Abstract
Every day, cells are faced with thousands of DNA lesions, which have to be repaired to preserve cell survival and function. DNA repair is more or less accurate and could result in genomic instability and cancer. We review here the current knowledge of the links between molecular features, treatment, and DNA repair in multiple myeloma (MM), a disease characterized by the accumulation of malignant plasma cells producing a monoclonal immunoglobulin. Genetic instability and abnormalities are two hallmarks of MM cells and aberrant DNA repair pathways are involved in disease onset, primary translocations in MM cells, and MM progression. Two major drugs currently used to treat MM, the alkylating agent Melphalan and the proteasome inhibitor Bortezomib act directly on DNA repair pathways, which are involved in response to treatment and resistance. A better knowledge of DNA repair pathways in MM could help to target them, thus improving disease treatment.
In multiple myeloma (MM), malignant plasma cells display marked genomic abnormalities that arise during pathogenesis of disease and accrue during progression, and implicate aberrant DNA repair pathways in both phases. Extrinsic and intrinsic factors initiate DNA repair. Extrinsic stimuli during germinal center transit in tumor origins result in recurrent chromosomal translocations, where double-strand DNA breaks are erroneously resolved. Distinctive features of ploidy arise early and associate with cell cycling and dysregulated DNA repair. Intrinsic cellular mechanisms mediate pervasive ongoing DNA damage at disease onset and during progression that recruits multiple DNA repair pathways. Further extrinsic challenges arise when DNA-modifying drugs, such as melphalan or Bortezomib, are delivered as therapy and contribute to disease progression and relapse. In this review, we examine the major cellular and molecular pathways that mediate DNA repair, and evaluate existing evidence for imbalanced mechanisms in MM cells. The evidence suggests that activity of DNA repair pathways in malignant plasma cells is highly likely to be pivotal in determining survival and therapeutic outcome. It specifically identifies key dysregulated pathways, the nature of cross-talk between central signaling pathways and DNA repair, polymorphisms in DNA repair genes, or deregulated expression of these genes, and shows that they frequently correlate with treatment response and patients’ survival. Furthermore, it reveals that therapeutic drugs used to treat MM can either activate or inhibit DNA repair pathways. The findings finally suggest that novel strategies to regulate DNA repair pathways have the potential to augment current therapies in resolving disease. Thus, understanding the DNA damage response in MM is important to improving patients’ survival and to progress the next phase of effective patient-specific therapies.
Multiple Myeloma
Etiology
MM remains in the main an incurable malignant plasma cell disease, with 22 000 new patients per year in the EU or in the USA and a median survival of 5 y.Citation1 Seminal observations have established that it is preceded by monoclonal gammopathy of undetermined significance (MGUS), a premalignant phaseCitation2-Citation6 that occurs in 3% of individuals over 50 y of age.Citation7 MGUS can persist before developing to MM at a 1% rate/year, independently of the duration of the MGUS phase.Citation2,Citation4,Citation6 MGUS or MM plasma cells are clonal cells, with molecular evidence that the same clone evolves during transformation between disease states.Citation8 In individuals with MGUS, proliferation of clonal plasma cells remains below detection levels.Citation9-Citation11 In patients, MM cells (MMCs) cycle poorly (median percentage of MMCs in the S phase = 0.4%),Citation12 and MMC proliferation is one of the strongest prognostic factors in MM, predicting for short overall survival.Citation10,Citation11,Citation13
Molecular abnormalities and genomic instability in MM
MMCs originate from post-follicular B cells, which have undergone multiple immunoglobulin (Ig) gene rearrangements during maturation: early recombination of V(D)J genes, and subsequent somatic hypermutation (SHM) of IGV genes that code for variable Ig domains, and deletional isotype class switch recombination (CSR) of Ig heavy chain (IGH) genes at the 14q32 locus.Citation14 SHM in IGV genes is also a feature of MGUS,Citation8 many of which undergo CSR. MGUS or MM can be grossly divided into non-hyperdiploid (< 48 and > 75 chromosomes, half of patients) or hyperdiploid tumors (48–75 chromosomes, trisomies of odd chromosomes 3, 5, 7, 9, 11, 19, 21).Citation3,Citation5,Citation6,Citation15-Citation17 Primary 14q32 chromosomal translocations (CTs) are found in the vast majority (> 70%) of non-hyperdiploid MM and less frequently in hyperdiploid tumors (15% of cases). These primary translocations are recurrent in subsets of disease, juxtaposing CCND1 gene – t(11q13;14q32), 15–18% of tumors; MMSET/FGFR3 genes – t(4p16;14q32), 10–15% of tumors; MAF, MAFB, or MAFA genes – t(14q32;16q23), t(14q32;20q11), t(8q23;14q32), 8% of tumors; or CCND3 gene – t(6p23;14q32), 2% of tumors, under the control of strong Eμ intronic or 3′ IGHα enhancers that dysregulate gene expression.Citation15-Citation17 Most 14q32 CTs map into the switch IgH region, implicating deletional CSR as a pathogenic mechanism. Resolution of double-strand DNA breaks during CSR requires DNA repair pathways, including non-homologous end joining (NHEJ) (discussed below).Citation3,Citation5,Citation14,Citation18 Double-strand DNA breaks can also occur during SHM, to generate deletions and insertions in IGV genes, which surprisingly occur in general as multiples of nucleotide triplets.Citation19 The aberrant resolution of SHM strand breaks by DNA repair specifically has been implicated in generating some CTs in MM.Citation20 Some14q32 CTs have as-yet-undefined partner chromosomes, suggesting that multiple transcriptionally active loci are captured and targeted for translocated lesions by dysregulated DNA repair. Importantly, these CTs are present in the MGUS phase,Citation8,Citation21 indicating that abnormalities in the DNA repair spectrum is manifest early in pathogenesis and may be a pre-requisite for malignant transformation.
A resulting hallmark of the CT abnormalities is a deregulated expression of at least 1 of the 3 cyclin D genes in virtually all patients with MGUS or MM, either by constitutive activation of CCND1 or CCND3 genes or by upregulation of pathways (FGFR3/MMSET or MAF) resulting in high CCND2 expression.Citation6,Citation22-Citation25 High cyclin D expression is found also in hyperdiploid MM, but the mechanisms are less clear and may involve gene amplification or downregulation of miRNA targeting genes (CCND1, FGFR3, MAF, TCC3), which are partners for IGH translocations in non-hyperdiploid tumors.Citation9,Citation26 This high expression of cyclin D may lead to deregulation of cell cycle checkpoints, as well as DNA repair pathways,Citation12,Citation27 to accentuate genetic instability, a hallmark of MM disease.Citation10,Citation11,Citation13,Citation28 Transposon-like elements can also integrate at abnormal sites to dysregulate cyclin D expression.Citation29 Little is known as yet of the role of DNA repair pathways in mediating these transposon-like events in MM. Other translocations and genetic alterations involve TP53, NFκB, MYC, and KRAS genes, whose gene products are critical players in DNA replication and repair, are detected in MM tumors, with an increased incidence with disease evolution.Citation14,Citation17,Citation20,Citation25,Citation30,Citation31 Monoallelic deletion of chromosome 17p, including TP53 gene, is found in 7–10% of newly diagnosed patients in association with reduced survival, in 50% of patients with extramedullary disease, and increases with disease activity.Citation6,Citation15-Citation17,Citation32 In 30% of patients with monoallelic del17p, TP53 mutations are found in the remaining allele.Citation33 P53 protein may also be degraded in MMCs by upregulation of MDM2 through methylation of the promoter of miRNAs targeting MDM2.Citation34 Constitutive NFκB activity is observed in primary MMCs from about 20% of newly diagnosed patients,Citation35-Citation38 in part due to alterations of various genes coding for proteins of the NFκB pathway.Citation36,Citation37,Citation39 Both canonical and non-canonical NFκB pathways are activated in MM,Citation40 in association with drug resistance and/or with relapse.Citation38 Finally, whereas translocations of MYC gene are found in MMCs of 15% of newly diagnosed patients,Citation41 amplifications of MYC gene are found in 70% of patients with hyperdiploid MM, and 20% of non-hyperdiploid ones.Citation42,Citation43 MYC translocations are found in the majority of MM cell lines (90%).Citation44 CTs can be late events in the 14q32 locus, notably involving MYC, and often appear as subclonal events.Citation21 These observations reveal an ongoing relevance of DNA repair pathway abnormalities for disease progression in MM. A role for these pathways appears to persist under therapy. Recent evidence from aCGH studies serially tracking MM progression has shown that subclonal competition marked by specific genomic abnormalities is a common feature of disease, and that therapeutic pressure may be an important driver.Citation45,Citation46 Some of these subclones may have existed at onset of therapy at below detection levels, and others reflect a further tumor diversification that recruits aberrant DNA repair. Comparable features are also emerging from next generation sequencing (NGS) of the tumor genome, where sets of gene mutations are identifiable only in relapse samples, also implicating dysregulated DNA repair pathways, and persist in progeny.Citation47,Citation48 High-resolution SNP arrays have delineated wide-ranging structural alterations in the MM genome, including loss of heterozygosity,Citation49 further highlighting a lack of corrective measures in the MM DNA repair machinery, or masking to generate these lesions. The propensity for the t(11;14) CT in MM in particular is also markedly increased by a CCND1 polymorphism from GWAS studies,Citation50 raising the question whether DNA repair mechanisms may differ in this constitutive genetic background and route to MM, and by extension in MM subsets segregated by specific CT abnormalities.
The molecular mechanisms that yield non-hyperdiploid or hyperdiploid states are as yet unknown, but are faithfully recapitulated in tumor progeny in MGUS and MM. Aneuploidy results from chromosomal instability, which can affect segregation of whole chromosomes or cause specific chromosomal abnormalities, including deletions, insertions, and translocations.Citation51 In relation to cancer, a question that has persisted is whether aneuploidy is a causal driver of transformation or a by-product, and is as yet not fully resolved. Whole chromosome segregation occurs during mitosis and begins during S phase, when DNA strands must be replicated precisely, involving specific components of the DNA repair pathway. Subsequently, in early mitosis, a spindle-assembly checkpoint ensures correct attachment of sister chromatids to the mitotic spindle prior to the initiation of chromosome segregation.Citation52 Several mechanisms can aberrantly impact on mitotic segregation of chromosomes, including mutations in spindle-assembly checkpoint genes which bypass p53-mediated cell arrest and lead to ploidy defects. These genes include the BUB and MAD spindle checkpoint gene families and have been found to be mutated in some cancers, but are extremely rare in MM, suggesting that comparable mutations do not play a role in chromosome instability in MM.Citation53 In contrast, genes regulating centrosome assembly, a microtubule-organizing structure that is essential for generating dual spindle poles in mitosis, have been implicated in MM-associated chromosome instability. By establishing a gene expression-based index (centrosome index) that correlated with centrosome amplification, it has been shown that a high centrosome index is associated with a markedly poor prognosis in MM.Citation54 Of note, genes regulating centrosome duplication and function and relevant to centrosome index include genes in DNA repair, such as ATM, ATR, RAD51, XRCC2, BRCA2, and others.Citation54
A further backdrop to the genomic landscape in MM is the essentially perpetual cascade of “intrinsic” molecular lesions that target DNA as part of normal physiological processes. About 105 DNA hits can occur on a daily basis in a cell, caused by spontaneous decay (depurination, deamination, and methylation reactions), replication errors, cellular metabolism, reactive oxygen species, and by environmental DNA damaging agents such as UV light, ionizing radiation, or chemicals.Citation55,Citation56 It is not clear whether the rate of these intrinsically generated defects are increased or decreased in MMCs. However, if unrepaired, these chemical modifications in the DNA may ultimately undermine genetic integrity and deregulate cell death inducing pathways, and may be selectively resolved for survival by cancer cells, for which they may be particularly well adapted.
