Abstract
The mechanisms that control proliferation, or lack thereof, in adult human β cells are poorly understood. Controlled induction of proliferation could dramatically expand the clinical application of islet cell transplantation and represents an important component of regenerative approaches to a functional cure of diabetes. Adult human β cells are particularly resistant to common proliferative targets and often dedifferentiate during proliferation. Here we show that expression of the transcription factor E2F3 has a role in regulating β-cell quiescence and proliferation. We found human islets have virtually no expression of the pro-proliferative G1/S transcription factors E2F1–3, but an abundance of inhibitory E2Fs 4–6. In proliferative human insulinomas, inhibitory E2Fs were absent, while E2F3 is expressed. Using this pattern as a “roadmap” for proliferation, we demonstrated that ectopic expression of nuclear E2F3 induced significant expansion of insulin-positive cells in both rat and human islets. These cells did not undergo apoptosis and retained their glucose-responsive insulin secretion, showing the ability to reverse diabetes in mice. Our results suggest that E2F4–6 may help maintain quiescence in human β cells and identify E2F3 as a novel target to induce proliferation of functional β cells. Refinement of this approach may increase the islets available for cell-based therapies and research and could provide important cues for understanding in vivo proliferation of β cells.
Introduction
Proliferation of insulin-secreting β cells is severely restricted in the endogenous adult pancreas, making established diabetes particularly difficult to treat. It has been shown that humans can regenerate endogenous insulin secretion after autologous hemopoetic stem cell transplantation.Citation1 However, the natural regenerative capacity is limited, and patients with more severe islet loss may never recuperate sufficient β-cell function.Citation2 At present, islet transplantation is the only minimally invasive therapy for type I diabetes that is able to provide endogenous insulin secretion sufficient to achieve normoglycemia without exogenous insulin administration. Most patients achieve euglycemia after the islet transplantation. However, over time a decline in islet function is observed, with long-term insulin-independence achieved in about 40–50% of patients after 3 to 5 y.Citation3,Citation4 Cadaver donors, the current source of islets for transplant, are too scarce to meet the demand of diabetic patients worldwide and provide for possible future re-transplants. Therefore, a stable renewable source of transplantable β cells is imperative for the advancement of a cell-based therapy for diabetes.
In adult islets, it is possible to drive quiescent cells to divide by altering the expression of cell cycle genes. The G1/S transition of the cell cycle is regulated in part by E2F transcription factors and is a tightly orchestrated process that culminates in DNA synthesis.Citation5,Citation6 E2Fs regulate expression of genes involved in many cellular process, including apoptosis, mitosis, differentiation, and development with E2F1, 2, and 3 as transcriptional activators and E2F4–8 as transcriptional repressors of these genes.Citation5,Citation7,Citation8 Regulation of E2F 1–3 activity is achieved by formation of a complex with the retinoblastoma protein (Rb). E2F is released from this regulatory complex by hyper-phosphorylation of Rb by cyclins D/A/E and cdks 2/4/6.Citation9,Citation10 E2F drives proliferation through the expression of target genes involved in DNA synthesis, including histone H2A, PCNA, DNA polymerase, RPA, Cdc6, and MCM.Citation5,Citation11 Aberrant E2F expression has been linked to most types of cancer, with E2F3 in particular being overexpressed in lung, bladder and prostate cancers.Citation12-Citation15 E2F4 and 5 regulate the expression of their target genes by occupying E2F1–3-responsive promoters, thereby preventing these activating E2Fs from binding.Citation8,Citation16 E2Fs 4 and 6 are thought to be a major influence on the maintenance of cellular quiescence.Citation8,Citation17 Expression of activating E2Fs is cyclic, with E2F1–3 highly expressed during G1/S and minimally expressed during the other phases of the cell cycle.Citation18 E2F6 also contributes to the cyclic expression of E2F by decreasing the pro-proliferative E2Fs after S-phase, when the expression of E2F1–3 is no longer needed.Citation19
Though it is a debated topic, endogenous islets are thought to maintain the β-cell mass, at least in part, through self-renewal by mitotic division.Citation20-Citation22 Additionally, different factors appear to be responsible for inducing embryonic expansion of β cells compared with post-weaning proliferation.Citation23 In adult β and progenitor cells, manipulation of several cell cycle proteins, including cyclin D, CDK4, and CDK6, have led to significant induction of β-cell proliferation.Citation24-Citation26 Additionally, E2F1 overexpression was recently shown to induce proliferation and apoptosis in mouse and human β cells.Citation27 E2F1 was also found to have a link to β-cell metabolism through its control overexpression of Kir6.2 and insulin secretion.Citation28
Here we use human insulinomas as a model for the induction of β-cell proliferation. By identifying E2F3 as a differentially expressed cell cycle transcription factor, we found that its overexpression in wild-type islets induces S-phase entry and functional proliferation of β cells in isolated human islets.
Results
Human adult islets have nuclear expression of anti-proliferative E2Fs
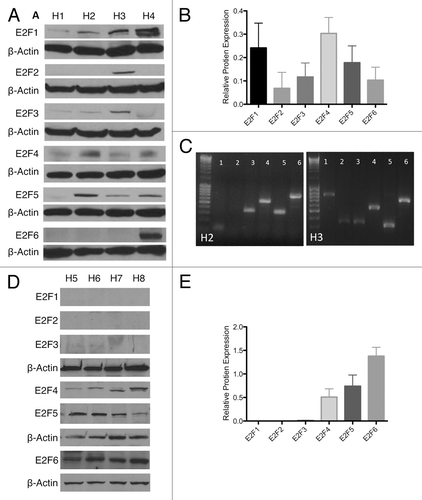
Since expression of E2F4–6 is known to inhibit proliferation, we sought to characterize their expression in wild-type human islets. Whole-cell and nuclear protein extracts from 8 human islet preparations (H1-H8) (Table S1) were probed for E2F1–6. In whole-cell extracts, islets contained all 6 E2Fs whose level of expression varied between donors (). This variable E2F pattern was partially confirmed at the RNA level, with some samples (H3) showing E2F2 at both protein or mRNA level, while other samples (H2) showed no E2F2 expression at either mRNA and protein (). Additionally, all samples showed E2F6 mRNA expression, while E2F6 protein was only detected in one whole-cell lysate. Since E2Fs are only able to transcriptionally regulate DNA if they are located in the nucleus, we probed for the presence of E2F1–6 in the nuclear protein extracts ().Citation29 While all E2Fs were detectable in the whole-cell lysate, only the anti-proliferative E2F4–6 were measurable in nuclear extracts. While whole-cell extracts displayed a variable E2F expression pattern, the functional E2Fs localized in nucleus had little variability between samples. Both western blots were semi-quantitatively normalized to β-actin to help illustrate virtual absence of some E2Fs in the nucleus (). The expression of repressive E2Fs in the nucleus may contribute to the slow proliferation rate seen in human islets.
Figure 1. Endogenous E2F expression. (A and B) Western blot and semi-quantification of protein isolated from whole islets of 4 human islet isolations (H1–H4) showing E2F 1–6 expression. (C) Reverse transcription PCR for E2F 1–6 in H2 and H3, showing variability of protein expression partially correlates with mRNA expression. (D and E) Western blot and semi-quantification of protein isolated from the nuclear fraction of islets of 4 human islet isolations (H5–H8) showing E2F 1–6 expression.
