 does not accumulate at IRIF. U2OS cells expressing GFP-Ezh2 were exposed to 2 Gy and left to recovery for 30 min. Cells were then fixed in 4% paraformaldehyde and co-immunostained with γ-H2AX and Ezh2 antibodies.
EZH2 recruits to Fok-1 endonuclease-induced DSBs
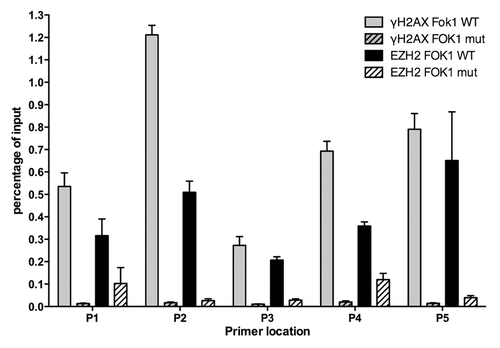
Laser micro-irradiation induces a multitude of DNA lesions at the laser tracks, including DNA single-strand breaks, UV damage, and base damage. Given that we cannot detect EZH2 enrichment in IRIF, we wanted to determine whether or not EZH2 recruits to DNA DSBs using a more sensitive chromatin immunoprecipitation technique. We employed a previously established system for targeted DSBs,Citation29 which targets the FOK-1 endonuclease to a specific integrated reporter site in the genome. FOK-1 generates a DSB at this site, enabling proteins to be mapped in the proximity of the DSB using ChIP. We transfected the transformed U2OS cells with either a functional FOK-1 construct or a mutated non-functional FOK-1 (D450A) construct and performed ChIP to determine the relative increase of EZH2. We saw increased levels of EZH2 only when we transfected with functional FOK-1 endonuclease, confirming that EZH2 is recruited and localized to DSBs (). We were able to confirm the functionality of the ChIP assay and the presence of DSBs by demonstrating that this correlated with a significant enrichment in γ-H2AX. These results complement the laser micro-irradiation data and demonstrate that DSBs are among the types of damage that recruit EZH2.
Figure 5. Ezh2 accumulates at Fok-1 induced DSBs. U2OS cells stably expressing the reporter locus were transfected with wild-type Fok-1 (FOK1 WT) or Fok1 catalytically inactive mutant (FOK1 mut) endonuclease. Cells were then fixed with 1% paraformaldehyde 18m h after transfection and harvested for chromatin immunoprecipitation (Ch-IP). Ch-IP was performed with Ezh2, γ-H2AX, and IgG antibodies. Quantitative PCR using 5 representative primers was done, and the Fok-1 DSB induced enrichment for each primer and antibody is plotted.
shRNA-mediated knockdown of EZH2 decreases DNA double-strand break repair
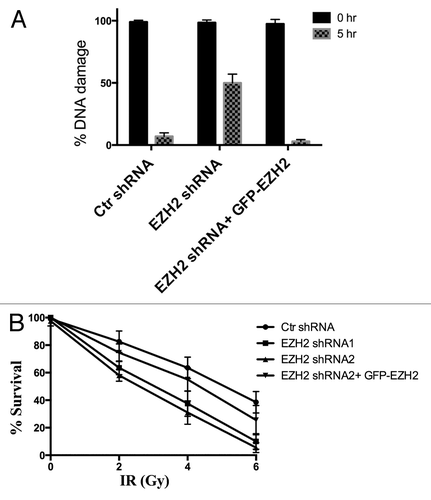
We wanted to determine whether or not the recruitment of EZH2 to sites of DSBs had an effect on the ability of cells to repair the damage. We used U2OS cells transfected with shRNA directed against EZH2 to determine the effect of EZH2 knockdown on DSB repair efficiency. Using the neutral constant field gel electrophoresis (CFGEP) assay, we quantified the proportion of DSBs that remained following gamma irradiation treatment at varying times after irradiation (). After treating U2OS cells with 40 Gy, they were immediately harvested or left for 5 h to allow DSB repair. Compared with control U2OS cells, where only 5% of the DSBs were not repaired after 5 h, more than 45% of the DSBs were not repaired in the EZH2-knockdown cells. To confirm this result, we reconstituted the EZH2-knockdown cells with GFP-EZH2 that is resistant to the shRNA to determine if this could re-establish the DNA DSB repair capacity back to control levels. We observed that GFP-EZH2 was able to restore DSB repair capacity back to control levels in the EZH2-knockdown cells. This indicates that PRC2 is necessary for efficient repair of the DNA damage.