Gene deregulation in MMCs may also due to epigenetic controls. Aberrant DNA methylation is observed in MMCs from patients and can affect tumor suppressor genes.Citation57,Citation58 In agreement, DNA methyltransferase (DNMT)3A and DNMT3B genes are overexpressed in MMCs compared with normal plasma cells.Citation59
To understand these genomic issues, we review the links between DNA repair pathways and genetic abnormalities in MM, which are likely to have a significant impact on disease occurrence, drug resistance, and/or relapse.
Normal DNA Repair Pathways
As described above, each cell must resolve up to 105 DNA lesions each day arising by spontaneous decays, replication errors, cellular metabolism, and exposure to irradiation or chemicals. To ensure cell survival and functionality, these lesions have to be repaired by specific pathways, which involve sensor proteins that nucleate the assembly of proteins and trigger a cascade of posttranslational modifications to initiate DNA repair.Citation60,Citation61 We begin with a brief overview of the essential features of the major DNA repair pathways that operate in normal cells.
Excision repair
Base excision repair (BER)
BER eliminates the 104 base damages that occur spontaneously and daily in each healthy cell due to hydrolysis, oxidation, or methylation reactions ().Citation55 Conventional BER is initiated by specific mono- or bi-functional DNA glycosylases, which recognize and hydrolyze the N-glycosylic bond between the damaged base and the sugar phosphate backbone, creating an apurinic/apyrimidinic (AP) intermediate site. The monofunctional glycosylases create a hydrolytic AP site, which is cleaved 5′ to the AP site by AP endonuclease 1 (APE1), and 3′ to the AP site by an AP lyase, to yield a single nucleotide gap that can be filled in by polymerase β and ligated by XRCC1/ligase 3
Figure 1. DNA repair pathways of single strand DNA damages. Every day, cells are faced with thousand of damages involving base damages or bulky adducts, which have to be repaired to preserve cell function. Base damages induced by depurination or deamination are repaired by base excision repair, bulky adducts (in particular pyrimidine dimers induced by UV radiation) by nucleotide excision repair and base mismatch occurring during DNA replication by mismatch repair. Repair pathways involve first sensing of damage, followed by cleavage of damaged DNA, and synthesis of the correct DNA sequence using complementary strand as template.
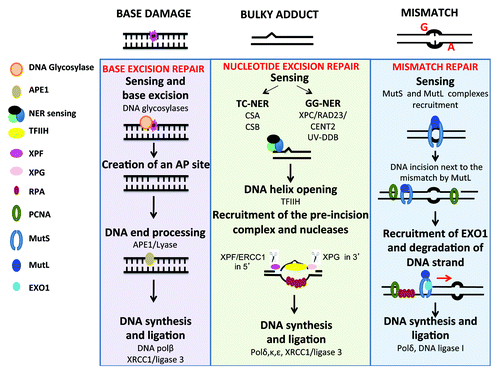
().Citation62 Following base removal by a bifunctional DNA glycosylase, the protein can incise the DNA backbone immediately 3′ to the AP site product via a β- or β,δ-elimination reaction, producing a single-strand break (SSB) with a 3′-phospho- α, β-unsaturated aldehyde (3′-PUA) or 3′-phosphate (3′-P) group. APE1 removes the 3′-PUA residue and polynucleotide kinase 3′-phosphatase (PNKP) excises the 3′-P moiety.
Nucleotide excision repair (NER)
NER corrects a wide spectrum of bulky helix-disturbing DNA lesions ().Citation63 Examples of lesions are cyclobutane pyrimidine dimers and pyrimidine-(6,4)-pyrimidone photoproducts generated by UV radiation, base adducts created by exogenous chemical agents such as melphalan, cisplatin, or benzopyrene, base lesions produced by reactive oxygen species (ROS) or endogenous lipid peroxidation products. NER can be coupled to transcription, a process known as transcription-coupled nucleotide excision repair (TC-NER), as opposed to global genome nucleotide excision repair (GG-NER), whereby the transcribed strand of active genes is repaired faster than in the genome in general and is triggered when the RNA polymerase is blocked at the lesion. In GG-NER, DNA lesions are sensed by XPC/RAD23/CENT2 and UV-DDB complexes, which recognize distortions of the DNA double helix. In TC-NER, the nucleotide excision repair process is coupled to transcription by CSA and CSB, 2 proteins implicated in Cockayne syndrome.Citation64 In both GG-NER and TC-NER, the damaged duplex DNA is opened around the lesion by TFIIH, a multi-subunit helicase complex that includes XPB, p62, p52, p44, p34, p8, XPD, and the CDK-activating kinase (CAK). Next, the structure-specific endonucleases XPG and XPF/ERCC1 excise DNA 3′ and 5′ to the lesion, respectively, to eliminate a 20–30 nt-long oligonucleotide that comprises the DNA lesion. Finally, new DNA is synthetized by polymerases δ, κ, and ε, using the intact strand as a template, and terminated by XRCC1/ligase3.
Mismatch repair (MMR)
MMR removes nucleotides that have been misincorporated during DNA replication and prevents promiscuous recombination between divergent sequences ().Citation65 Mispaired bases are sensed by the MutSα heterodimer (MSH2-MSH6), whereas MutSβ (MSH2-MSH3) recognizes insertions/deletions of more than 2 extra bases. Lesion recognition is followed by the recruitment of MutLα (MLH1/PMS2) or MutLβ (MLH1/MLH3), which have endonuclease activity that can incise DNA near the mismatch. The nick is used by the 5′ exonuclease Exo1 as an entry point to degrade DNA past the mismatch, and the resulting single-stranded DNA gap is filled in by polymerase δ and sealed with DNA ligase I.Citation65,Citation66
Double-strand break (DSB) repair
DSBs can be generated by DNA replication when cells enter S phase before completing SSB repair. They can also be generated by reactive oxygen species, ionizing radiation, or chemicals such as inhibitor of topoisomerase I or during the repair of interstrand DNA crosslinks (ICLs).Citation60,Citation67 The choice between NHEJ and homologous recombination (HR) repair pathways is regulated by complex regulatory mechanisms and involves competition between 53BP1 and BRCA1. 53BP1 favors NHEJ and is antagonized by the product of the breast cancer susceptibility gene BRCA1, which promotes HRCitation68,Citation69 (Figs. Two and 3). Methylation of histone H4 by MMSET leads to recruitment of 53BP1,Citation70 which blocks DNA end resection by the Mre11/Rad50/Nbs1 (MRN) complex, CtIP, and BRCA1.Citation69 In contrast, histone H4 acetylation by Tip60 blocks 53BP1 recruitment and promotes BRCA1 occupancy and, subsequently, HR.Citation71 HR is also promoted by PARP1 and by proteins implicated in the inherited disorder, Fanconi anemia, from the toxic interference of NHEJ proteins.Citation72-Citation74 Cell cycle-regulated proteins such as cyclin-dependent kinases also play a key role in choice of pathway to resolve DSBs.Citation60
Non-homologous end joining (NHEJ)
NHEJ is a process that is active in all phases of the cell cycle, whereby broken ends are simply spliced back together (). As NHEJ functions independently of a DNA template for repair, it is intrinsically prone to altering the genetic information of the repaired DNA molecules. NHEJ starts with the recognition of DNA ends by Ku70/80 and is followed by the recruitment and activation of the DNA-dependent protein kinase DNA-PKcs, and of DNA end-processing enzymes such as ARTEMIS and template-independent polymerases (polymerases λ and μ) that might be necessary for end ligation by the XLF-XRCC4-DNA ligase 4 complex.Citation18,Citation60 DSBs are also generated during normal SHM and CSR modifications of B-lymphocyte Ig genes and repaired mainly by NHEJ.Citation75
Figure 2. DNA repair pathways of double-strand DNA breaks. DNA double-strand breaks (DSB) occur in transcriptionally active or replicating cells in the presence of reactive oxygen species, ionizing radiation, or when the replication machinery encounters unrepaired single-strand damage. DSBs also enable DNA rearrangements in order to select for antigen-specific T or B lymphocytes. In addition, the main drugs used in cancer target cell replication machinery resulting in DSB and cell death. Two major pathways compete to repair DSBs. The accurate homologous recombination (HR) requires a duplicated sister chromatid and is active only at the end of S phase or in the G2 phase. Non-homologous end joining (NHEJ) is active in all phases of cell cycle and involved partial resection and ligation of DNA ends. The choice between HR and NHEJ in replicating cells is dictated by 53BP1 and BRCA1, which compete to bind DNA ends and block mutually. alt-NHEJ, alternative less accurate pathways, and SSA, single-strand annealing, can repair DSBs and may yield to DNA deletion, amplification, or translocation.