Human insulinomas have an E2F expression pattern that may drive proliferation
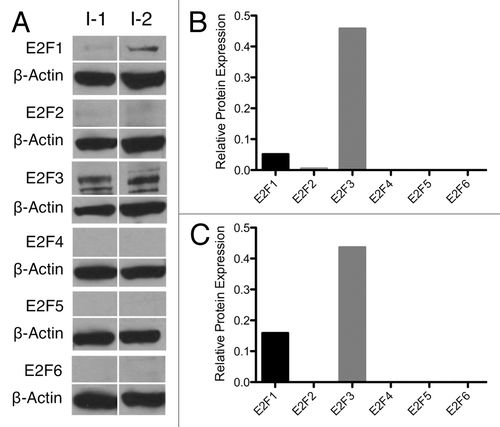
To examine the E2F expression in a “positive control” for β-cell proliferation, we repeated the E2F protein screen on whole-cell lysates of human insulinomas obtained from 2 patients, one benign (I-1) and the other malignant (I-2), at the University of Illinois at Chicago Hospital. These patients were hyperinsulinemic, hypoglycemic, and gained a significant amount of weight. While our access was limited to only 2 fresh insulinoma samples, making generalizations difficult, the results obtained with these samples appeared reproducible. E2F1–6 protein expression was again probed by western blot () and normalized to β actin to help visualize any loss of E2F expression (). In these insulinomas, the pro-proliferative E2Fs 1 and 3 were expressed compared with the virtual absence of E2F2, 4, 5, and 6. Unlike wild-type islets, none of the anti-proliferative E2F4–6 were observed in either of these samples. Comparison of E2F expression levels between western blots may not be appropriate, as the strength of a band depends on several factors. However, the comparison of the presence or absence of a given E2F in wild-type vs. insulinoma samples may give insight into the mechanism of proliferation in β cells. The expression of proliferative E2Fs in insulinomas may contribute to their increased proliferation rate.
Figure 2. Insulinoma E2F expression. (A) Whole-cell protein samples from benign (I-1) and malignant (I-2) human insulinomas probed for E2F1–6 by western blot. (B and C) Insulinoma protein expression normalized to β-actin by densitomerty.
Adenoviruses are able to mediate overexpression of E2F3 in human
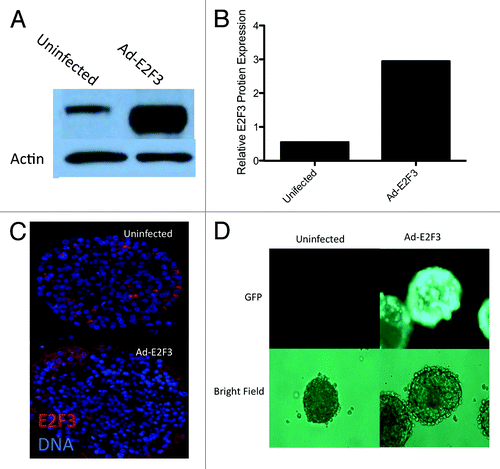
Since E2F3 was differentially expressed in proliferating insulinomas and wild-type islets, we wanted to see if ectopic induction of E2F3 would enhance proliferation in native islets. To this end, we infected adult human islets with adenoviruses expressing E2F3-GFP to assess their ability to induce transgene expression in whole islets. This was confirmed by detection of E2F3 by western blot (). More than a 3-fold increase in E2F3 expression was observed when these immunoblots were normalized to β actin (). Fluorescent staining of E2F3 in an infected islet revealed that the adenovirus only penetrated about 1–5 cell layers deep, but did not infect the core of the islet (). Visualization of the GFP tag further confirmed the viruses’ ability to infect the mantle of whole human islets ().
Figure 3. Adenovirus function and infection. (A) Western blot for whole-cell protein extracts following ad-E2F3 infection at a MOI of 500. (B) Quantification of western blots for E2F3 in uninfected and ad-E2F3 infected islets, normalized to β-actin by densitometry. (C) Human islets infected with ad-E2F3 and fluorescently stained for E2F3 showing the level of penetration the adenovirus achieves in whole human islets. (D) Expression of GFP-tagged ad-E2F3 in infected and uninfected human islets after 3 d of culture.
Recapitulation of insulinoma-E2F3 expression induces proliferation of otherwise quiescent adult β cells
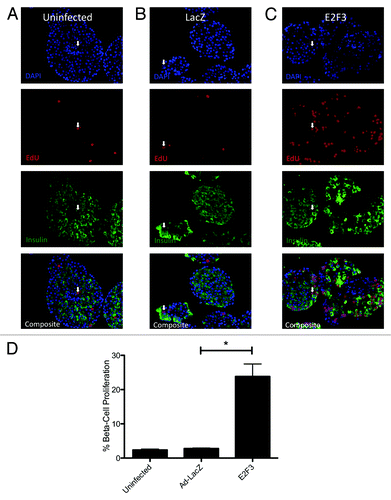
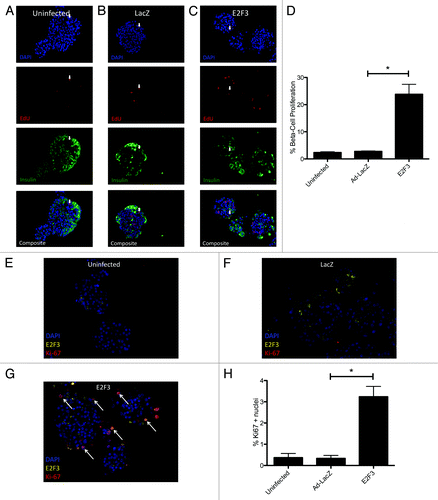
To determine if ectopic E2F3 enhanced β-cell proliferation, S-phase entry was examined by EdU incorporation in rat islets transfected with ad-E2F3. Uninfected () and ad-LacZ () infected islets showed no significant difference in EdU incorporation, with only 2.638% ± 0.2688 and 2.879% ± 0.2122 proliferation of insulin-positive cells. The addition of the E2F3 adenovirus significantly increased β-cell-specific proliferation to 23.83% ± 3.659, P = 0.004 (). These results are quantified for 3 rat preparations, with a range of 700–1000 insulin-positive cells counted per preparation ().