Figure 6. Ezh2 knockdown decreases DSB repair efficiency and increases cellular sensitivity to IR. (A) U2OS cells stably expressing Ezh2-shRNA, and U2OS cells stably expressing Ezh2-shRNA reconstituted with shRNA-resistant GFP-Ezh2 construct were exposed to 40 Gy ionizing radiation. Cells were harvested immediately to determine total damage and after 5 hrs to determine the DNA damage remaining. Total DNA damage immediately after IR is referred to as 100% DNA damage. (B) U2OS cells transfected with control shRNA, two different Ezh2- shRNAs, or Ezh2-shRNA reconstituted with shRNA -esistant GFP-Ezh2 construct were plated at low density. Cells were then exposed to 0, 2, 4, or 6 Gy and left to recover for 2 wk. The cells were then fixed and stained and colonies were counted. Each colony represents a surviving cell, and the percent survival is plotted for each dose of IR.
EZH2 knockdown decreases cellular survival after exposure to ionizing radiation
After determining that PRC2 is recruited to DSB and is necessary for efficient DSB repair, we wanted to determine whether or not PRC2 is necessary for survival in response to ionizing radiation. A previous study demonstrated a 20–40 percent decrease in IR-resistance upon knockdown of PRC2 subunits.Citation13 We used a colony formation cellular survival assay to determine whether EZH2 plays an essential role in the cellular response to ionizing radiation induced damage. Using the colony formation assay and sequentially increasing doses of radiation from 0 Gy to 6 Gy, we determined the proportion of cells that survived each dose and continued to proliferate into a viable colony (). Using U2OS transfected with shRNA directed against EZH2, we determined that knockdown of EZH2 decreased cellular viability by 50% after a 4 Gy dose compared with control cells. Reconstituting EZH2-knockdown cells with shRNA-resistant GFP-EZH2 construct restored cellular viability back to control levels. This result supports the constant field gel electrophoresis assay results, and suggests that EZH2 is required for the efficient repair of DNA double-strand breaks, and that EZH2 participates in radiation resistance.
Discussion
Polycomb group proteins have recently been shown to play a role in the DNA damage response in addition to the previously established roles in development and stem-cell maintenance. Members of the PRC1 complex, BMI-1, RING-2, and CBX4, all recruit to sites of DSBs and are responsible for post-translational modifications that promote DNA damage signaling and DSB repair.Citation23,Citation25,Citation30 During gene repression, PRC2 is thought to promote the recruitment of PRC1. A previous study had reported that EZH2 recruitment to sites of laser micro-irradiation increased lysine 27 methylation at the DNA damage sites, and it was proposed that PRC2-mediated methylation was responsible for the recruitment of PRC1 to sites of DNA damage. However, we have previously shown that PRC1 recruitment is PARP-dependent but does not require PRC2 activity to recruit to DSBs. Therefore, we further investigated the role of PRC2 in DSB repair. GFP-tagged EZH2, SUZ12, and EED all recruit to the laser-induced DNA damage tracks with similar kinetics to other early DNA damage response proteins. In addition, we observed that EZH2 recruits to FOK-1 endonuclease-induced DSBs. Furthermore, knockdown of the enzymatic component of PRC2, EZH2, impaired the ability of cells to repair DSBs and increased their cellular sensitivity to ionizing radiation. These results are consistent with PRC2 functioning in the DNA damage response pathway. Surprisingly, and in contrast to recent publications,Citation13,Citation14 we observed no increase in H3K27me3 at sites of DNA damage. While this contradicts previous reports, it is consistent with our finding that PRC1 does not require PRC2 activity to recruit to sites of DNA damage.Citation23