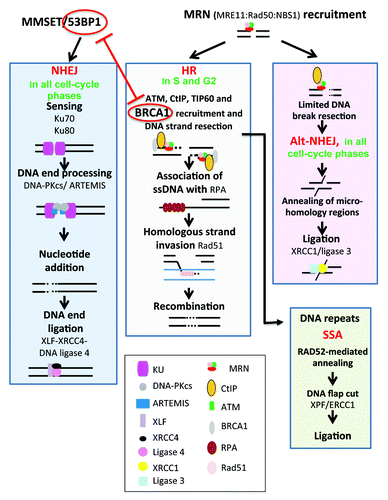
Homologous recombination (HR)
HR is active in the S or G2 phases of the cell cycle, when sister chromatids templates are available for repair (). HR begins with nucleolytic resection of DNA ends, mediated by the MRN complex, CtIP, and BRCA1, which yields 3′ single-stranded DNA tails that are stabilized by replication protein A (RPA). Next, the breast cancer susceptibility gene product BRCA2 catalyzes the displacement of RPA and the formation of a RAD51 nucleoprotein filament. The RAD51 filament promotes homology search and catalyzes an exchange strand between the broken duplex and the intact sister chromatid. The 3′ end of the invading strand is extended by DNA synthesis using the sister chromatid as a template, and intact duplexes are eventually restored using one of multiple resolution mechanisms.Citation76
Single strand annealing (SSA)
Extensive DNA resection within repetitive DNA sequences can result in RAD52-mediated annealing of resected ends, followed by removal of DNA flaps by XPF-ERCC1 nucleases (). SSA is inaccurate because it results in the deletion of one of the repeats and the intervening sequence. SSA may also lead to translocations when 2 DSBs occur within or near repeats located on different chromosomes.Citation60,Citation77
Alternative NHEJ (alt NHEJ)
Defects in NHEJ promote DSB repair by a robust but less accurate alternative NHEJ pathway, whereby DNA ends are resected up to regions of microhomology, annealed, and ligated by XRCC1/ligase 3 ().Citation60,Citation66 The 53BP1 protein protects DNA ends from nucleolytic degradation and thereby prevents microhomology mediated repair.Citation78
Interstrand-crosslink (ICL) repair
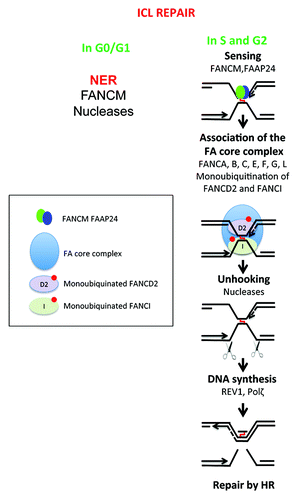
ICLs are covalent bounds between the 2 antiparallel strands in duplex DNA. These lesions are induced by treatment with bifunctional alkylating agents, such as melphalan, which is frequently used to treat patients with MM.Citation67 In non-replicating cells, the repair of mono- and di-adducts is mediated by the nucleotide excision repair machineryCitation79 and by the DNA translocase FANCM, which facilitates the access of nucleases to the lesion ().Citation80 In S-phase cells, crosslink repair is coupled to DNA replication, features DSBs as repair intermediates, and depends on the homologous recombination machinery ).Citation67 The products of genes that are defective in the chromosomal instability disorder Fanconi anemia (FA) orchestrate the coordinated action of the multiple enzymatic activities that are necessary to couple the recombination-dependent ICL repair with DNA replication.Citation81 In response to genotoxic insults, a complex of 8 FA proteins in the nucleus (FANCA, -B, -C, -E, -F, -G, -L, and -M) catalyzes the conjugation of an ubiquitin moiety to FANCD2 and FANCI protein to activate their DNA repair functions. Ubiquitinated FANCD2 and FANCI are necessary for nucleolytic incisions to unhook the ICLs and for the insertion of nucleotides opposite the unhooked crosslinks.Citation82,Citation83 However, it is not yet entirely clear which of the structure-specific endonuclease FAN1, MUS81/EME1, SLX1-SLX4 (FANCP), and ERCC1/XPF are responsible for ICL unhooking; translesion DNA synthesis across the unhooked ICL depends on REV1 and polymerases ζ.Citation67
Figure 3. DNA repair of interstrand crosslinks (ICLs). Toxic compounds can create DNA Interstrand Crosslinks (ICLs), which impair transcription and replication machinery, and yield to cell death if unrepaired. ICL formation is the active principle of many cancer drugs, in particular alkylating agents. The removal of ICL involves the Fanconi anemia pathway, with sensing of ICL by FANCM, and then recruitment of protein complex resulting in ICL removal, creation of DSB, which is repaired by homologous recombination. In non-cycling cells, the initial sensing by FANCM is followed by ICL removal and DSB repair by poorly known mechanisms.
Deregulation of DNA Repair in Mul tiple Myeloma Cells
DNA repair pathways are critical to repair both the developmental impact of DSBs generated by SHM and CSR in Ig genes in MM origins and resolve the plethora of DNA lesions that occur each day in cells due to endogenous and exogenous stress. We review here the current knowledge on changes in DNA repair pathways in MM that could trigger disease development, perpetuate genomic instability, and affect response to diverse treatment regimen.
Unresolved data on NHEJ deregulation in multiple myeloma cells
Although several reports have documented polymorphisms or deregulation in genes/proteins involved in NHEJ pathway in MMCs, no definitive conclusion can be drawn as to whether the NHEJ pathway is deregulated in MMCs as compared with MGUS or healthy plasma cells (). Association between polymorphisms or deregulated expression of the XRCC5 (encoding Ku80), XRCC6 (encoding Ku70), ARTEMIS, LIG4 (encoding DNA ligase 4), or XRCC4 genes and the potential risk of developing MM has been described (). The polymorphic XRCC5 (Ku80) sequence is located within an exon splicing enhancer sequence and may result in exon skipping, yielding splicing errors and affecting mRNA stability and protein translation.Citation84 The alanine-3-valine or threonine-9-isoleucine DNA ligase 4 allele is associated with a decreased risk of developing MM.Citation85 A 4–5-fold increase in XRCC4 gene expression was found in MMCs compared with other lymphoma cells (mantle cell lymphoma, follicular cell lymphoma, diffuse large B-cells lymphoma, marginal zone lymphomas) or normal B cells from excised benign reactive lymph nodes.Citation86 In that study, XRCC6 (Ku70) gene expression was found to be decreased in MMCs and in other lymphoma cells compared with normal B cells. Finally, a recent study points out an increased expression of XRCC5 and ARTEMIS genes in MMCs cells compared with MGUS plasma cells, in association with shorter patients’ overall survival.Citation87
Figure 4. Double-strand DNA repair pathways in multiple myeloma. Genes coding for homologous recombination or non-homologous end joining pathways, whose promoter methylation (digit 1), polymorphism (digit 2), expression (digit 3), pathway activity (digit 4) are modified in multiple myeloma cells, are indicated. They are highlighted in red if the change is associated with a bad prognosis, in green with a good one, and blue in case of no prognostic value. The upwards or downwards arrows indicate an increased or decreased gene or protein expression or pathway activity. The existence of a truncated variant is indicated by digit 5.
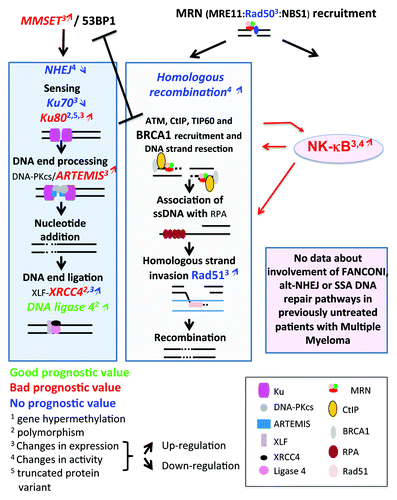
How these polymorphisms or gene expression variations affect NHEJ activity in MMC, however, remains elusive. One study reported the existence of a 69 kDa Ku86 variant (Ku86v) in a majority of patients’ primary MMCs. This short variant of Ku80 has reduced affinity for DNA ends and is unable to associate with and activate DNA-PKcs, which renders cells hypersensitive to chemotherapeutic drugs or to irradiation.Citation88 These data, however, were challenged by Kato et al., who failed to detect the variant Ku86v in primary MMCs or MM cell lines.Citation89 Subsequent studies pointed out that this variant could be generated by proteolytic cleavage when manipulating cells in vitro, both in human lymphocytes or MM cell lines.Citation90-Citation92 Using an in vitro DNA repair assay, NHEJ activity was found to be corrupted in one (RPMI8226) and functional in two (U266, OPM2) MM cell lines.Citation93 A link between NHEJ defect and radiation sensitivity was unclear, since both RPMI8266 and U266 cells were radiation-sensitive, and addition of a DNA-PKcs inhibitor was toxic in untreated cells but remained innocuous to irradiated cells. On the contrary, OPM2 cells were radiation-resistant, and the DNA-PKcs inhibitor abrogated radiation resistance while exhibiting no toxicity to untreated OPM2 cells.Citation93 Presumably, the inactivation of a specific DNA repair pathway may be compensated by the activity of alternative pathways, as discussed above. A recent report shows an increased affinity of Ku80 for DNA ends, and constitutive activation of NHEJ in MM cell lines compared with healthy peripheral blood mononuclear cells.Citation87 Surprisingly, however, MM cell lines showed little or no ability to repair radiation-induced damages, suggesting that constitutive activation of NHEJ pathway in these MM cell lines constitutes a response to endogenous stress.Citation87 As reviewed above (), NHEJ is favored by 53BP1, which blocks DNA end resection.Citation69 Recruitment of 53BP1 to DSBs is dependent on histone H4 methylation by MMSET,Citation70 the product of WHSC1 gene that is translocated and highly expressed in 15% of patients with MM and t(4p16;14q32) translocation in association with poor prognosisCitation15(). Of note, the 5′ region of WHSC1 (MMSET) gene is deleted in 30–40% of these patients with t(4p16;14q32) translocation,Citation25,Citation94 resulting in production of a MMSET protein lacking the serine in position 102. This residue is phosphorylated by ATM, which is a necessary event for the recruitment of MMSET to DSB.Citation70 How WHSC1 (MMSET) translocation and spiked expression impact on HR and NHEJ pathways in MM cells has yet to be investigated. Given the association of t(4;14) with poor outcome in MM, this potential perturbation of DNA repair may prove highly relevant to disease progression.
Increased homologous recombination activity in multiple myeloma cells
HR activity is higher in primary MMCs from patients and MM cell lines compared with healthy plasma cells, as evidenced by higher recombination activity in plasmid assays and increased expression of genes/proteins involved in homologous recombination such as RAD50 and RAD51 ().Citation86,Citation95 Increased HR activity in MM cell lines resulted in resistance to dexamethasone and progressive accumulation of loss of heterozygosity.Citation95 Unlike in healthy plasma cells, nuclear foci identified by labeling of phosphorylated histone H2AX (γH2AX), which are indicators of DSBs,Citation96 are detected in primary MMCs of untreated patients or in MM cell lines, and ATR is constitutively phosphorylated in MM cell lines.Citation97 Since HR is active in the S and G2 phases of the cell cycle, HR-mediated repair in MM cell lines or primary MMCs may occur in only a minor fraction of cycling MMCs. In the vast majority of patients, only a small percentage of myeloma cells can be found in S phase, so other DNA repair pathways may predominate and influence MMC development, and survival after drug treatment. However, as discussed above, even if just a small fraction of MMCs are engaged in the cell cycle, their abundance is highly correlated with adverse prognosis. Therefore, DNA repair pathways involved in this minority of “feeder” cells in S and G2 might be of particular importance for MM evolution, prognosis, and response to treatment.
Crosstalk between NFκB pathway and DNA repair pathway
The NFκB pathway is frequently deregulated in MM and is selectively targeted for somatic mutations in MM as defined by NGS.Citation39 This may affect the interplay between NFκB signals and activation of DNA repair pathways. Conversely, it may be feasible for DNA repair mechanisms to promote NFκB activation and MMC survival. These significant possibilities arise, as NFκB activation has been shown to stimulate HR, increase ATM and BRCA2 expression, and directly associate with and stimulate CtIP and BRCA1, thus enhancing DSB end resection and RPA coating, which are mandatory steps for BRCA2-mediated RPA replacement by RAD51 and homologous strand invasionCitation98 (). Consequently, it is conceptually feasible that DNA damage and especially DSBs may induce NFκB activation in MMCs to promote tumor survival. Additional mechanisms may facilitate this cross-talk in MMCs. Irradiation or topoisomerase inhibitors induce NFκB through ATM recruitment and activation at DSBs. ATM binds and phosphorylates NEMO, inducing its ubiquitination and further translocation to the cytoplasm, where NEMO binds to and activates IKKα and IKKβ kinases, which, in turn, activates the NFκB pathway.Citation99,Citation100 Another mechanism involves association of PIDD (p53-induced protein with a death domain) with RIP1 (receptor-interacting protein 1) and NEMO, which leads to NEMO sumoylation. NEMO then associates with ATM resulting in NFκB activation.Citation101 Finally, PARP1 may induce association between ATM and the SUMO-1 ligase PIASy, which, in turn, sumoylates and activates IKKγ.Citation98,Citation102 These data are illuminating. They reveal cross-talk that links central conduits for signal transduction to mechanisms that regulate genomic structural integrity, suggesting that somatic mutations in key genes that transduce external stimuli (e.g., RAS) may now need to be considered for their impact on genomic instability in MM.