Figure 4.E2F3 induced proliferation in rat islets. (A–C) Representative pictures of paraffin embedded sections for uninfected (MOI 0) (A), ad-LacZ infected (MOI 500) (B) and ad-E2F3 (MOI 500) (C) stained for DNA stained with DAPI (blue) EdU (red), and insulin (green). (D) Quantification of immunohistochemistry for EdU+/insulin+ cells. % β-cell proliferation represents the number of cells that co-stained for EdU and insulin over the total number of insulin-positive cells, *P = 0.004. White arrows, sample EdU/Insulin positive nuclei. n = 3 islet preparations
Many methods of proliferating rodent β cells do not translate or translate poorly into humans.Citation30 To confirm the proliferative effect of E2F3 seen in rodents, human islets were infected and assayed for EdU incorporation and Ki67. Representative pictures of uninfected () and ad-LacZ () showed little proliferation by EdU incorporation, 0.5208% ± 0.0579 and 1.504% ± 0.22, respectively, with P = 0.04. Others have noted that adenovirus infection with transgenes typically used as viral controls can activate Akt expression and produce small but statistically significant levels of proliferation, similar to what we see here.Citation31 Infection with ad-E2F3 significantly raised the β-cell proliferation rate to 7.158% ± 0.4776, P < 0.001 (). There results were confirmed by Ki67 staining in uninfected and ad-LacZ-infected islets, showing modest proliferation rates 0.36% ± 0.2 and 0.34% ± 0.14 (). Similar to the EdU assay, the addition of ad-E2F3 significantly increased proliferation to 3.24% ± 0.48, P = 0.004 (). Little nuclear expression of E2F3 was seen in the uninfected and LacZ-infected islets. Conversely, most Ki67-positive nuclei in the E2F3-infected group also co-stained for nuclear E2F3 (white arrows, ). These results were quantified for 3 human islet preparations with 700–1000 insulin-positive cells counted per preparation ().
Figure 5.E2F3 induced proliferation in human islets. (A–C) Representative pictures of uninfected (MOI 0) (A) human islets, ad-LacZ infected (MOI 500) (B), and ad-E2F3 infected (MOI 500) (C) cultured in the presence of EdU for 4 d and stained for DNA stained with DAPI (blue), EdU (red), and insulin (green). (D) Quantification of immunohistochemistry for EdU and insulin-positive cells, (*) p = 0.04 (uninfected to ad-LacZ) **P < 0.001 (ad-LacZ to ad-E2F3). % β-cell proliferation represents the number of cells that co-stained for EdU and insulin over the total number of insulin-positive cells. (E–G) White arrows, sample EdU/Insulin positive nuclei. Sample images showing Ki67 and E2F3 staining in uninfected, ad-LacZ and ad-E2F3 infected human islet. Arrows indicate co-stained Ki67-positive and E2F3-positive nuclei. (H) Quantification of Ki67 nuclei. n = 3 islet preparations
Overexpression of E2F3 does not impair glucose-responsive insulin secretion or induce apoptosis
Efforts to proliferate β cells often result in dedifferentiation and apoptosis as they grow.Citation27,Citation32 To ensure that E2F3-proliferated islets retained their phenotype and glucose-sensitivity, we performed a static glucose incubation on rat and human islets. Rat () and human () islets showed that insulin secretion in response to both high (28 mM) and low (2.8 mM) glucose were normal in E2F3-infected islets. Additionally, overexpression of E2F3 did not affect the total insulin content of the cells (). Previous efforts to proliferate β cells using E2F1 resulted in activation of apoptosis and cell death.Citation27 To investigate the possibility that overexpression of E2F3 induced apoptosis, we assayed infected human islets for caspase-3 activity. Uninfected, ad-LacZ, and ad-E2F3 infected islets all showed similar levels of caspase-3 activity, indicating that unlike E2F1, E2F3-induced proliferation does not induce apoptosis ().
Figure 6. Assessing β-cell function: static glucose incubation and apoptosis. (A and B) Rat (A) and human (B) islets were incubated in low (2.8 mM) and high (28 mM) glucose for 1 h, P = ns (C and D): Total insulin content of uninfected, ad-LacZ (MOI 500) and ad-E2F3 (MOI 500) islets, P = ns, n = 3 islet preparations (E): Caspase-3 activity in uninfected, ad-LacZ and ad-E2F3 infected human islets, P = ns, n = 4 islet preparations.
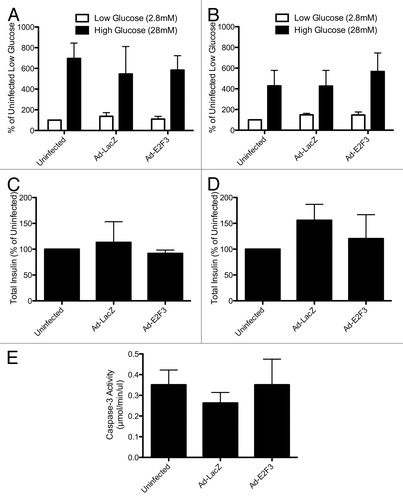
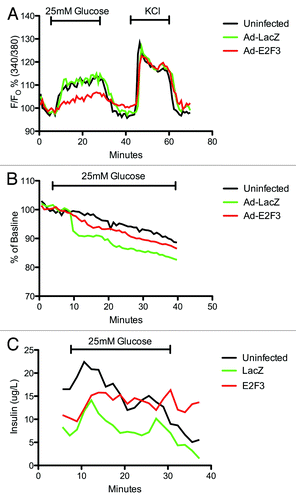
To further characterize the β-cell function, we assayed E2F3 infected islets for: (1) intracellular calcium concentrations in response to glucose or KCl, an indicator of the glucose-stimulated insulin secretion kinetics determined mainly by KATP and calcium ion channels and (2) hyperpolarization of the mitochondrial membrane, an indicator of glucose sensitivity and ATP production. Despite a small decrease in intracellular calcium levels in E2F3 infected islets, all 3 conditions showed statistically similar calcium levels in response to glucose and KCl, area under curve P = 0.1543 (). Mitochondrial potential and dynamic insulin secretion also revealed no impairment or loss of function (). Overall, no negative effects on insulin secretion, glucose sensitivity, total insulin content, caspase-3 activity or KATP/calcium ion channels were observed with the overexpression of E2F3, and the islets appear to retain their phenotype.
Figure 7. Assessing β-cell function: microfluidic assay. (A) Dynamic measurement of intra-cellular calcium levels in uninfected, ad-LacZ and ad-E2F3 infected human islets. Islets were incubated with Fura-2 then loaded into a microfluidic device mounted on an epifluorescence microscope and perifused by continuous flow of 2 mM glucose and 25 mM glucose in krebs-ringer buffer. KCl was then added to inhibit K+ efflux and depolarize the cells. Fura-2/AM was excited at 340 and 380 nm and emission was detected at 510 nm. Results are presented as percent of baseline. p = ns, n = 3 (B) Rh123 excitation at 490 nm and emission at 530 nm was measured to assess mitochondrial potential during exposure to 25 mM glucose. Results are presented as percent of baseline. P = ns, n = 3 (C): Dynamic insulin secretion during the intracellular calcium measurements in (A), P = ns. Black bar, uninfected; green bar, Ad-LacZ infected; red bar, Ad-E2F3 infected islets.
Human and rat E2F3 infected islets reverse diabetes in a minimal mass mouse model
To determine if the in vitro proliferative effect of E2F3 resulted in functional β cells, we used a minimal mass islet transplant model. Similar numbers of rat or human islets were infected with E2F3, LacZ, or no virus, cultured and transplanted under the kidney capsule of STZ-induced diabetic nude mice. Any changes in blood glucose levels were believed to be from proliferation of insulin-secreting cells rather than an enhancement of insulin secretion from existing cells, since we saw in vitro evidence of proliferation, but not of improved insulin secretion or insulin secretion kinetics. We found E2F3 infected rat islets were able to reverse diabetes (reversal defined as blood glucose <210 mg/dL), obtaining an average blood glucose of 184.2 ± 30.4 mg/dL, compared with 280.1 ± 30.4 mg/dL with uninfected islets, P = 0.009 (). All mice gained weight throughout the 21-d period (). After 21 d, to further characterize the graft function, we performed IPGTTs on the transplanted mice, revealing the E2F3-infected mice had an enhanced ability to clear glucose, with an AUC of 24 390 mg/dL/min ± 2547 compared with 42 654 mg/dL/min ± 10684 for uninfected, P = 0.04 (). Following the IPGTT, a unilateral nephrectomy of the grafted kidney showed the transplanted islets restored normoglycemia, rather than a spontaneous regeneration of the native pancreas (). This suggests that despite an initial culture of 250 IEq in each condition, the overexpression of E2F3 drove proliferation of insulin-secreting cells, such that the effective number of β cells was higher in E2F3-treated islets.