Using laser microirradiation to induce DNA damage causes multiple types of DNA lesions, including base damage, UV damage, single-strand breaks as well as DSBs. To better define whether DSB damage recruits EZH2, we utilized a construct that generates only DSBs via an endonuclease. We found that PRC2 recruits specifically to these endonuclease induced DSBs. We were also able to show that EZH2 does not recruit to UV damage. As well, neither UV damage nor the alkylating agent MNNG changed the kinetics of EZH2. This indicates that EZH2 does not respond to the UV damage response or SSBs caused during base excision repair. We also found that DSB repair efficiency is decreased when EZH2 is knocked down in U2OS cells, which is consistent with the knockdown of PRC1 members. These results support the current working hypothesis that PRC2 is involved in the DNA damage response, and further establishes PRC2’s role as an early, essential DSB repair complex.
Histone post-translational modifications are essential to the DNA damage response. They are responsible for the recruitment and retention of DSB proteins, as well as, propagation of the DSB signaling cascade. Histone methylation has been shown to be necessary for the recruitment and retention of 53BP1. We propose that the histone methyltransferase complex, PRC2, is also essential to the DSB repair pathway. It is still unclear what role PRC2 plays in facilitating the repair of DSBs. It will be important to determine the importance of PRC2-mediated methylation and the substrates that are targeted, as well as the DSB repair proteins that may require PRC2 methylation within the repair pathway.
In response to DSBs, ATM mediates transcriptional silencing near the break site partly through the induction of histone ubiquitylation.Citation29 These post-translational modifications observed at DSBs are consistent with a role for polycomb proteins in DSB-induced gene repression. It may be that one role of PRC2 at DSB sites is to contribute to DSB-induced gene silencing through the methylation of H3K27. However, in the case of laser micro-irradiation, we failed to observe any increase in H3 lysine 27 methylation at the DNA damage site. Given that we employed the same antibody as previous publications,Citation13 we cannot explain this difference. However, it is consistent with our previous finding that EZH2 is not required for the recruitment of PRC1 to DSBs.Citation23
In addition to PRC2 acting directly at sites of DNA damage, PRC2 has been implicated in several aspects of the DNA damage response. Overexpression of EZH2 has been seen to confer resistance to cisplatin in ovarian cancer cells;Citation31 moreover, overexpression of EZH2 downregulates RAD51 expression in breast epithelial cells, thereby impairing DNA repair.Citation32 EZH2 has also been implicated in regulating cancer cell apoptosis in response to genotoxic stresses by regulating CHK1 activation.Citation19 The use of PRC2 inhibitors has been suggested for adjuvant chemotherapy, but the mechanistic role PRC2 plays in DNA damage signaling and repair must be elucidated before PRC2 inhibitors can be used effectively. Our data, in combination with the previous studies,Citation13-Citation15,Citation19 suggests a role for PRC2 in DSB repair and supports the hypothesis that PRC2 inhibitors could be used to sensitize undifferentiated cancers overexpressing PRC2 proteins to DNA damaging agents. The failure to detect PRC2 at sites of IR-induced DSBs while being able to detect it by chromatin IP at a defined DSB may mean that PRC2 is only required in low stoichiometric amounts, similar to what has been observed for Ku70 and Ku80, which are essential in DSB repair, recruit