Base excision repair
Very little data exists to implicate BER in MM. A polymorphism in OGG1 gene, linked to low BER activity, is associated with an increased risk for MM.Citation103 Polymorphisms in APE1 or MUTYH () genes are associated with shorter survival of patients with MM.Citation104 In addition, a higher expression of APE1, using immunofluorescence staining, was detected in MMCs as compared with controls.Citation105
Figure 5. Single-strand DNA repair pathways in multiple myeloma. The changes in genes implicated in single-strand damage repair (BER, NER, or MMR) and modified in MM cells compare with healthy cells are highlighted. As indicated in , digit 1 indicates changes in promoter methylation, digit 2 the existence of polymorphisms, digit 3 changes in protein expression and digit 4 changes in activity. The upwards or downwards arrows indicate an increased or decreased gene expression or activity.
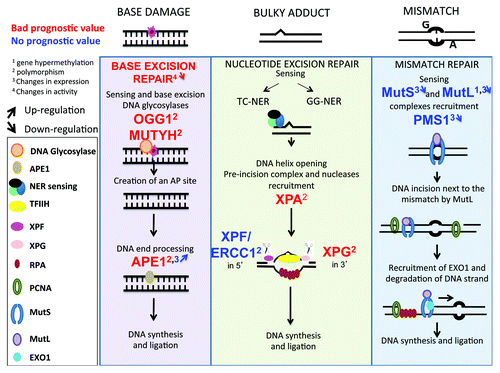
Nucleotide excision repair
Polymorphisms within 3 genes encoding the NER proteins XPA, XPG (ERCC5 gene), and ERCC1 were recently reported in MMCs in a large cohort of patients, with a shorter survival associating with ERCC5 rs1047768 and XPA rs1800975Citation106 (), and emphasizing the importance of NER to remove ICLs that are likely to be induced by alkylating drugs, in particular melphalan, in non-replicating but transcriptionally active cells.Citation67,Citation107 This is of importance for MMCs, which are malignant plasma cells with a constitutively high transcriptional activity to produce Ig.
Mismatch repair
Relatively few observations on the mismatch repair pathway in MM have been published, but are discordant. In this pathway, the DNA lesion is sensed and recognized by the MutSα heterodimer (MSH2-MSH6) or by the MutSβ (MSH2-MSH3) complex, followed by the recruitment of MutLα (MLH1/PMS2) or MutLβ (MLH1/MLH3) to the repair foci. In a study of 30 patients with MM and 13 with MGUS, MLH1 promoter was found methylated in plasma cells of 10% of patients with MM but not from MGUS patients.Citation108 MLH1 promoter methylation was also associated with a decrease or loss of MLH1 gene expression in MMCs. These data suggest that a decrease in MLH1 expression follows promoter hypermethylation, and may play a role in the transition of MGUS to MM. MLH1 promoter hypermethylation, however, was not confirmed in another study of 53 MM patients. Decreased expression of MSH2, MLH1, PMS1 genes, coding for proteins involved in MMR, was reported in another study on MMCs from a small cohort of patients and requires further confirmatory studies.Citation109
Somatic mutations in DNA repair genes
Interestingly, acquired somatic mutations in MMCs, excluding p53, appear to be markedly limited in the spectrum of genes that mediate DNA repair, as exemplified in the genome NGS data of a large cohort of 38 MM cases.Citation39,Citation110
DNA Repair Pathways: Targets For Drugs Used to Treat MM and Role in Resistance
Melphalan and DNA repair pathways
Melphalan is used in patients receiving high-dose therapy (i.e., high dose melphalan 2 × 100 mg/m2) and autologous stem cell transplantation, as well as in non-transplantable patients in combination with steroids and either a proteasome inhibitor Bortezomib (VMP-regimen) or an immunomodulatory drug Thalidomide (MPT-regimen).Citation4 Melphalan is a nitrogen mustard (4-[bis{2-chloroethyl}amino]-1-phenylalanine), which induces alkylation of the guanine in N-7 and adenine in N-3 and interstrand DNA crosslinks between guanine and guanine or guanine and adenine.Citation111 Ninety-five percent of the lesions are monoadducts and 5% interstrand DNA crosslinks (ICLs),Citation112 and these lesions impair DNA transcription and replication, yielding cell death if unrepaired. ICL number in MMCs increases with the melphalan concentration used to treat MMCs in vitro,Citation113 and this is likely to also occur in vivo (). Although ICLs are infrequent, they are highly toxic for cells. The mechanisms of resistance to melphalan therapy have been extensively examined in MMC. One mechanism of resistance to alkylating drugs is the ability of MMCs to export the drug through upregulation of membrane efflux proteins. This has been well documented for many cancersCitation114 and in particular for MM.Citation115 A second resistance mechanism is the ability of MMCs to excise melphalan monoadducts and ICLs through DNA repair pathways, supported by 3 lines of evidence. (1) MMCs from patients relapsing from high-dose or continuous melphalan therapy are able to efficiently repair ICLs in vitro, whereas MMCs from untreated patients are not.Citation113 (2) Upon in vivo melphalan treatment, the efficiency of ICL repair within the TP53 gene in patients’ non-tumor leukocytes can efficiently predict for patient sensitivity to melphalan.Citation116 (3) Associations between melphalan resistance and polymorphisms in genes encoding DNA repair proteins have been documented: in a study of 169 patients treated with vincristine-adriamycine-dexamethasone then high-dose melphalan and stem cell transplantation, patients’ outcome was significantly associated with SNPs in OGG1, PARP, PCNA, RAD51, and XPC genes and patients’ survival with BARD1, BRCA1, ERCC1, PCNA, and TP53BP1 genes (). Moreover SNPs in BRCA1, CDKN1A, and XRCC1 genes were associated with therapy-associated mucositis.Citation103 Many of these genes encode for proteins implicated in the HR or NER pathways that are necessary for melphalan-induced ICL repair. In another study, polymorphisms in the ERCC2 and XRCC3 genes were implicated in the repair of bulky adducts and DSBs and associated with treatment failure.Citation117
Figure 6. DNA repair pathways and drugs used to treat patients with multiple myeloma. (A) Velcade and Melphalan, 2 of the main drugs used to treat MM patients, modify DNA repair pathways in 2 opposite ways. Melphalan induces adducts and ICLs that need to be repaired mainly by the Fanconi anemia pathway and homologous recombination. Base excision repair (BER), non-homologous end joining and nucleotide excision repair (NER) pathways are also involved in ICLs repair. Changes in activity or expression of genes involved in these pathways or the existence of polymorphisms may activate and thus ease the repair of the lesions leading to cell survival and resistance to Melphalan. Some polymorphisms are associated with a better response after treatment. Changes in BER pathway can affect ICLs repair: an increased activity could help to repair the lesions but a decreased activity may also facilitate ICLs repair by the Fanconi anemia pathway by improving DNA accessibility. Velcade inhibits Fanconi anemia pathway, homologous recombination and NER. In red are summarized the features and deregulations associated with disease progression and resistance to melphalan, in green the features and deregulations associated with response to treatment and a better outcome after treatment with melphalan. (B) Very few data show a possible association between treatment with IMiDs and dexamethasone and DNA repair pathways. In green are summarized polymorphisms and changes in protein expression associated with a better outcome after treatment with dexamethasone or IMiDs (lenalidomide, thalidomide, and pomalidomide).
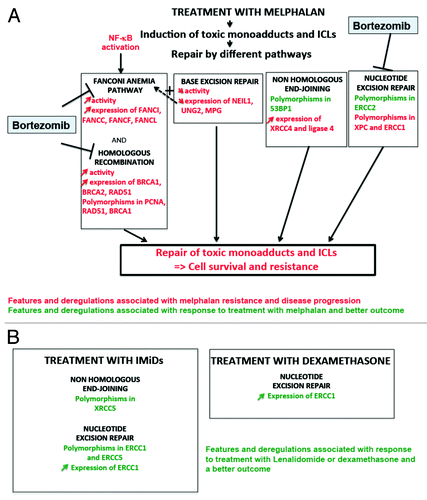
More detailed studies of mechanisms of resistance to melphalan have utilized MM cell lines, which proliferate highly unlike primary MMCs in the majority of patients. Increased expressions of genes coding for Fanconi anemia (FA) or HR pathways such as BRCA1, BRCA2, FANCA, FANCC, FANCF, FANCL, and RAD51C and upregulation of FA and HR pathways were found in melphalan-resistant myeloma cell lines.Citation118 FANCF is a key player in melphalan resistance, since knockdown of FANCF can reverse resistance and overexpression of FANCF reduces melphalan sensitivityCitation118 (). Constitutive NFκB activation is in part responsible for transcriptional activation of genes encoding FA proteins and for melphalan resistance (further highlighting cross-talk with DNA repair).Citation119 Thus, the FA pathway confers resistance of MMCs to melphalan and presents a potential therapeutic target. Curcumin, for instance, inhibits both the NFκB and the FA pathwaysCitation120,Citation121 and increases drug concentration inside MMCs.Citation121 Hence, combinatorial therapies using curcumin may overcome resistance of MM cell lines to melphalan. Blocking autophagy may also improve the efficiency of melphalan in MM cell lines and patients’ primary MMCs.Citation122 Autophagy is activated after DNA damage by melphalan, and several studies show that autophagy could have some protective effect in part by increasing ATP production.Citation123
As BER is involved in the repair of psoralen-induced ICLs,Citation124 it may contribute to melphalan resistance (Figs. One and 6A). A siRNA to APE1 sensitized MMCs to melphalan.Citation105 Overexpression of a dominant-negative form of APE1, however, impaired the ability of CHO cells to repair DNA lesions induced by alkylating agents such as decarbazine, thiotepa, busulfan carmustine, but, surprisingly, not by melphalan.Citation125 On the contrary, a decrease in the expression of 3 DNA glycosylases (NEIL1, UNG2, MPG) associated with a decrease repair of 8-oxodG and acquisition of melphalan resistance in a cell line (). The authors hypothesized that decreased activity of these glycosylases in resistant cells induced less AP sites caused by endogenous reactive oxygen species. Therefore, fewer proteins involved in AP site repair were mobilized closed to the sites of ICLs, facilitating the accessibility of ICL repair proteins to ICLs.Citation126 Finally, an increase expression of 2 proteins involved in NHEJ (XRCC4 and ligase 4) was also observed in a melphalan-resistant cell lineCitation126 (Figs. Two and 6A). Defining the precise array of DNA repair proteins with a role in melphalan resistance may allow additional targeted drugs to be developed that can block culprit proteins.