Figure 8. In vivo function of E2F3 infected rat and human islets. (A) Blood glucose measurements for mice transplanted with minimal mass (250 IEq) of ad-E2F3 infected (MOI 500) or uninfected (MOI 0) rat islets, AUC P = 0.009 (B) Weight of mice transplanted with rat islets infected with ad-E2F3 and uninfected. Nephrectomy performed at day 21. (C and D) Intraperitoneal glucose tolerance testing in transplanted mice 21 d after minimal rat islet mass transplant and area under the curve of glucose clearance, *P = 0.04, n = 4. Blue bar, uninfected; red bar, Ad-E2F3 infected islets. (E) Blood glucose measurements for mice transplanted with 1500 IEq of uninfected (MOI 0), ad-LacZ (MOI 500), and ad-E2F3 (MOI 500) infected human islets, AUC P = ns. (F) Change in weight over the 21 d transplant follow up. Nephrectomy performed at day 21. (G and H) 21-d IPGTT of mice transplanted with human islets and area under the curve of glucose clearance, AUC P = ns, n = 5. Back bar with circles, uninfected islets (2000 IEq); black bar with squares, uninfected islets (1000 IEq); blue bar, uninfected islets (1500 IEq); green bar, Ad-Lacz infected islets (1500 IEq); red bar, Ad-E2F3 infected islets (1500 IEq); NFX, unilateral nephrectomy.
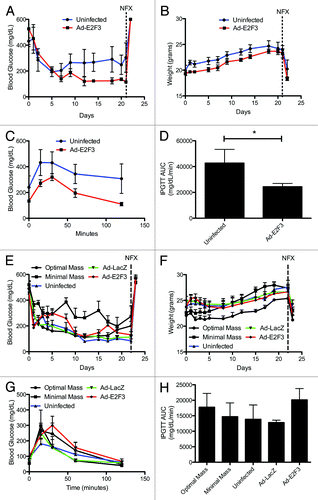
To test the in vivo function of expanded human islets, we transplanted 2000 IEq, of uninfected islets as a positive control and 1000 IEq of uninfected islets as a negative control. We found mice transplanted with 1500 IEq of E2F3-infected islets were able to reverse diabetes, with an average blood glucose level of 203.2 mg/dL ± 19.2 compared with LacZ, 157.3 mg/dL ± 20.2, and uninfected islets, 158.1 mg/dL ± 19.8, revealing no significant difference (). Additionally, all groups showed weight gains during the 21 d (). Glucose tolerance test further revealed no impairment of β-cell function with the addition of E2F3, with an area under the curve of 13 868 mg/dL/min ± 4629 for uninfected islets, 12 792 mg/dL/min ± 776 for ad-LacZ and, 20 138 mg/dL/min ± 3652 for ad-E2F3-infected, P = ns (). These results show that E2F3 induces proliferation in human β cells, but under these conditions, the proliferation is not significant enough to affect transplant outcomes. However, E2F3 infected islets do not display an impaired function and are able to reverse diabetes in mice.
Discussion
We report several new observations of human β-cell proliferation. First, we show that wild-type islets contain repressive E2Fs in their nucleus. Second, we show that proliferative human insulinomas have virtually no expression of these repressive E2Fs, but do express the proliferation-inducing E2F3. Third, when E2F3 is overexpressed in wild-type rat and human islets, it induces significant β-cell proliferation. Lastly, we show that this expansion does not impair the β-cell phenotype, and the proliferation occurs without an increase in apoptosis. These observations add to the growing picture of the human β-cell cycle, both in vitro for cell replacement therapies and in vivo for regeneration of islets.
Replication of adult human β cells is a rare event, with multiple mechanisms limiting proliferation and maintaining quiescence. Our observation of expression of E2F4–6 in the nucleus of wild-type islets may represent one such mechanism. During G0 of the cell cycle, E2F4/5-p107/130 complexes and pocket protein-independent E2F6 occupy the promoter region of genes responsible for controlling DNA synthesis, inhibiting their transcription and preventing transition from G1 to S-phase.Citation8,Citation10,Citation16,Citation17,Citation29 E2F4 and 5 lack a nuclear location signal and are therefore sequestered in the cytoplasm unless bound to a pocket protein.Citation10,Citation29 This implies the nuclear expression of E2F4 and 5 we see is p107/130 complexed and functionally repressing E2F1–3 target genes, which may impair proliferation and promote quiescence. The presence of nuclear E2F3 in our Ki67 stain suggests that the constitutively expressed E2F3 is able to overcome endogenous Rb regulation. This free E2F3 is then able to enter the nucleus and affect target genes. The Ki67 stains also show co-staining of E2F3 and Ki67+ nuclei, which implies the nuclear location of E2F3 is a strong proliferative signal, perhaps independent of expression level. Methods to move the E2F3 normally found in the cytoplasm of islets into the nucleus may be a means to induce proliferation without overexpression of cell cycle genes.
Debate over whether proliferation or differentiation of new β cells maintains the adult β-cell mass is ongoing, but evidence suggests that mitotic division is the predominate pathway.Citation20-Citation22 Insulinomas show that cell cycle mutations are capable of proliferating insulin-producing β cells and could provide a model for their expansion for research and cell therapies. Rodent insulinomas have been used to reveal D cyclins and cdks, upstream effectors of E2F, as potent inducers of proliferation.Citation33,Citation34 For the first time, we describe expression of pro-proliferative E2F3 and no repressive E2F4–6 expression in human insulinomas. Since E2F lies downstream in the G1/S checkpoint, mutations of the many upstream regulatory genes can profoundly effect it, resulting in aberrant expression in almost all forms of cancer.Citation12 E2F3 in particular is highly expressed in lung, bladder, and prostate cancers.Citation13-Citation15 Additionally, E2F3 is required for normal proliferation and is rate limiting in the proliferation of tumor cell lines.Citation35 Surprisingly, though E2F1 and 3 are necessary to start quiescent cells cycling, only E2F3 is needed for subsequent rounds of division.Citation36 This may explain why both E2F1 and 3 were expressed in our insulinomas, with E2F3 being the more prevalent one. E2F1–3 are maximally expressed during G1/S and minimally expressed during G0, while E2F4–6 remains relatively constant.Citation18 This implies a decrease in E2F4–6 is not required for E2F1–3 to drive proliferation, as pro-proliferative E2Fs are able to displace the occupying E2F4–6.Citation8 A reduction of E2F4–6 does not appear necessary to induce mitosis, but could further enhance E2F3-induced proliferation.