to sites of laser micro-irradiation, but cannot be detected in IR-induced foci. The first association between the PcG gene family and cancer development arose from the functional characterization of mouse BMI-1. BMI-1 was identified as a proto-oncogene that cooperates with MYC to promote the generation of B- and T-cell lymphomas.Citation33-Citation35 Although BMI-1 is arguably the PcG protein most strongly associated with neoplastic development, several other PcG members are also linked to tumorigenesis. For example, SUZ12 is overexpressed in both colon and breast cancers.Citation36,Citation37 Furthermore, EZH2 is upregulated in several human tumors, such as multiple types of lymphoma, prostate and breast cancer.Citation35,Citation38,Citation39 EZH2 expression is associated with poor prognosis and is indicative of a metastatic character of the disease.Citation38,Citation39Overexpression of EZH2 and its aberrant recruitment on target gene sites have been implicated in cancer initiation and progression. In prostate cancer, overexpression of EZH2 can be driven by the upregulation of MYC protein via transcriptional (direct targeting of EZH2 promoter) and post-transcriptional (downregulation miR-26a and -26b) mechanisms.Citation40 In acute myeloid leukemia cells, Evi1 physically interacts with EZH2 and recruits EZH2 and other PcG proteins to the PTEN promoter region.Citation33,Citation40,Citation41 This induces histone modifications (H3K27me3), which represses PTEN transcription and activates the PI3KAKT/mTOR signaling pathway. This mechanism also contributes to leukemogenesis.Citation40,Citation41 Interestingly, Evi1 is expressed in 10% of cases of acute myeloid leukemias associated with extremely poor prognosis.Citation42 A recent report found that downregulation of miR-31 was a common event in melanoma tumors and cell lines and was associated with genomic loss in a subset of samples.Citation43 Furthermore, miR-31 overexpression resulted in downregulation of EZH2 and a de-repression of its target gene rap1GAP; increased expression of EZH2 was associated with melanoma progression and overall patient survival.Citation42 Since PcG are linked to malignant transformation in various hematological and epithelial cancers, understanding how these proteins function may enable the exploitation of this pathway in the treatment of human cancer.
Material and Methods
Cell culture and transfections
U2OS, H2AX WT, and H2AX KO cells (a gift from Dr Andre Nussenszeig) were cultured in DMEM supplemented with 10% FBS at 37 °C and 5% CO2. U2OS cells with the stably integrated p3216PECMS2β insert (gift from Dr Susan Janicki) were cultured in High Glucose DMEM with 10% FBS and 100 μg/mL hygromycin B. EBS, YZ5 (ATM deficient and proficient, respectively) and MO59J, MO59K (DNA-PKcs deficient and proficient, respectively) were cultured in 50% DMEM/50% F12 supplemented with 10% FBS. All plasmid transfections were performed with Effectene using the manufacturer’s protocol.
Laser micro-irradiation
Cells were cultured on 35-mm glass bottom culture dishes (MatTek Corporation) 24 h before the experiment. Cells were treated with 1 μg/ml Hoechst 33258 for 15 min and then placed on a heated stage (37 °C) of a laser-scanning confocal microscope (LSM510; Carl Zeiss, Inc). DNA damage was generated along a 0.2–1 μm-wide region across the nucleus by excitation of the Hoechst 33258 dye using 20 mW a near infrared 750-nm titanium-sapphire laser line. The laser output was set to 15% and we applied 10 iterations to generate localized DNA damage using a Plan-Neofluar 40×/1.3 N.A. oil immersion objective. GFP fluorescence imaging was recorded after excitation with a 488-nm argon laser using a 515–540 nm band-pass filter. Mean and standard error for a minimum of ten cells were plotted.