Proteasome inhibitor (PI) and DNA repair pathways
Bortezomib is a proteasome inhibitor (PI) that reversibly targets the catalytic β5 and β1 proteasome subunits and inhibits chymotrypsin-like and peptidylglutamyl peptide hydrolyzing activities.Citation127 Bortezomib can thus induce proteotoxic stress by preventing misfolded protein degradation, and its efficiency can be improved by co-treatment with inducers of misfolded protein such as puromycine.Citation128 Bortezomib preferentially targets proliferating cells but is also active in quiescent cells in the G0 phase of the cell cycle.Citation129 As protein ubiquitination is critical for building DNA repair foci through protein recruitment and degradation, it is not altogether surprising that PI affects DNA repair, as has been shown in the main for FA and HR pathways. PIs block NFκB mediated activation of FA pathway,Citation119 decrease expression of FANC proteins, and inhibit FA pathway by preventing FANCD2 monoubiquitination and FANCD2 focus formation, and also inhibit HR by blocking recruitment of NBS1, phospho-ATM, BRCA1, and Rad51 ().Citation130,Citation131 The decrease in FANCD2 and H2Ax ubiquination by PI is explained by the accumulation of poly-ubiquitinated proteins and, consequently, the decrease of the pool of available nuclear ubiquitin.Citation132 Bortezomib treatment may prevent DNA resection through inhibiting proteasomal degradation of proteins involved in chromatin relaxation, thus preventing recruitment of RPA onto ssDNA.Citation130,Citation131 This effect of Bortezomib on blocking DNA repair could explain why a phase II study has shown that the combination of Bortezomib together with high-dose melphalan before autologous stem cell transplantation could increase by 3-fold the complete remission rate (35% vs. 11%).Citation133 The underlying mechanisms inferred here from improved remission rates may include a reduced potential of DNA repair pathways to clear lesions, thereby increasing susceptibility to apoptotic clearance and/or reducing the efficacy of DNA repair in genomic adaptations to resist therapy. The clinical benefit of combination of Bortezomib and high-dose melphalan is currently being tested in large clinical trials according to clinicaltrial.gov. New proteasome inhibitors entering clinical development are Carfilzomib or MLN2238. Carfilzomib bind irreversibly to proteasome subunits and can overcome resistance to Bortezomib.Citation134 These new PIs targeting the proteasome are expected to also target DNA repair pathways. The effect of PIs on other DNA repair pathways is less studied. It is noteworthy that in human fibroblasts, PIs block the repair of UV-induced lesions by NER.Citation135
Immunodulatory drugs, dexamethasone, and DNA repair pathways
IMiDs (Thalidomide, Lenalidomide, or Pomalidomide) are immunomodulatory drugs that bind to an F-box protein Cereblon, a member of an ubiquitin ligase complex.Citation136,Citation137 Dexamethasone, a major drug in MM, is a corticosteroid that binds to intracellular glucocorticoid receptors, which migrate to the nucleus and act as a transcription factor. Resistance to lMiDs involves mostly mutations or downregulation of cereblon expression.Citation47,Citation137 Various mechanisms have been described for dexamethasone resistance: expression of a truncated glucocorticoid receptor,Citation138 mutations in receptor gene,Citation47 epigenetic silencing of RASD1, cell growth inhibitor induced by dexamethasone treatment,Citation139 overexpression of Hsp27,Citation140 or change in the secretion of cytokines and chemokines in the microenvironment.Citation141 Overexpression of Bruton tyrosine kinase could also play a role in resistance according to a recent study.Citation142 But very few data report a possible association between DNA repair pathway features or deregulations and sensitivity or resistance to dexamethasone and IMiDs. Polymorphisms in ERCC1, ERCC5, or XRCC5 (Ku70) genes (coding for NER or NHEJ proteins) (, , and ) are associated with a better response to Thalidomide as a single agent in MM patients, and polymorphisms in ERCC1 and XRCC5 with a longer overall survival.Citation143 Gene expression profiling of MMCs from patients harvested before and immediately (48 h) after treatment with dexamethasone (40 mg/day) or with thalidomide (400 mg/day) has revealed an overall increase in ERCC1 expression (NER pathway) after dexamethasone treatment and a downregulation by thalidomide. An increased ERCC1 expression after treatment with dexamethasone or Thalidomide was associated with better patients’ outcome.Citation144 Of note, IMIDs can increase ROS production, DNA oxidation that will require DNA repair. In MMC, polymorphic variants of NER and NHEJ proteins, and modulation of their levels of expression by specific drugs will affect functional activities that appear to associate with antitumor effects: again, the precise mechanisms remain to be elucidated. The expression of heme oxydase 1 HO-1, an enzyme implicated in cellular response to oxidative stress, was enhanced after 3 mo of treatment with Lenalidomide and Bortezomib,Citation145 and treatment with Thalidomide or Lenalidomide induces upregulation of the oxidative-stress responsive 1 (OXSR1) gene. This upregulation is associated with a poor outcome and could therefore be a mechanism of resistance after prolonged exposure to drugs.Citation144
New therapeutic strategies: Synthetic lethality
As reviewed above, DNA repair pathways can be deregulated in MMCs and modulate the activity of the major drugs currently used in disease. Thus, one can speculate that targeting DNA repair pathways may potentiate the efficacy of current drugs and reverse drug resistance. This strategy, called “synthetic lethality” is currently being developed in breast cancers with mutated BRCA1 or BRCA2 genes. In these cancer cells, DSBs repair by HR fails due to a lack of functional BRCA1 or BRCA2 proteins. Treatment of these cancer cells with PARP1 inhibitors such as Olaparib inhibits SSB repair, yielding DSBs, which cannot be repaired by a defective HR and are consequently highly toxic for cancer cells.Citation66,Citation146 Such a strategy has been investigated for MMCs, in which HR was inhibited by Bortezomib treatment, as co-treatment of MMCs with Bortezomib and PARP1 inhibitor resulted in DSB accumulation and MMC killing.Citation132 Importantly, many inhibitors targeting DNA repair pathways (PARP1, DNA-PK, ATM, ATR, MGMT, APE) or cell cycle checkpoints (CHK1, CHK2) have now been developedCitation66,Citation146-Citation148 and could be useful to induce the death of MMCs in combination with DNA damage inducing drugs to develop therapeutic synthetic lethality.Citation148-Citation150 These inhibitors could be of particular interest for improving response and survival of patients treated with high-dose melphalan and autologous stem cell transplantation. In these patients, only few MMCs (5.5 MMCs/μL after 7 d) may survive in an almost empty bone marrow 9 d after melphalan treatment.Citation151 These few resistant MMCs likely harbor melphalan monoadducts or crosslinks, which have to be repaired to get MMC survival and tumor regrowth. Targeting DNA repair pathways with specific drugs could help killing MMCs, preventing or delaying relapse.
Concluding Remarks
This review maps the complexity of molecular lesions that impact on MMCs during disease pathogenesis, progression, and response to therapy, where genomic instability is met by an array of DNA repair pathways. A large body of evidence suggests that deregulation of these DNA repair pathways in malignant plasma cells mediates onset of disease and survival. The precise nature of dysregulated DNA repair and how it improves MMC survival or is co-opted during antitumor therapy is as yet underexplored and requires and warrants extensive study. Existing data already support the development of new targeted therapies, such as synthetic lethality. If fully understood, DNA repair dysregulations could be exploited to selectively kill MMCs by targeting the remaining repair pathways that are critical for their survival.
| Abbreviations: | ||
| 3′-P | = | 3′-phosphate |
| 3′-PUA | = | 3′-phospho-α,β-unsaturated aldéhyde |
| alt NHEJ | = | alternative non-homologous end joining |
| AP | = | apurinic/apyrimidinic |
| APE1 | = | AP endonuclease 1 |
| BER | = | base excision repair |
| CAK | = | CDK-activating kinase |
| CSR | = | class switch recombination |
| CT | = | chromosomal translocation |
| FA | = | Fanconi anemia |
| GG-NER | = | global genome nucleotide excision repair |
| HR | = | homologous recombination |
| ICL | = | interstrand DNA crosslink |
| Ig | = | immunoglobulin |
| IGH | = | immunoglobulin heavy chain |
| MGUS | = | monoclonal gammopathy of undetermined significance |
| MM | = | multiple myeloma |
| MMCs | = | multiple myeloma cells |
| MMR | = | mismatch repair |
| MRN | = | Mre11, Rad50, Nbs1 |
| NER | = | nucleotide excision repair |
| NGS | = | next generation sequencing |
| NHEJ | = | non-homologous end joining |
| PI | = | proteasome inhibitor |
| PIDD | = | p53-induced protein with a death domain |
| PNKP | = | polynucleotide kinase 3′-phosphate |
| RIP1 | = | receptor-interacting protein 1 |
| ROS | = | reactive oxygen species |
| RPA | = | replication protein A |
| SHM | = | somatic hypermutation |
| SSA | = | single strand annealing |
| SSB | = | single-strand break |
| TC-NER | = | transcription-coupled nucleotide excision repair |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by grants from the European community (FP7-OVERMYR project), the INCA (Institut National du Cancer) institute (2012–109/087437, Paris, France) and from the Association pour la Recherche sur le Cancer (ARC) (SL220110603450, Paris France). CG is supported by a French Labex EpiGenMed fellowship.