Extracellular signals of proliferation that have dramatic effects in other cell types often do not elicit a similar response in human β cells.Citation37-Citation39 In contrast, efforts to manipulate genes controlling the G1/S checkpoint have led to robust proliferation of insulin-positive cells.Citation24,Citation25,Citation27,Citation33 These nutrient and extracellular signals of proliferation may be linked to nuclear cell cycle proteins through, among many others, AKT and mTORC1 pathways.Citation40,Citation41 This signal likely activates DNA synthesis and proliferation through E2F, since they are upstream effectors of E2F.Citation30 E2F expression has been shown to control proliferation through activation or repression of genes involved in DNA synthesis.Citation5,Citation6,Citation11 However, we do not uncover specific E2F3 target genes involved in either maintaining quiescence or inducing proliferation. Our finding that E2F3 drives proliferation in islets is previously unreported, but recently it has been shown that E2F1 overexpression is capable of inducing proliferation and considerable apoptosis in mouse and human β cells.Citation27 In contrast, here we show that overexpression of E2F3 induces significant proliferation without apoptosis. Induction of apoptosis may be unique to E2F1, as its target genes include p53, p73, several caspases, and APAF1, while E2F3 is not an activator of these genes.Citation7,Citation42 Similarly, E2F3 is able to positively regulate a unique set of pro-proliferative target genes, such as DNA polymerase A, DHFR, and cdc6 (among others), while E2F1 is not.Citation35,Citation43
Whereas many attempts to proliferate β cells result in the loss of glucose-sensitive insulin section and the β-cell phenotype, E2F3 proliferated islets retained these abilities.Citation32 These cells displayed unimpaired intracellular calcium levels, mitochondrial potential, and dynamic insulin secretion, confirming the presence and integrity of glucose-stimulated insulin secretion coupling by the presence of functional KATP and voltage-gated calcium channels. The Cdk4-E2F1 pathway regulates the expression of Pdx1, a gene expressed in the mature pancreas.Citation44 Additionally, E2F1 was recently found to regulate expression of the potassium channel subunit Kir6.2.Citation28 Despite significant homology between E2F1 and E2F3, it is not known if E2F3 also regulates Kir6.2 expression and the secretion of insulin, though our results imply that the pathway allowing for insulin secretion remain intact. Maintenance of Pdx1 and Kir6.2 expression could be one mechanism by which E2F3-proliferated islets retain their function during culture, since reduced Pdx1 expression has been linked to loss of β-cell function associated with aging.Citation45,Citation46 These features make E2F3 a favorable target for increasing human β-cell mass and lend to its ability to induce proliferation independent of apoptosis while retaining the β-cell phenotype.
E2F3-proliferated rat islets showed robust function both in vitro and in vivo through functional increases in β-cell numbers. In humans, islets infected with E2F3 showed normal function and retained the β-cell phenotype both in vitro and in vivo, but did not increase the β-cell mass sufficiently to improve transplant outcomes. This is likely due to the difference between E2F3-induced proliferation rates between rats (23.8%) and humans (7.2%). The disparity in knowledge between the rodent and human β-cell cycle has been emphasized recently for this exact reason.Citation30 Importantly, the overall function of the islets was not seemingly impaired by ectopic delivery of E2F3. We propose that either improving the infection efficiency or concomitant E2F4–6 knockout could increase E2F3-induced proliferation sufficiently to functionally impact islet grafts and reduce the islets required to reverse diabetes in this model. Decreased expression of inhibitory cell cycle proteins, such as E2F4–6, could release the cell cycle “brake” and allow overexpression of genes driving proliferation to have a more profound effect on the expansion of β cells.Citation47 Future strategies for increasing the infection efficiency could include dissociation of islets, infection and expansion, then re-aggregation before transplantation, or the use of an alternative, non-viral DNA delivery system, such as gold nanoparticles, that infect the whole islets rather than merely the mantle.Citation48,Citation49
One concern with this approach is the uncontrolled expression of cell cycle genes, leading to the formation of tumors. While this is a consideration for long-term culture of permanently mutated cells, the use of a transient non-incorporating adenovirus lessens these concerns in the experiments shown here. Any cell type overexpressing cell cycle proteins should be carefully monitored and assessed for the possible formation of tumors during long-term culture in vivo. In addition to possible oncogenic formation, long-term function of transfected islets also remains to be investigated. Islets transplanted under the kidney capsule have shown function for up to 1 y, but how the proliferative stimulus of E2F3 may effect this is unknown.Citation50 However, no negative effects of E2F3 overexpression were observed in transplanted islets after 21 d, but long-term studies are needed to assess their function after sustained in vivo culture.
Further understanding of the β-cell cycle is critical for the advancement of islet transplantation and the understanding of in vivo development, growth, and death of insulin-secreting cells. Here, we identify expression of a key G1/S checkpoint regulator, E2F in the wild-type islets and human insulinomas. Mimicking this expression in human islets induces proliferation of β cells without induction of apoptosis. Furthermore, these proliferated β cells retain their phenotype with no impairment of glucose-sensitive insulin secretion. This identifies E2F3 as a novel target to induce expansion of insulin-secreting cells in vitro and may one day allow for in vivo pancreatic regeneration.
Materials and Methods
Islet isolation and culture
Human islet isolation was preformed according to previously described protocols using a modified Ricordi semi-automated method and purified by a continuous gradient.Citation51,Citation52 Following isolation, human islets were cultured overnight in CMRL 1066 (Cellgro) supplanted with 2.5% human albumin, 0.25% sodium bicarbonate, 0.02 mg/mL ciprofloxacin, 0.2% ITS, and 10 mM HEPES. One islet equivalent (IEq) was defined as an islet of 150 um diameter. Rat pancreata were isolated from 8–12-wk-old male Lewis rats as previously described.Citation53 Pancreatic tissue was digested using 2.2 mg/ml Collagenase XI (Sigma). Islets were cultured for 24 h in RPMI 1640 with GlutaMAX (Invitrogen) supplemented with 10% FBS, 100 U/mL penicillin and 100 µg/mL streptomycin. All experiments were done in accordance with protocols approved by the University of Illinois at Chicago Office of Animal Care and Institutional Biosafety Committee.
Immunoblotting and RT-PCR for E2F 1–6
Equivalent amounts of protein were separated on 4–20% polyacrylamide gels (Bio-Rad) and transferred to nitrocellulose membranes. Membranes were incubated with E2F antibodies (E2F1–3 Santa Cruz Biotechnology and E2F4–6 Abcam), 1:200 and visualized with 1:200 horseradish–peroxidase-coupled secondary antibodies (Cell Signaling). Reverse transcription PCR was performed on RNA samples isolated by RNeasy Mini Kit (Qiagen) and combined with oligonucleotide primers by heat shock in a 70 °C water bath followed by 5 min in ice. cDNA was synthesized by reverse transcription of 1 μg of whole islet RNA with the ImProm-II Reverse Transcription System (Promega) following the manufacturer's instructions. Tubes were annealed for 5 min at 25 °C, extended for 1 h at 42 °C, and inactivation of the reverse transcriptase occurred at 70°C for 15 min. PCR was performed with 3 μL of cDNA from the heat-inactivated PCR tube. Tubes contained a final volume of 25 μL: 12.5 μL PCR Master Mix (Promega), 8.7 μL nuclease-free water, 3 μL cDNA, 0.4 μL (20nM) forward primer, 0.4 μL (20 nM) reverse primer for E2F 1–6. Tubes were run at 95 °C for 4 min, cycled 30 times (95 °C for 30 s, 52 °C for 30 s, 72 °C for 1.5 min), and finally 72 °C for 7 min. Next, the PCR products were separated on 2% agarose gels and visualized with SyberSafe at 1:10 000 (Invitrogen), a UV transilluminator, and a digital camera.