Immunofluorescence microscopy
Immunofluroescence was performed as previously described.Citation7. Cells were fixed with 4.0% paraformaldehyde (PFA) in PBS, pH 7.5, for 5 min at room temperature. Cells were then permeablized with PBS containing 0.5% Triton X-100 for 5 min, and then washed with PBS 3 times. Cells were then incubated with primary antibody for 30 min at room temperature. Cells were then rinsed with PBS containing 0.1% Triton X-100 and then washed 3 times with PBS. Cells were subsequently incubated with a secondary antibody for 30 min at room temperature. Next, cells were rinsed with PBS containing 0.1% Triton X-100 and washed three times with PBS. Finally, cells were mounted onto slides with a 90% glycerol/PBS-based medium containing 0.5 μg/mL DAPI. Cells were observed using a Axiovert 200M; Carl Zeiss Inc microscope.
Chromatin Immunoprecipitation
Cells were transfected 24 h prior to harvest with either Fok-1 or catalytically dead Fok-D480A. Cells were then fixed in suspension at room temp with 1% PFA for 10 min, and then the PFA was quenched with 0.125 M glycine for 5 min. Cells were then centrifuged at 1400× g and the nuclei were released in Nuclei Isolation Buffer (250 mM sucrose, 150 mM NaCl, 20 mM Tris pH 8.0, 1.5 mM MgCl2, 0.2 mM CaCl2, 0.1% IGEPAL CA-630). Nuclei were centrifuged at 3200× g and resuspended in a modified RIPA buffer (50 mM TRIS-HCl pH 7.4, 150 mM NaCl, 1% IGEPAL CA-630). Micrococcal nuclease was added for 10 min at room temp to digest the DNA (2000 gel units in modified RIPA plus 5 mM CaCl2). The digested chromatin was then incubated with Dynabeads and preincubated with the appropriate antibody according to the manufacturer’s protocol. After elution, PFA crosslinks were reversed for 6 h at 65 °C, and proteinase K (3 mg/mL) was added. Quantitative PCR was performed using 6 previously described primers (Shanbhag et al. 2010) and SYBR-green master mix. Calculations based on Ct values were done as stated by invitrogen methods.
Constant field gel electrophoresis (CFGEP) assay
The amount of DSBs remaining after exposure to IR was measured using a modified constant field gel electrophoresis (CFGE) protocol previously described.Citation7 In brief, cells were exposed to 40 Gy IR and harvested immediately to measure initial DNA damage levels and after 5 h at 37 °C to measure DNA damage remaining after repair. Cells were harvested using trypsin, centrifuged and washed with 1× PBS. 150 000 cells were mixed with low melting point agarose (1.25% type VII in PBS with 5 mM EDTA) and transferred to a plug mold (Biorad cat# 170-3713). The cells were then lysed in the plug mold at 4 °C for 24 h in lysis buffer (25 mM EDTA pH 8.5, 0.5% SDS, with 3 mg/mL proteinase K added just before lysis). The cells were resolved using agarose gel electrophoresis (0.7%) in TAE (0.04 mM TRIS-acetate, and 1 mM EDTA, pH 8) at 4 °C for 18 h at 1 V/cm. Laser scanning equipment (Typhoon 9200 Variable Mode Imager; ImageQuant 5.2 software; GE Healthcare) was used to quantify the relative amounts of DNA that migrated into the gel. This was used to calculate the number of DSBs.
Colony formation assay
Cellular sensitivity to IR was determined using a previously described protocol.Citation23 In brief, U2OS cells were transfected with either control shRNA or Ezh2-shRNA 24 h prior to the experiment. Cells were plated at low density and exposed to different doses of IR (2, 4, or 6 Gy). Cells were then left to grow at 37 °C for approximately 10 d (or until colonies of 40–50 cells were present). The cells were then fixed and stained with crystal violet. Colonies were counted to determine cellular viability. Error bars are from duplicate samples.