Related Research Data
References
- Siegel R, Naishadham D, Jemal A. Cancer statistics, 2012. CA Cancer J Clin 2012; 62:10 - 29; http://dx.doi.org/10.3322/caac.20138; PMID: 22237781
- Kyle RA, Therneau TM, Rajkumar SV, Offord JR, Larson DR, Plevak MF, Melton LJ 3rd. A long-term study of prognosis in monoclonal gammopathy of undetermined significance. N Engl J Med 2002; 346:564 - 9; http://dx.doi.org/10.1056/NEJMoa01133202; PMID: 11856795
- Landgren O, Kyle RA, Pfeiffer RM, Katzmann JA, Caporaso NE, Hayes RB, Dispenzieri A, Kumar S, Clark RJ, Baris D, et al. Monoclonal gammopathy of undetermined significance (MGUS) consistently precedes multiple myeloma: a prospective study. Blood 2009; 113:5412 - 7; http://dx.doi.org/10.1182/blood-2008-12-194241; PMID: 19179464
- Rajkumar SV. Treatment of multiple myeloma. Nat Rev Clin Oncol 2011; 8:479 - 91; http://dx.doi.org/10.1038/nrclinonc.2011.63; PMID: 21522124
- Weiss BM, Abadie J, Verma P, Howard RS, Kuehl WM. A monoclonal gammopathy precedes multiple myeloma in most patients. Blood 2009; 113:5418 - 22; http://dx.doi.org/10.1182/blood-2008-12-195008; PMID: 19234139
- Rajkumar SV, Gupta V, Fonseca R, Dispenzieri A, Gonsalves WI, Larson D, Ketterling RP, Lust JA, Kyle RA, Kumar SK. Impact of primary molecular cytogenetic abnormalities and risk of progression in smoldering multiple myeloma. Leukemia 2013; 27:1738 - 44; http://dx.doi.org/10.1038/leu.2013.86; PMID: 23515097
- Kyle RA, Therneau TM, Rajkumar SV, Larson DR, Plevak MF, Offord JR, Dispenzieri A, Katzmann JA, Melton LJ 3rd. Prevalence of monoclonal gammopathy of undetermined significance. N Engl J Med 2006; 354:1362 - 9; http://dx.doi.org/10.1056/NEJMoa054494; PMID: 16571879
- Zojer N, Ludwig H, Fiegl M, Stevenson FK, Sahota SS. Patterns of somatic mutations in VH genes reveal pathways of clonal transformation from MGUS to multiple myeloma. Blood 2003; 101:4137 - 9; http://dx.doi.org/10.1182/blood-2002-09-2825; PMID: 12531815
- Witzig TE, Timm M, Larson D, Therneau T, Greipp PR. Measurement of apoptosis and proliferation of bone marrow plasma cells in patients with plasma cell proliferative disorders. Br J Haematol 1999; 104:131 - 7; http://dx.doi.org/10.1046/j.1365-2141.1999.01136.x; PMID: 10027725
- Hose D, Rème T, Hielscher T, Moreaux J, Messner T, Seckinger A, Benner A, Shaughnessy JD Jr., Barlogie B, Zhou Y, et al. Proliferation is a central independent prognostic factor and target for personalized and risk-adapted treatment in multiple myeloma. Haematologica 2011; 96:87 - 95; http://dx.doi.org/10.3324/haematol.2010.030296; PMID: 20884712
- Greipp PR, Katzmann JA, O’Fallon WM, Kyle RA. Value of beta 2-microglobulin level and plasma cell labeling indices as prognostic factors in patients with newly diagnosed myeloma. Blood 1988; 72:219 - 23; PMID: 3291982
- Greipp PR, Leong T, Bennett JM, Gaillard JP, Klein B, Stewart JA, Oken MM, Kay NE, Van Ness B, Kyle RA. Plasmablastic morphology--an independent prognostic factor with clinical and laboratory correlates: Eastern Cooperative Oncology Group (ECOG) myeloma trial E9486 report by the ECOG Myeloma Laboratory Group. Blood 1998; 91:2501 - 7; PMID: 9516151
- Paiva B, Vídriales M-B, Montalbán M-Á, Pérez JJ, Gutiérrez NC, Rosiñol L, Martínez-López J, Mateos MV, Cordón L, Oriol A, et al. Multiparameter flow cytometry evaluation of plasma cell DNA content and proliferation in 595 transplant-eligible patients with myeloma included in the Spanish GEM2000 and GEM2005<65y trials. Am J Pathol 2012; 181:1870 - 8; http://dx.doi.org/10.1016/j.ajpath.2012.07.020; PMID: 22974582
- Nussenzweig A, Nussenzweig MC. Origin of chromosomal translocations in lymphoid cancer. Cell 2010; 141:27 - 38; http://dx.doi.org/10.1016/j.cell.2010.03.016; PMID: 20371343
- Fonseca R, Barlogie B, Bataille R, Bastard C, Bergsagel PL, Chesi M, Davies FE, Drach J, Greipp PR, Kirsch IR, et al. Genetics and cytogenetics of multiple myeloma: a workshop report. Cancer Res 2004; 64:1546 - 58; http://dx.doi.org/10.1158/0008-5472.CAN-03-2876; PMID: 14989251
- Kumar S, Fonseca R, Ketterling RP, Dispenzieri A, Lacy MQ, Gertz MA, Hayman SR, Buadi FK, Dingli D, Knudson RA, et al. Trisomies in multiple myeloma: impact on survival in patients with high-risk cytogenetics. Blood 2012; 119:2100 - 5; http://dx.doi.org/10.1182/blood-2011-11-390658; PMID: 22234687
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer 2012; 12:335 - 48; http://dx.doi.org/10.1038/nrc3257; PMID: 22495321
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181 - 211; http://dx.doi.org/10.1146/annurev.biochem.052308.093131; PMID: 20192759
- Wilson PC, de Bouteiller O, Liu YJ, Potter K, Banchereau J, Capra JD, Pascual V. Somatic hypermutation introduces insertions and deletions into immunoglobulin V genes. J Exp Med 1998; 187:59 - 70; http://dx.doi.org/10.1084/jem.187.1.59; PMID: 9419211
- Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest 2012; 122:3456 - 63; http://dx.doi.org/10.1172/JCI61188; PMID: 23023717
- Chng WJ, Glebov O, Bergsagel PL, Kuehl WM. Genetic events in the pathogenesis of multiple myeloma. Best Pract Res Clin Haematol 2007; 20:571 - 96; http://dx.doi.org/10.1016/j.beha.2007.08.004; PMID: 18070707
- Hurt EM, Wiestner A, Rosenwald A, Shaffer AL, Campo E, Grogan T, Bergsagel PL, Kuehl WM, Staudt LM. Overexpression of c-maf is a frequent oncogenic event in multiple myeloma that promotes proliferation and pathological interactions with bone marrow stroma. Cancer Cell 2004; 5:191 - 9; http://dx.doi.org/10.1016/S1535-6108(04)00019-4; PMID: 14998494
- Kyle RA, Rajkumar SV. Monoclonal gammopathy of undetermined significance and smouldering multiple myeloma: emphasis on risk factors for progression. Br J Haematol 2007; 139:730 - 43; http://dx.doi.org/10.1111/j.1365-2141.2007.06873.x; PMID: 18021088
- Bergsagel PL, Kuehl WM, Zhan F, Sawyer J, Barlogie B, Shaughnessy J Jr.. Cyclin D dysregulation: an early and unifying pathogenic event in multiple myeloma. Blood 2005; 106:296 - 303; http://dx.doi.org/10.1182/blood-2005-01-0034; PMID: 15755896
- Chesi M, Bergsagel PL. Many multiple myelomas: making more of the molecular mayhem. Hematology Am Soc Hematol Educ Program 2011; 2011:344 - 53; http://dx.doi.org/10.1182/asheducation-2011.1.344; PMID: 22160056
- Rio-Machin A, Ferreira BI, Henry T, Gómez-López G, Agirre X, Alvarez S, Rodriguez-Perales S, Prosper F, Calasanz MJ, Martínez J, et al. Downregulation of specific miRNAs in hyperdiploid multiple myeloma mimics the oncogenic effect of IgH translocations occurring in the non-hyperdiploid subtype. Leukemia 2013; 27:925 - 31; http://dx.doi.org/10.1038/leu.2012.302; PMID: 23174883
- Li Z, Jiao X, Wang C, Shirley LA, Elsaleh H, Dahl O, Wang M, Soutoglou E, Knudsen ES, Pestell RG. Alternative cyclin D1 splice forms differentially regulate the DNA damage response. Cancer Res 2010; 70:8802 - 11; http://dx.doi.org/10.1158/0008-5472.CAN-10-0312; PMID: 20940395
- Kuehl WM, Bergsagel PL. Molecular pathogenesis of multiple myeloma and its premalignant precursor. J Clin Invest 2012; 122:3456 - 63; http://dx.doi.org/10.1172/JCI61188; PMID: 23023717
- Gabrea A, Bergsagel PL, Chesi M, Shou Y, Kuehl WM. Insertion of excised IgH switch sequences causes overexpression of cyclin D1 in a myeloma tumor cell. Mol Cell 1999; 3:119 - 23; http://dx.doi.org/10.1016/S1097-2765(00)80180-X; PMID: 10024885
- Munshi NC, Avet-Loiseau H. Genomics in multiple myeloma. Clin Cancer Res 2011; 17:1234 - 42; http://dx.doi.org/10.1158/1078-0432.CCR-10-1843; PMID: 21411439
- Chng WJ, Gonzalez-Paz N, Price-Troska T, Jacobus S, Rajkumar SV, Oken MM, Kyle RA, Henderson KJ, Van Wier S, Greipp P, et al. Clinical and biological significance of RAS mutations in multiple myeloma. Leukemia 2008; 22:2280 - 4; http://dx.doi.org/10.1038/leu.2008.142; PMID: 18528420
- Avet-Loiseau H, Attal M, Campion L, Caillot D, Hulin C, Marit G, Stoppa AM, Voillat L, Wetterwald M, Pegourie B, et al. Long-term analysis of the IFM 99 trials for myeloma: cytogenetic abnormalities [t(4;14), del(17p), 1q gains] play a major role in defining long-term survival. J Clin Oncol 2012; 30:1949 - 52; http://dx.doi.org/10.1200/JCO.2011.36.5726; PMID: 22547600
- Lodé L, Eveillard M, Trichet V, Soussi T, Wuillème S, Richebourg S, Magrangeas F, Ifrah N, Campion L, Traullé C, et al. Mutations in TP53 are exclusively associated with del(17p) in multiple myeloma. Haematologica 2010; 95:1973 - 6; http://dx.doi.org/10.3324/haematol.2010.023697; PMID: 20634494
- Pichiorri F, Suh S-S, Rocci A, De Luca L, Taccioli C, Santhanam R, Zhou W, Benson DM Jr., Hofmainster C, Alder H, et al. Downregulation of p53-inducible microRNAs 192, 194, and 215 impairs the p53/MDM2 autoregulatory loop in multiple myeloma development. Cancer Cell 2010; 18:367 - 81; http://dx.doi.org/10.1016/j.ccr.2010.09.005; PMID: 20951946
- Bharti AC, Shishodia S, Reuben JM, Weber D, Alexanian R, Raj-Vadhan S, Estrov Z, Talpaz M, Aggarwal BB. Nuclear factor-kappaB and STAT3 are constitutively active in CD138+ cells derived from multiple myeloma patients, and suppression of these transcription factors leads to apoptosis. Blood 2004; 103:3175 - 84; http://dx.doi.org/10.1182/blood-2003-06-2151; PMID: 15070700