Viral infections
Full-length human E2F3 cDNA obtained from Origene was packaged at Vector Biolabs into an adenovirus-type 5 (dE1/E3) with a CMV promoter and GFP tag. Control adenovirus for ad-CMV-LacZ was purchased Vector Biolabs. Infections of human and rat islets were performed in ultra-low attachment plates overnight in a minimal volume of media. The following day media supplemented with 20 μM of EdU was added to the plates. Following 4 d of culture the islets were collected, fixed, and embedded, as described below. MOI for adenoviruses were as described in figure legends. For determination of multiplicity of infection (MOI), 1 IEq was estimated to contain 1000 cells.
Immunohistochemistry
Following infection and culture, islets were collected fixed in Bouin solution. The islets were then suspended in 2% agarose, dehydrated in a series of 70–100% alcohol and embedded in paraffin blocks. Paraffin blocks were serially sectioned with 5 µm sections taken every 30 µm. For EdU incorporation staining, the slides were de-waxed, rehydrated, and stained with the Click-IT EdU labeling kit (Invitrogen) according to manufacturer's instructions. Following the EdU staining, slides were co-stained for insulin with 1:200 polyclonal guinea pig anti-insulin primary antibody (Dako) and 1:200 anti-guinea pig 594 nm secondary antibody (Invitrogen). Slides contained fluorescence emitting at 461 nm (DAPI) 594 nm (insulin), and 647 nm. For Ki67 stains, heat-treated antigen retrieval was followed by incubation in Triton-X 0.1%, and overnight incubation in Ki67 antibody (1:50, Dako) and E2F3 (1:200, Santa Cruz Biotechnology). The following day, slides were completed with 1:200 anti-mouse 647 nm and anti-rabbit 488nm secondary antibodies (Invitrogen), as well as DAPI to label DNA. Digital photographs were taken using an inverted fluorescence microscope.
Microfludic assays
Islets were labeled with ratiometric fluorescent dye fura-2/AM (Invitrogen) to determine intracellular calcium levels and fluorescent rhodamine 123 dye (Sigma) to determine changes in mitochondrial potentials, as previously described.Citation54 Both fluorescence signals were expressed as “change-in-percentage” after being normalized against basal intensity levels established before stimulation. The perifusates were collected at 1 mL/min to determine insulin secretory kinetics via ELISA kit (Mercodia AB).
Glucose-static incubations
Following infection and 4 d culture, static glucose incubations were performed as described previously, with the exception of the low and high glucose concentrations, which were changed to 2.8 mM and 28 mM.Citation55 Quantification of insulin concentrations performed by ELISA (Mercodia) per manufacture's instruction.
Islet transplants
Islets were infected as described above and cultured overnight prior to transplant. Eight–12-wk-old male nude mice were rendered diabetic by a single intraperitoneal injection of 220 mg/kg streptozotocin (STZ). Diabetes was considered established after 2 consecutive days of blood glucose readings over 350 mg/dL. Sub-kidney capsule transplantation of islets was performed as previously described.Citation56 Mice were then monitored daily for the first 5 d and 3 times weekly thereafter for weight and non-fasting blood glucose for 21 d.
IPGTT and nephrectomy
At 21 d post-transplantation, mice were assayed for glucose tolerance by intraperitoneal glucose tolerance test. The animals were fasted overnight and intraperitoneally injected with a 3 g glucose/kg solution. Blood glucose was measured at: 0, 15, 30, 60, and 120 min following injection. After IPGTT, unilateral nephrectomy removed the grafted islets to confirm the absence of in situ pancreatic regeneration.
Statistical analysis
Two-tailed unpaired t-tests were performed for pairs of data. One-way ANOVA was used to assess differences between multiple groups of data. The area under the curve was determined by Mann-Whitney U test. P values < 0.05 were considered significant. Standard error bars were used in the appropriate figures.
| Abbreviations: | ||
| Rb | = | retinoblastoma protein |
| IEq | = | islet equivalent |
Additional material
Download Zip (66.2 KB)Aknowledgements
Author contributions: experimental concept, design and execution, analysis and interpretation of data, drafting article, critical revisions of article, and final approval of version to be published by BAR; assistance with western blots YC; assistance with islet transplants, critical revisions of article, and final approval of article to be published PVS and YW, microfluidic assay performed by QW and YW, experimental concept and design, and final approval of article to be published by PS and JO. BAR and JO are the guarantors of this work and, as such, had full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. We report no potential conflicts of interest related to this article.
We would like to acknowledge the Chicago Diabetes Project, Washington Square Health Foundation, the Christopher Family Foundation, and NIH R01 grant DK091526 for financial support, Andrew Stewart (Mount Sinai Hospital) for assistance with viral infections and proliferation assays and Julie Kerr-Conte (Université de Lille 2) for assistance with proliferation assays.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Related Research Data
References
- Couri CE, Oliveira MC, Stracieri AB, Moraes DA, Pieroni F, Barros GM, et al. C-peptide levels and insulin independence following autologous nonmyeloablative hematopoietic stem cell transplantation in newly diagnosed type 1 diabetes mellitus. JAMA 2009; 301:1573 - 9; http://dx.doi.org/10.1001/jama.2009.470; PMID: 19366777
- Gu W, Hu J, Wang W, Li L, Tang W, Sun S, et al. Diabetic ketoacidosis at diagnosis influences complete remission after treatment with hematopoietic stem cell transplantation in adolescents with type 1 diabetes. Diabetes Care 2012; 35:1413 - 9; http://dx.doi.org/10.2337/dc11-2161; PMID: 22723579
- Ichii H, Ricordi C. Current status of islet cell transplantation. J Hepatobiliary Pancreat Surg 2009; 16:101 - 12; http://dx.doi.org/10.1007/s00534-008-0021-2; PMID: 19110649