Acknowledgments
The Alberta Cancer Foundation (to MH) and the Canadian Institute of Health Research (CIHR) (to GP and MJH); an Alberta Innovates Health Solutions (AIHS) Senior Scholar (to MJH); Alberta Cancer Foundation postdoctoral fellowships (to IHI) and Alberta Cancer Foundation graduate student scholarship (to SC). We thank Drs Andre Nussenzweig, Mien-Chie Hung, and Xiaochun Yu for providing valuable reagents. We also thank the Cellular Imaging Facility at the Cross Cancer Institute for the use of microscopes and image analysis software.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Gieni RS, Hendzel MJ. Polycomb group protein gene silencing, non-coding RNA, stem cells, and cancer. Biochem Cell Biol 2009; 87:711 - 46; http://dx.doi.org/10.1139/O09-057; PMID: 19898523
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298:1039 - 43; http://dx.doi.org/10.1126/science.1076997; PMID: 12351676
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev Cancer 2006; 6:846 - 56; http://dx.doi.org/10.1038/nrc1991; PMID: 17060944
- Fischle W, Wang Y, Jacobs SA, Kim Y, Allis CD, Khorasanizadeh S. Molecular basis for the discrimination of repressive methyl-lysine marks in histone H3 by Polycomb and HP1 chromodomains. Genes Dev 2003; 17:1870 - 81; http://dx.doi.org/10.1101/gad.1110503; PMID: 12897054
- Min J, Zhang Y, Xu RM. Structural basis for specific binding of Polycomb chromodomain to histone H3 methylated at Lys 27. Genes Dev 2003; 17:1823 - 8; http://dx.doi.org/10.1101/gad.269603; PMID: 12897052
- Wang H, Wang L, Erdjument-Bromage H, Vidal M, Tempst P, Jones RS, et al. Role of histone H2A ubiquitination in Polycomb silencing. Nature 2004; 431:873 - 8; http://dx.doi.org/10.1038/nature02985; PMID: 15386022
- Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 2010; 191:45 - 60; http://dx.doi.org/10.1083/jcb.201003034; PMID: 20921134
- Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol 2011; 31:1972 - 82; http://dx.doi.org/10.1128/MCB.00981-10; PMID: 21383063
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 26; http://dx.doi.org/10.1016/j.cell.2005.09.038; PMID: 16377563
- Doil C, Mailand N, Bekker-Jensen S, Menard P, Larsen DH, Pepperkok R, et al. RNF168 binds and amplifies ubiquitin conjugates on damaged chromosomes to allow accumulation of repair proteins. Cell 2009; 136:435 - 46; http://dx.doi.org/10.1016/j.cell.2008.12.041; PMID: 19203579
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 2010; 107:18475 - 80; http://dx.doi.org/10.1073/pnas.1012946107; PMID: 20937877
- Gieni RS, Ismail IH, Campbell S, Hendzel MJ. Polycomb group proteins in the DNA damage response: a link between radiation resistance and “stemness”. Cell Cycle 2011; 10:883 - 94; http://dx.doi.org/10.4161/cc.10.6.14907; PMID: 21346409
- Chou DM, Adamson B, Dephoure NE, Tan X, Nottke AC, Hurov KE, et al. A chromatin localization screen reveals poly (ADP ribose)-regulated recruitment of the repressive polycomb and NuRD complexes to sites of DNA damage. Proc Natl Acad Sci U S A 2010; 107:18475 - 80; http://dx.doi.org/10.1073/pnas.1012946107; PMID: 20937877
- O’Hagan HM, Mohammad HP, Baylin SB. Double strand breaks can initiate gene silencing and SIRT1-dependent onset of DNA methylation in an exogenous promoter CpG island. PLoS Genet 2008; 4:e1000155; http://dx.doi.org/10.1371/journal.pgen.1000155; PMID: 18704159