- Annunziata CM, Davis RE, Demchenko Y, Bellamy W, Gabrea A, Zhan F, Lenz G, Hanamura I, Wright G, Xiao W, et al. Frequent engagement of the classical and alternative NF-kappaB pathways by diverse genetic abnormalities in multiple myeloma. Cancer Cell 2007; 12:115 - 30; http://dx.doi.org/10.1016/j.ccr.2007.07.004; PMID: 17692804
- Keats JJ, Fonseca R, Chesi M, Schop R, Baker A, Chng W-J, Van Wier S, Tiedemann R, Shi CX, Sebag M, et al. Promiscuous mutations activate the noncanonical NF-kappaB pathway in multiple myeloma. Cancer Cell 2007; 12:131 - 44; http://dx.doi.org/10.1016/j.ccr.2007.07.003; PMID: 17692805
- Markovina S, Callander NS, O’Connor SL, Kim J, Werndli JE, Raschko M, Leith CP, Kahl BS, Kim K, Miyamoto S. Bortezomib-resistant nuclear factor-kappaB activity in multiple myeloma cells. Mol Cancer Res 2008; 6:1356 - 64; http://dx.doi.org/10.1158/1541-7786.MCR-08-0108; PMID: 18708367
- Chapman MA, Lawrence MS, Keats JJ, Cibulskis K, Sougnez C, Schinzel AC, Harview CL, Brunet JP, Ahmann GJ, Adli M, et al. Initial genome sequencing and analysis of multiple myeloma. Nature 2011; 471:467 - 72; http://dx.doi.org/10.1038/nature09837; PMID: 21430775
- Demchenko YN, Glebov OK, Zingone A, Keats JJ, Bergsagel PL, Kuehl WM. Classical and/or alternative NF-kappaB pathway activation in multiple myeloma. Blood 2010; 115:3541 - 52; http://dx.doi.org/10.1182/blood-2009-09-243535; PMID: 20053756
- Avet-Loiseau H, Gerson F, Magrangeas F, Minvielle S, Harousseau JL, Bataille R, Intergroupe Francophone du Myélome. Rearrangements of the c-myc oncogene are present in 15% of primary human multiple myeloma tumors. Blood 2001; 98:3082 - 6; http://dx.doi.org/10.1182/blood.V98.10.3082; PMID: 11698294
- Chng WJ, Huang GF, Chung TH, Ng SB, Gonzalez-Paz N, Troska-Price T, Mulligan G, Chesi M, Bergsagel PL, Fonseca R. Clinical and biological implications of MYC activation: a common difference between MGUS and newly diagnosed multiple myeloma. Leukemia 2011; 25:1026 - 35; http://dx.doi.org/10.1038/leu.2011.53; PMID: 21468039
- Bergsagel PL, Affer M, Glebov OK, Chen W-DD, Keats JJ, Brents LA, et al. Promiscuous cryptic rearrangements of the MYC locus cis-dysregulate MYC expression and are present in the majority of patients with hyperdiploid myeloma. Blood ASH meeting abstracts 2012; 120:724
- Dib A, Gabrea A, Glebov OK, Bergsagel PL, Kuehl WM. Characterization of MYC translocations in multiple myeloma cell lines. J Natl Cancer Inst Monogr 2008; 2008:25 - 31; http://dx.doi.org/10.1093/jncimonographs/lgn011; PMID: 18647998
- Keats JJ, Chesi M, Egan JB, Garbitt VM, Palmer SE, Braggio E, Van Wier S, Blackburn PR, Baker AS, Dispenzieri A, et al. Clonal competition with alternating dominance in multiple myeloma. Blood 2012; 120:1067 - 76; http://dx.doi.org/10.1182/blood-2012-01-405985; PMID: 22498740
- Magrangeas F, Avet-Loiseau H, Gouraud W, Lodé L, Decaux O, Godmer P, Garderet L, Voillat L, Facon T, Stoppa AM, et al. Minor clone provides a reservoir for relapse in multiple myeloma. Leukemia 2013; 27:473 - 81; http://dx.doi.org/10.1038/leu.2012.226; PMID: 22874878
- Egan JB, Kortuem KM, Kurdoglu A, Izatt T, Aldrich J, Reiman R, Phillips L, Baker A, Shi CX, Schmidt J, et al. Extramedullary myeloma whole genome sequencing reveals novel mutations in Cereblon, proteasome subunit G2 and the glucocorticoid receptor in multi drug resistant disease. Br J Haematol 2013; 161:748 - 51; http://dx.doi.org/10.1111/bjh.12291; PMID: 23480694
- Weston-Bell N, Gibson J, John M, Ennis S, Pfeifer S, Cezard T, Ludwig H, Collins A, Zojer N, Sahota SS. Exome sequencing in tracking clonal evolution in multiple myeloma following therapy. Leukemia 2013; 27:1188 - 91; http://dx.doi.org/10.1038/leu.2012.287; PMID: 23147253
- Walker BA, Morgan GJ. Use of single nucleotide polymorphism-based mapping arrays to detect copy number changes and loss of heterozygosity in multiple myeloma. Clin Lymphoma Myeloma 2006; 7:186 - 91; http://dx.doi.org/10.3816/CLM.2006.n.057; PMID: 17229333
- Weinhold N, Johnson DC, Chubb D, Chen B, Försti A, Hosking FJ, Broderick P, Ma YP, Dobbins SE, Hose D, et al. The CCND1 c.870G>A polymorphism is a risk factor for t(11;14)(q13;q32) multiple myeloma. Nat Genet 2013; 45:522 - 5; http://dx.doi.org/10.1038/ng.2583; PMID: 23502783
- Ricke RM, van Deursen JM. Aneuploidy in health, disease, and aging. J Cell Biol 2013; 201:11 - 21; http://dx.doi.org/10.1083/jcb.201301061; PMID: 23547028
- Siegel JJ, Amon A. New insights into the troubles of aneuploidy. Annu Rev Cell Dev Biol 2012; 28:189 - 214; http://dx.doi.org/10.1146/annurev-cellbio-101011-155807; PMID: 22804579
- Chng WJ, Fonseca R. Centrosomes and myeloma; aneuploidy and proliferation. Environ Mol Mutagen 2009; 50:697 - 707; http://dx.doi.org/10.1002/em.20528; PMID: 19739237
- Chng WJ, Braggio E, Mulligan G, Bryant B, Remstein E, Valdez R, Dogan A, Fonseca R. The centrosome index is a powerful prognostic marker in myeloma and identifies a cohort of patients that might benefit from aurora kinase inhibition. Blood 2008; 111:1603 - 9; http://dx.doi.org/10.1182/blood-2007-06-097774; PMID: 18006703
- Lindahl T. Instability and decay of the primary structure of DNA. Nature 1993; 362:709 - 15; http://dx.doi.org/10.1038/362709a0; PMID: 8469282
- Hoeijmakers JHJ. DNA damage, aging, and cancer. N Engl J Med 2009; 361:1475 - 85; http://dx.doi.org/10.1056/NEJMra0804615; PMID: 19812404
- Galm O, Wilop S, Reichelt J, Jost E, Gehbauer G, Herman JG, Osieka R. DNA methylation changes in multiple myeloma. Leukemia 2004; 18:1687 - 92; http://dx.doi.org/10.1038/sj.leu.2403434; PMID: 15318245
- Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, Davies FE, Ross FM, Morgan GJ. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011; 117:553 - 62; http://dx.doi.org/10.1182/blood-2010-04-279539; PMID: 20944071
- Amodio N, Leotta M, Bellizzi D, Di Martino MT, D’Aquila P, Lionetti M, Fabiani F, Leone E, Gullà AM, Passarino G, et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012; 3:1246 - 58; PMID: 23100393
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179 - 204; http://dx.doi.org/10.1016/j.molcel.2010.09.019; PMID: 20965415
- Iyama T, Wilson DM 3rd. DNA repair mechanisms in dividing and non-dividing cells. DNA Repair (Amst) 2013; 12:620 - 36; http://dx.doi.org/10.1016/j.dnarep.2013.04.015; PMID: 23684800
- Caldecott KW. Single-strand break repair and genetic disease. Nat Rev Genet 2008; 9:619 - 31; PMID: 18626472
- Friedberg EC. How nucleotide excision repair protects against cancer. Nat Rev Cancer 2001; 1:22 - 33; http://dx.doi.org/10.1038/35094000; PMID: 11900249
- Hanawalt PC, Spivak G. Transcription-coupled DNA repair: two decades of progress and surprises. Nat Rev Mol Cell Biol 2008; 9:958 - 70; http://dx.doi.org/10.1038/nrm2549; PMID: 19023283
- Jiricny J. The multifaceted mismatch-repair system. Nat Rev Mol Cell Biol 2006; 7:335 - 46; http://dx.doi.org/10.1038/nrm1907; PMID: 16612326
- Shaheen M, Allen C, Nickoloff JA, Hromas R. Synthetic lethality: exploiting the addiction of cancer to DNA repair. Blood 2011; 117:6074 - 82; http://dx.doi.org/10.1182/blood-2011-01-313734; PMID: 21441464
- Deans AJ, West SC. DNA interstrand crosslink repair and cancer. Nat Rev Cancer 2011; 11:467 - 80; http://dx.doi.org/10.1038/nrc3088; PMID: 21701511
- Bunting SF, Callén E, Wong N, Chen H-T, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243 - 54; http://dx.doi.org/10.1016/j.cell.2010.03.012; PMID: 20362325
- Chapman JR, Taylor MRG, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497 - 510; http://dx.doi.org/10.1016/j.molcel.2012.07.029; PMID: 22920291
- Pei H, Zhang L, Luo K, Qin Y, Chesi M, Fei F, Bergsagel PL, Wang L, You Z, Lou Z. MMSET regulates histone H4K20 methylation and 53BP1 accumulation at DNA damage sites. Nature 2011; 470:124 - 8; http://dx.doi.org/10.1038/nature09658; PMID: 21293379
- Tang J, Cho NW, Cui G, Manion EM, Shanbhag NM, Botuyan MV, Mer G, Greenberg RA. Acetylation limits 53BP1 association with damaged chromatin to promote homologous recombination. Nat Struct Mol Biol 2013; 20:317 - 25; http://dx.doi.org/10.1038/nsmb.2499; PMID: 23377543
- Hochegger H, Dejsuphong D, Fukushima T, Morrison C, Sonoda E, Schreiber V, Zhao GY, Saberi A, Masutani M, Adachi N, et al. Parp-1 protects homologous recombination from interference by Ku and Ligase IV in vertebrate cells. EMBO J 2006; 25:1305 - 14; http://dx.doi.org/10.1038/sj.emboj.7601015; PMID: 16498404
- Adamo A, Collis SJ, Adelman CA, Silva N, Horejsi Z, Ward JD, Martinez-Perez E, Boulton SJ, La Volpe A. Preventing nonhomologous end joining suppresses DNA repair defects of Fanconi anemia. Mol Cell 2010; 39:25 - 35; http://dx.doi.org/10.1016/j.molcel.2010.06.026; PMID: 20598602
- Pace P, Mosedale G, Hodskinson MR, Rosado IV, Sivasubramaniam M, Patel KJ. Ku70 corrupts DNA repair in the absence of the Fanconi anemia pathway. Science 2010; 329:219 - 23; http://dx.doi.org/10.1126/science.1192277; PMID: 20538911
- Lieber MR. NHEJ and its backup pathways in chromosomal translocations. Nat Struct Mol Biol 2010; 17:393 - 5; http://dx.doi.org/10.1038/nsmb0410-393; PMID: 20368722
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4:435 - 45; http://dx.doi.org/10.1038/nrm1127; PMID: 12778123