- Ryan EA, Shandro T, Green K, Paty BW, Senior PA, Bigam D, et al. Assessment of the severity of hypoglycemia and glycemic lability in type 1 diabetic subjects undergoing islet transplantation. Diabetes 2004; 53:955 - 62; http://dx.doi.org/10.2337/diabetes.53.4.955; PMID: 15047610
- Bracken AP, Ciro M, Cocito A, Helin K. E2F target genes: unraveling the biology. Trends Biochem Sci 2004; 29:409 - 17; http://dx.doi.org/10.1016/j.tibs.2004.06.006; PMID: 15362224
- Johnson DG, Schwarz JK, Cress WD, Nevins JR. Expression of transcription factor E2F1 induces quiescent cells to enter S phase. Nature 1993; 365:349 - 52; http://dx.doi.org/10.1038/365349a0; PMID: 8377827
- DeGregori J, Johnson DG. Distinct and Overlapping Roles for E2F Family Members in Transcription, Proliferation and Apoptosis. Curr Mol Med 2006; 6:739 - 48; PMID: 17100600
- Takahashi Y, Rayman JB, Dynlacht BD. Analysis of promoter binding by the E2F and pRB families in vivo: distinct E2F proteins mediate activation and repression. Genes Dev 2000; 14:804 - 16; PMID: 10766737
- Nevins JR. E2F: a link between the Rb tumor suppressor protein and viral oncoproteins. Science 1992; 258:424 - 9; http://dx.doi.org/10.1126/science.1411535; PMID: 1411535
- Dyson N. The regulation of E2F by pRB-family proteins. Genes Dev 1998; 12:2245 - 62; http://dx.doi.org/10.1101/gad.12.15.2245; PMID: 9694791
- Blais A, Dynlacht BD. Hitting their targets: an emerging picture of E2F and cell cycle control. Curr Opin Genet Dev 2004; 14:527 - 32; http://dx.doi.org/10.1016/j.gde.2004.07.003; PMID: 15380244
- Sherr CJ, McCormick F. The RB and p53 pathways in cancer. Cancer Cell 2002; 2:103 - 12; http://dx.doi.org/10.1016/S1535-6108(02)00102-2; PMID: 12204530
- Cooper CS, Nicholson AG, Foster C, Dodson A, Edwards S, Fletcher A, et al. Nuclear overexpression of the E2F3 transcription factor in human lung cancer. Lung Cancer 2006; 54:155 - 62; http://dx.doi.org/10.1016/j.lungcan.2006.07.005; PMID: 16938365
- Olsson AY, Feber A, Edwards S, Te Poele R, Giddings I, Merson S, et al. Role of E2F3 expression in modulating cellular proliferation rate in human bladder and prostate cancer cells. Oncogene 2007; 26:1028 - 37; http://dx.doi.org/10.1038/sj.onc.1209854; PMID: 16909110
- Foster CS, Falconer A, Dodson AR, Norman AR, Dennis N, Fletcher A, et al. Transcription factor E2F3 overexpressed in prostate cancer independently predicts clinical outcome. Oncogene 2004; 23:5871 - 9; http://dx.doi.org/10.1038/sj.onc.1207800; PMID: 15184867
- Stevens C, La Thangue NB. E2F and cell cycle control: a double-edged sword. Arch Biochem Biophys 2003; 412:157 - 69; http://dx.doi.org/10.1016/S0003-9861(03)00054-7; PMID: 12667479
- Ogawa H, Ishiguro K-i, Gaubatz S, Livingston DM, Nakatani Y. A complex with chromatin modifiers that occupies E2F- and Myc-responsive genes in G0 cells. Science 2002; 296:1132 - 6; http://dx.doi.org/10.1126/science.1069861; PMID: 12004135
- Zhu WW, Giangrande PH, Nevins JR. Temporal control of cell cycle gene expression mediated by E2F transcription factors. Cell Cycle 2005; 4:633 - 6; http://dx.doi.org/10.4161/cc.4.5.1650; PMID: 15876877
- Giangrande PH, Zhu W, Schlisio S, Sun X, Mori S, Gaubatz S, et al. A role for E2F6 in distinguishing G1/S- and G2/M-specific transcription. Genes Dev 2004; 18:2941 - 51; http://dx.doi.org/10.1101/gad.1239304; PMID: 15574595
- Teta M, Rankin MM, Long SY, Stein GM, Kushner JA. Growth and regeneration of adult beta cells does not involve specialized progenitors. Dev Cell 2007; 12:817 - 26; http://dx.doi.org/10.1016/j.devcel.2007.04.011; PMID: 17488631
- Dor Y, Brown J, Martinez OI, Melton DA. Adult pancreatic beta-cells are formed by self-duplication rather than stem-cell differentiation. Nature 2004; 429:41 - 6; http://dx.doi.org/10.1038/nature02520; PMID: 15129273
- Bonner-Weir S, Li W-C, Ouziel-Yahalom L, Guo L, Weir GC, Sharma A. Beta-cell growth and regeneration: replication is only part of the story. Diabetes 2010; 59:2340 - 8; http://dx.doi.org/10.2337/db10-0084; PMID: 20876724
- Gunasekaran U, Hudgens CW, Wright BT, Maulis MF, Gannon M. Differential regulation of embryonic and adult β cell replication. Cell Cycle 2012; 11:2431 - 42; http://dx.doi.org/10.4161/cc.20545; PMID: 22659844
- Cozar-Castellano I, Takane KK, Bottino R, Balamurugan AN, Stewart AF. Induction of beta-cell proliferation and retinoblastoma protein phosphorylation in rat and human islets using adenovirus-mediated transfer of cyclin-dependent kinase-4 and cyclin D1. Diabetes 2004; 53:149 - 59; http://dx.doi.org/10.2337/diabetes.53.1.149; PMID: 14693709
- Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, et al. Induction of human beta-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 2010; 59:1926 - 36; http://dx.doi.org/10.2337/db09-1776; PMID: 20668294
- Chen S, Shimoda M, Chen J, Matsumoto S, Grayburn PA. Transient overexpression of cyclin D2/CDK4/GLP1 genes induces proliferation and differentiation of adult pancreatic progenitors and mediates islet regeneration. Cell Cycle 2012; 11:695 - 705; http://dx.doi.org/10.4161/cc.11.4.19120; PMID: 22373529
- Grouwels G, Cai Y, Hoebeke I, Leuckx G, Heremans Y, Ziebold U, et al. Ectopic expression of E2F1 stimulates beta-cell proliferation and function. Diabetes 2010; 59:1435 - 44; PMID: 20299467
- Annicotte J-S, Blanchet E, Chavey C, Iankova I, Costes S, Assou S, et al. The CDK4-pRB-E2F1 pathway controls insulin secretion. Nat Cell Biol 2009; 11:1017 - 23; http://dx.doi.org/10.1038/ncb1915; PMID: 19597485
- Müller H, Moroni MC, Vigo E, Petersen BO, Bartek J, Helin K. Induction of S-phase entry by E2F transcription factors depends on their nuclear localization. Mol Cell Biol 1997; 17:5508 - 20; PMID: 9271426
- Kulkarni RN, Mizrachi E-B, Ocana AG, Stewart AF. Human β-cell proliferation and intracellular signaling: driving in the dark without a road map. Diabetes 2012; 61:2205 - 13; http://dx.doi.org/10.2337/db12-0018; PMID: 22751699
- Icyuz M, Bryant SM, Fortinberry HK, Molakandov K, Siegal GP, Contreras JL, et al. Adenovirus infection activates akt1 and induces cell proliferation in pancreatic islets1. Transplantation 2009; 87:821 - 4; http://dx.doi.org/10.1097/TP.0b013e318199c686; PMID: 19300183