- Hong Z, Jiang J, Lan L, Nakajima S, Kanno S, Koseki H, et al. A polycomb group protein, PHF1, is involved in the response to DNA double-strand breaks in human cell. Nucleic Acids Res 2008; 36:2939 - 47; http://dx.doi.org/10.1093/nar/gkn146; PMID: 18385154
- Sustáčková G, Kozubek S, Stixová L, Legartová S, Matula P, Orlova D, et al. Acetylation-dependent nuclear arrangement and recruitment of BMI1 protein to UV-damaged chromatin. J Cell Physiol 2012; 227:1838 - 50; http://dx.doi.org/10.1002/jcp.22912; PMID: 21732356
- He A, Shen X, Ma Q, Cao J, von Gise A, Zhou P, et al. PRC2 directly methylates GATA4 and represses its transcriptional activity. Genes Dev 2012; 26:37 - 42; http://dx.doi.org/10.1101/gad.173930.111; PMID: 22215809
- Martin C, Cao R, Zhang Y. Substrate preferences of the EZH2 histone methyltransferase complex. J Biol Chem 2006; 281:8365 - 70; http://dx.doi.org/10.1074/jbc.M513425200; PMID: 16431907
- Wu Z, Lee ST, Qiao Y, Li Z, Lee PL, Lee YJ, et al. Polycomb protein EZH2 regulates cancer cell fate decision in response to DNA damage. Cell Death Differ 2011; 18:1771 - 9; http://dx.doi.org/10.1038/cdd.2011.48; PMID: 21546904
- Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer 2012; 106:243 - 7; http://dx.doi.org/10.1038/bjc.2011.551; PMID: 22187039
- Chang CJ, Hung MC. The role of EZH2 in tumour progression. Br J Cancer 2012; 106:243 - 7; http://dx.doi.org/10.1038/bjc.2011.551; PMID: 22187039
- Gieni RS, Ismail IH, Campbell S, Hendzel MJ. Polycomb group proteins in the DNA damage response: a link between radiation resistance and “stemness”. Cell Cycle 2011; 10:883 - 94; http://dx.doi.org/10.4161/cc.10.6.14907; PMID: 21346409
- Ismail IH, Andrin C, McDonald D, Hendzel MJ. BMI1-mediated histone ubiquitylation promotes DNA double-strand break repair. J Cell Biol 2010; 191:45 - 60; http://dx.doi.org/10.1083/jcb.201003034; PMID: 20921134
- Kim JS, Heale JT, Zeng W, Kong X, Krasieva TB, Ball AR Jr., et al. In situ analysis of DNA damage response and repair using laser microirradiation. Methods Cell Biol 2007; 82:377 - 407; http://dx.doi.org/10.1016/S0091-679X(06)82013-3; PMID: 17586265
- Ismail IH, Gagné JP, Caron MC, McDonald D, Xu Z, Masson JY, et al. CBX4-mediated SUMO modification regulates BMI1 recruitment at sites of DNA damage. Nucleic Acids Res 2012; 40:5497 - 510; http://dx.doi.org/10.1093/nar/gks222; PMID: 22402492
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, et al. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011; 470:124 - 8; http://dx.doi.org/10.1038/nature09658; PMID: 21293379
- Fnu S, Williamson EA, De Haro LP, Brenneman M, Wray J, Shaheen M, et al. Methylation of histone H3 lysine 36 enhances DNA repair by nonhomologous end-joining. Proc Natl Acad Sci U S A 2011; 108:540 - 5; http://dx.doi.org/10.1073/pnas.1013571108; PMID: 21187428
- Kong X, Mohanty SK, Stephens J, Heale JT, Gomez-Godinez V, Shi LZ, et al. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res 2009; 37:e68; http://dx.doi.org/10.1093/nar/gkp221; PMID: 19357094
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010; 141:970 - 81; http://dx.doi.org/10.1016/j.cell.2010.04.038; PMID: 20550933
- Ginjala V, Nacerddine K, Kulkarni A, Oza J, Hill SJ, Yao M, et al. BMI1 is recruited to DNA breaks and contributes to DNA damage-induced H2A ubiquitination and repair. Mol Cell Biol 2011; 31:1972 - 82; http://dx.doi.org/10.1128/MCB.00981-10; PMID: 21383063
- Hu S, Yu L, Li Z, Shen Y, Wang J, Cai J, et al. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol Ther 2010; 10:788 - 95; http://dx.doi.org/10.4161/cbt.10.8.12913; PMID: 20686362
- Zeidler M, Varambally S, Cao Q, Chinnaiyan AM, Ferguson DO, Merajver SD, et al. The Polycomb group protein EZH2 impairs DNA repair in breast epithelial cells. Neoplasia 2005; 7:1011 - 9; http://dx.doi.org/10.1593/neo.05472; PMID: 16331887
- van Lohuizen M, Verbeek S, Scheijen B, Wientjens E, van der Gulden H, Berns A. Identification of cooperating oncogenes in E mu-myc transgenic mice by provirus tagging. Cell 1991; 65:737 - 52; http://dx.doi.org/10.1016/0092-8674(91)90382-9; PMID: 1904008
- Lindström MS, Klangby U, Wiman KG. p14ARF homozygous deletion or MDM2 overexpression in Burkitt lymphoma lines carrying wild type p53. Oncogene 2001; 20:2171 - 7; http://dx.doi.org/10.1038/sj.onc.1204303; PMID: 11360201
- van Kemenade FJ, Raaphorst FM, Blokzijl T, Fieret E, Hamer KM, Satijn DP, et al. Coexpression of BMI-1 and EZH2 polycomb-group proteins is associated with cycling cells and degree of malignancy in B-cell non-Hodgkin lymphoma. Blood 2001; 97:3896 - 901; http://dx.doi.org/10.1182/blood.V97.12.3896; PMID: 11389032
- Kirmizis A, Bartley SM, Farnham PJ. Identification of the polycomb group protein SU(Z)12 as a potential molecular target for human cancer therapy. Mol Cancer Ther 2003; 2:113 - 21; PMID: 12533679
- Kirmizis A, Bartley SM, Kuzmichev A, Margueron R, Reinberg D, Green R, et al. Silencing of human polycomb target genes is associated with methylation of histone H3 Lys 27. Genes Dev 2004; 18:1592 - 605; http://dx.doi.org/10.1101/gad.1200204; PMID: 15231737
- Kleer CG, Cao Q, Varambally S, Shen R, Ota I, Tomlins SA, et al. EZH2 is a marker of aggressive breast cancer and promotes neoplastic transformation of breast epithelial cells. Proc Natl Acad Sci U S A 2003; 100:11606 - 11; http://dx.doi.org/10.1073/pnas.1933744100; PMID: 14500907
- Varambally S, Dhanasekaran SM, Zhou M, Barrette TR, Kumar-Sinha C, Sanda MG, et al. The polycomb group protein EZH2 is involved in progression of prostate cancer. Nature 2002; 419:624 - 9; http://dx.doi.org/10.1038/nature01075; PMID: 12374981
- Yoshimi A, Kurokawa M. Evi1 forms a bridge between the epigenetic machinery and signaling pathways. Oncotarget 2011; 2:575 - 86; PMID: 21795762
- Yoshimi A, Goyama S, Watanabe-Okochi N, Yoshiki Y, Nannya Y, Nitta E, et al. Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 2011; 117:3617 - 28; http://dx.doi.org/10.1182/blood-2009-12-261602; PMID: 21289308
- Valk PJ, Verhaak RG, Beijen MA, Erpelinck CA, Barjesteh van Waalwijk van Doorn-Khosrovani S, Boer JM, et al. Prognostically useful gene-expression profiles in acute myeloid leukemia. N Engl J Med 2004; 350:1617 - 28; http://dx.doi.org/10.1056/NEJMoa040465; PMID: 15084694
- Asangani IA, Harms PW, Dodson L, Pandhi M, Kunju LP, Maher CA, et al. Genetic and epigenetic loss of microRNA-31 leads to feed-forward expression of EZH2 in melanoma. Oncotarget 2012; 3:1011 - 25; PMID: 22948084