- Weinberg N, Ouziel-Yahalom L, Knoller S, Efrat S, Dor Y. Lineage tracing evidence for in vitro dedifferentiation but rare proliferation of mouse pancreatic beta-cells. Diabetes 2007; 56:1299 - 304; http://dx.doi.org/10.2337/db06-1654; PMID: 17303800
- Fiaschi-Taesch N, Bigatel TA, Sicari B, Takane KK, Salim F, Velazquez-Garcia S, et al. Survey of the human pancreatic beta-cell G1/S proteome reveals a potential therapeutic role for cdk-6 and cyclin D1 in enhancing human beta-cell replication and function in vivo. Diabetes 2009; 58:882 - 93; http://dx.doi.org/10.2337/db08-0631; PMID: 19136653
- Cozar-Castellano I, Harb G, Selk K, Takane K, Vasavada R, Sicari B, et al. Lessons from the first comprehensive molecular characterization of cell cycle control in rodent insulinoma cell lines. Diabetes 2008; 57:3056 - 68; http://dx.doi.org/10.2337/db08-0393; PMID: 18650366
- Humbert PO, Verona R, Trimarchi JM, Rogers C, Dandapani S, Lees JA. E2f3 is critical for normal cellular proliferation. Genes Dev 2000; 14:690 - 703; PMID: 10733529
- Kong LJ, Chang JT, Bild AH, Nevins JR. Compensation and specificity of function within the E2F family. Oncogene 2007; 26:321 - 7; http://dx.doi.org/10.1038/sj.onc.1209817; PMID: 16909124
- Rutti S, Sauter NS, Bouzakri K, Prazak R, Halban PA, Donath MY. In vitro proliferation of adult human beta-cells. PLoS One 2012; 7:e35801; http://dx.doi.org/10.1371/journal.pone.0035801; PMID: 22563404
- Cozar-Castellano I, Weinstock M, Haught M, Velázquez-Garcia S, Sipula D, Stewart AF. Evaluation of beta-cell replication in mice transgenic for hepatocyte growth factor and placental lactogen: comprehensive characterization of the G1/S regulatory proteins reveals unique involvement of p21cip. Diabetes 2006; 55:70 - 7; http://dx.doi.org/10.2337/diabetes.55.01.06.db05-0632; PMID: 16380478
- Guney MA, Petersen CP, Boustani A, Duncan MR, Gunasekaran U, Menon R, et al. Connective tissue growth factor acts within both endothelial cells and beta cells to promote proliferation of developing beta cells. Proceedings of the National Academy of Sciences 2011;
- Balcazar N, Sathyamurthy A, Elghazi L, Gould A, Weiss A, Shiojima I, et al. mTORC1 activation regulates beta-cell mass and proliferation by modulation of cyclin D2 synthesis and stability. J Biol Chem 2009; 284:7832 - 42; http://dx.doi.org/10.1074/jbc.M807458200; PMID: 19144649
- Blandino-Rosano M, Chen AY, Scheys JO, Alejandro EU, Gould AP, Taranukha T, et al. mTORC1 signaling and regulation of pancreatic β-cell mass. Cell Cycle 2012; 11:1892 - 902; http://dx.doi.org/10.4161/cc.20036; PMID: 22544327
- DeGregori J, Leone G, Miron A, Jakoi L, Nevins JR. Distinct roles for E2F proteins in cell growth control and apoptosis. Proc Natl Acad Sci U S A 1997; 94:7245 - 50; http://dx.doi.org/10.1073/pnas.94.14.7245; PMID: 9207076
- Schlisio S, Halperin T, Vidal M, Nevins JR. Interaction of YY1 with E2Fs, mediated by RYBP, provides a mechanism for specificity of E2F function. EMBO J 2002; 21:5775 - 86; http://dx.doi.org/10.1093/emboj/cdf577; PMID: 12411495
- Kim SY, Rane SG. The Cdk4-E2f1 pathway regulates early pancreas development by targeting Pdx1+ progenitors and Ngn3+ endocrine precursors. Development 2011; 138:1903 - 12; http://dx.doi.org/10.1242/dev.061481; PMID: 21490060
- Vetterli L, Maechler P. Resveratrol-activated SIRT1 in liver and pancreatic β-cells: a Janus head looking to the same direction of metabolic homeostasis. Aging (Albany NY) 2011; 3:444 - 9; PMID: 21483037
- Gunasekaran U, Gannon M. Type 2 diabetes and the aging pancreatic beta cell. Aging (Albany NY) 2011; 3:565 - 75; PMID: 21765202
- Tavana O, Zhu C. Too many breaks (brakes): pancreatic β-cell senescence leads to diabetes. Cell Cycle 2011; 10:2471 - 84; http://dx.doi.org/10.4161/cc.10.15.16741; PMID: 21750406
- Jeong JH, Lee JI, Ju MK, Joo DJ, Huh KH, Kim MS, et al. Proliferation of pancreatic endocrine cells using disaggregation-expansion-reaggregation technology in isolated rat islets. Transplant Proc 2010; 42:907 - 10; http://dx.doi.org/10.1016/j.transproceed.2010.02.044; PMID: 20430201
- Vega RA, Wang Y, Harvat T, Wang S, Qi M, Adewola AF, et al. Modified gold nanoparticle vectors: a biocompatible intracellular delivery system for pancreatic islet cell transplantation. Surgery 2010; 148:858 - 65, discussion 865-6; http://dx.doi.org/10.1016/j.surg.2010.07.036; PMID: 20800254
- Hiller WFA, Klempnauer J Jr., Lück R, Steiniger B. Progressive deterioration of endocrine function after intraportal but not kidney subcapsular rat islet transplantation. Diabetes 1991; 40:134 - 40; http://dx.doi.org/10.2337/diabetes.40.1.134; PMID: 2015968
- Gangemi A, Salehi P, Hatipoglu B, Martellotto J, Barbaro B, Kuechle JB, et al. Islet transplantation for brittle type 1 diabetes: the UIC protocol. Am J Transplant 2008; 8:1250 - 61; http://dx.doi.org/10.1111/j.1600-6143.2008.02234.x; PMID: 18444920
- Shapiro AMJ, Lakey JRT, Ryan EA, Korbutt GS, Toth E, Warnock GL, et al. Islet transplantation in seven patients with type 1 diabetes mellitus using a glucocorticoid-free immunosuppressive regimen. N Engl J Med 2000; 343:230 - 8; http://dx.doi.org/10.1056/NEJM200007273430401; PMID: 10911004
- Gotoh M, Maki T, Kiyoizumi T, Satomi S, Monaco AP. An improved method for isolation of mouse pancreatic islets. Transplantation 1985; 40:437 - 8; http://dx.doi.org/10.1097/00007890-198510000-00018; PMID: 2996187
- Adewola AF, Lee D, Harvat T, Mohammed J, Eddington DT, Oberholzer J, et al. Microfluidic perifusion and imaging device for multi-parametric islet function assessment. Biomed Microdevices 2010; 12:409 - 17; http://dx.doi.org/10.1007/s10544-010-9398-1; PMID: 20300858
- Maedler K, Fontana A, Ris F, Sergeev P, Toso C, Oberholzer J, et al. FLIP switches Fas-mediated glucose signaling in human pancreatic β cells from apoptosis to cell replication. Proceedings of the National Academy of Sciences 2002; 99:8236 - 41; http://dx.doi.org/10.1073/pnas.122686299
- Oberholzer J, Yu D, Triponez F, Cretin N, Andereggen E, Mentha G, et al. Decomplementation with cobra venom factor prolongs survival of xenografted islets in a rat to mouse model. Immunology 1999; 97:173 - 80; http://dx.doi.org/10.1046/j.1365-2567.1999.00742.x; PMID: 10447729