- Hartlerode AJ, Scully R. Mechanisms of double-strand break repair in somatic mammalian cells. Biochem J 2009; 423:157 - 68; http://dx.doi.org/10.1042/BJ20090942; PMID: 19772495
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 2010; 207:855 - 65; http://dx.doi.org/10.1084/jem.20100244; PMID: 20368578
- Williams HL, Gottesman ME, Gautier J. Replication-independent repair of DNA interstrand crosslinks. Mol Cell 2012; 47:140 - 7; PMID: 22658724
- Wang Y, Leung JW, Jiang Y, Lowery MG, Do H, Vasquez KM, Chen J, Wang W, Li L. FANCM and FAAP24 maintain genome stability via cooperative as well as unique functions. Mol Cell 2013; 49:997 - 1009; http://dx.doi.org/10.1016/j.molcel.2012.12.010; PMID: 23333308
- Kim N, Jinks-Robertson S. Transcription as a source of genome instability. Nat Rev Genet 2012; 13:204 - 14; PMID: 22330764
- Knipscheer P, Räschle M, Smogorzewska A, Enoiu M, Ho TV, Schärer OD, Elledge SJ, Walter JC. The Fanconi anemia pathway promotes replication-dependent DNA interstrand cross-link repair. Science 2009; 326:1698 - 701; http://dx.doi.org/10.1126/science.1182372; PMID: 19965384
- Constantinou A. Rescue of replication failure by Fanconi anaemia proteins. Chromosoma 2012; 121:21 - 36; http://dx.doi.org/10.1007/s00412-011-0349-2; PMID: 22057367
- Hayden PJ, Tewari P, Morris DW, Staines A, Crowley D, Nieters A, Becker N, de Sanjosé S, Foretova L, Maynadié M, et al. Variation in DNA repair genes XRCC3, XRCC4, XRCC5 and susceptibility to myeloma. Hum Mol Genet 2007; 16:3117 - 27; http://dx.doi.org/10.1093/hmg/ddm273; PMID: 17901044
- Roddam PL, Rollinson S, O’Driscoll M, Jeggo PA, Jack A, Morgan GJ. Genetic variants of NHEJ DNA ligase IV can affect the risk of developing multiple myeloma, a tumour characterised by aberrant class switch recombination. J Med Genet 2002; 39:900 - 5; http://dx.doi.org/10.1136/jmg.39.12.900; PMID: 12471202
- Roddam PL, Allan JM, Dring AM, Worrillow LJ, Davies FE, Morgan GJ. Non-homologous end-joining gene profiling reveals distinct expression patterns associated with lymphoma and multiple myeloma. Br J Haematol 2010; 149:258 - 62; http://dx.doi.org/10.1111/j.1365-2141.2010.08088.x; PMID: 20148879
- Calimeri T, Fulcineti M, Lin J, Samur MK, Calkins AS, Vahia AV, et al. Aberrant non-homologous end joining in multiple myeloma: a role in genomic instability and as a potential prognostic marker. Blood ASH meeting abstracts 2012; 120:2932
- Tai YT, Teoh G, Lin B, Davies FE, Chauhan D, Treon SP, Raje N, Hideshima T, Shima Y, Podar K, et al. Ku86 variant expression and function in multiple myeloma cells is associated with increased sensitivity to DNA damage. J Immunol 2000; 165:6347 - 55; PMID: 11086072
- Kato M, Iida S, Komatsu H, Ueda R. Lack of Ku80 alteration in multiple myeloma. Jpn J Cancer Res 2002; 93:359 - 62; http://dx.doi.org/10.1111/j.1349-7006.2002.tb01264.x; PMID: 11985783
- Han Z, Johnston C, Reeves WH, Carter T, Wyche JH, Hendrickson EA. Characterization of a Ku86 variant protein that results in altered DNA binding and diminished DNA-dependent protein kinase activity. J Biol Chem 1996; 271:14098 - 104; http://dx.doi.org/10.1074/jbc.271.24.14098; PMID: 8662896
- Łanuszewska J, Widłak P. The truncation of Ku86 in human lymphocytes. Cancer Lett 2004; 205:197 - 205; http://dx.doi.org/10.1016/j.canlet.2003.10.016; PMID: 15036652
- Gullo CA, Ge F, Cow G, Teoh G. Ku86 exists as both a full-length and a protease-sensitive natural variant in multiple myeloma cells. Cancer Cell Int 2008; 8:4; http://dx.doi.org/10.1186/1475-2867-8-4; PMID: 18442416
- Yang C, Betti C, Singh S, Toor A, Vaughan A. Impaired NHEJ function in multiple myeloma. Mutat Res 2009; 660:66 - 73; http://dx.doi.org/10.1016/j.mrfmmm.2008.10.019; PMID: 19028508
- Keats JJ, Reiman T, Belch AR, Pilarski LM. Ten years and counting: so what do we know about t(4;14)(p16;q32) multiple myeloma. Leuk Lymphoma 2006; 47:2289 - 300; http://dx.doi.org/10.1080/10428190600822128; PMID: 17107900
- Shammas MA, Shmookler Reis RJ, Koley H, Batchu RB, Li C, Munshi NC. Dysfunctional homologous recombination mediates genomic instability and progression in myeloma. Blood 2009; 113:2290 - 7; http://dx.doi.org/10.1182/blood-2007-05-089193; PMID: 19050310
- Mah L-J, El-Osta A, Karagiannis TC. gammaH2AX: a sensitive molecular marker of DNA damage and repair. Leukemia 2010; 24:679 - 86; http://dx.doi.org/10.1038/leu.2010.6; PMID: 20130602
- Walters DK, Wu X, Tschumper RC, Arendt BK, Huddleston PM, Henderson KJ, Dispenzieri A, Jelinek DF. Evidence for ongoing DNA damage in multiple myeloma cells as revealed by constitutive phosphorylation of H2AX. Leukemia 2011; 25:1344 - 53; http://dx.doi.org/10.1038/leu.2011.94; PMID: 21566653
- Volcic M, Karl S, Baumann B, Salles D, Daniel P, Fulda S, Wiesmüller L. NF-κB regulates DNA double-strand break repair in conjunction with BRCA1-CtIP complexes. Nucleic Acids Res 2012; 40:181 - 95; http://dx.doi.org/10.1093/nar/gkr687; PMID: 21908405
- Janssens S, Tschopp J. Signals from within: the DNA-damage-induced NF-kappaB response. Cell Death Differ 2006; 13:773 - 84; http://dx.doi.org/10.1038/sj.cdd.4401843; PMID: 16410802
- Miyamoto S. Nuclear initiated NF-κB signaling: NEMO and ATM take center stage. Cell Res 2011; 21:116 - 30; http://dx.doi.org/10.1038/cr.2010.179; PMID: 21187855
- Habraken Y, Piette J. NF-kappaB activation by double-strand breaks. Biochem Pharmacol 2006; 72:1132 - 41; http://dx.doi.org/10.1016/j.bcp.2006.07.015; PMID: 16965765
- Stilmann M, Hinz M, Arslan SC, Zimmer A, Schreiber V, Scheidereit C. A nuclear poly(ADP-ribose)-dependent signalosome confers DNA damage-induced IkappaB kinase activation. Mol Cell 2009; 36:365 - 78; http://dx.doi.org/10.1016/j.molcel.2009.09.032; PMID: 19917246
- Dumontet C, Landi S, Reiman T, Perry T, Plesa A, Bellini I, Barale R, Pilarski LM, Troncy J, Tavtigian S, et al. Genetic polymorphisms associated with outcome in multiple myeloma patients receiving high-dose melphalan. Bone Marrow Transplant 2010; 45:1316 - 24; http://dx.doi.org/10.1038/bmt.2009.335; PMID: 19966851
- Ushie C, Saitoh T, Iwasaki A, Moriyama N, Murakami H.. The polymorphisms of base excision repair genes influence the prognosis of multiple myeloma. Blood ASH meeting abstracts 2012; 120
- Yang Z-Z, Chen X-H, Wang D. Experimental study enhancing the chemosensitivity of multiple myeloma to melphalan by using a tissue-specific APE1-silencing RNA expression vector. Clin Lymphoma Myeloma 2007; 7:296 - 304; http://dx.doi.org/10.3816/CLM.2007.n.006; PMID: 17324338
- de Larrea CF, Navarro A, Tovar N, Pedrosa F, Díaz T, Cibeira MAT, et al. Impact of single nucleotide polymorphisms in genes involved in DNA repair and drug metabolism on survival after autologous stem cell transplantation in patients with multiple myeloma. Blood ASH meeting abstracts 2012; 120:2934
- Episkopou H, Kyrtopoulos SA, Sfikakis PP, Fousteri M, Dimopoulos MA, Mullenders LHF, Souliotis VL. Association between transcriptional activity, local chromatin structure, and the efficiencies of both subpathways of nucleotide excision repair of melphalan adducts. Cancer Res 2009; 69:4424 - 33; http://dx.doi.org/10.1158/0008-5472.CAN-08-3489; PMID: 19417135
- Martin P, Santón A, García-Cosio M, Bellas C. hMLH1 and MGMT inactivation as a mechanism of tumorigenesis in monoclonal gammopathies. Mod Pathol 2006; 19:914 - 21; http://dx.doi.org/10.1038/modpathol.3800590; PMID: 16607377
- Kotoula V, Hytiroglou P, Kaloutsi V, Barbanis S, Kouidou S, Papadimitriou CS. Mismatch repair gene expression in malignant lymphoproliferative disorders of B-cell origin. Leuk Lymphoma 2002; 43:393 - 9; http://dx.doi.org/10.1080/10428190290006215; PMID: 11999575
- Walker BA, Wardell CP, Melchor L, Hulkki S, Potter NE, Johnson DC, Fenwick K, Kozarewa I, Gonzalez D, Lord CJ, et al. Intraclonal heterogeneity and distinct molecular mechanisms characterize the development of t(4;14) and t(11;14) myeloma. Blood 2012; 120:1077 - 86; http://dx.doi.org/10.1182/blood-2012-03-412981; PMID: 22573403
- Osborne MR, Wilman DE, Lawley PD. Alkylation of DNA by the nitrogen mustard bis(2-chloroethyl)methylamine. Chem Res Toxicol 1995; 8:316 - 20; http://dx.doi.org/10.1021/tx00044a018; PMID: 7766817
- Muniandy PA, Liu J, Majumdar A, Liu S-T, Seidman MM. DNA interstrand crosslink repair in mammalian cells: step by step. Crit Rev Biochem Mol Biol 2010; 45:23 - 49; http://dx.doi.org/10.3109/10409230903501819; PMID: 20039786
- Spanswick VJ, Craddock C, Sekhar M, Mahendra P, Shankaranarayana P, Hughes RG, Hochhauser D, Hartley JA. Repair of DNA interstrand crosslinks as a mechanism of clinical resistance to melphalan in multiple myeloma. Blood 2002; 100:224 - 9; http://dx.doi.org/10.1182/blood.V100.1.224; PMID: 12070031
- Gottesman MM, Fojo T, Bates SE. Multidrug resistance in cancer: role of ATP-dependent transporters. Nat Rev Cancer 2002; 2:48 - 58; http://dx.doi.org/10.1038/nrc706; PMID: 11902585
- Yang HH, Ma MH, Vescio RA, Berenson JR. Overcoming drug resistance in multiple myeloma: the emergence of therapeutic approaches to induce apoptosis. J Clin Oncol 2003; 21:4239 - 47; http://dx.doi.org/10.1200/JCO.2003.06.001; PMID: 14615454
- Dimopoulos MA, Souliotis VL, Anagnostopoulos A, Bamia C, Pouli A, Baltadakis I, Terpos E, Kyrtopoulos SA, Sfikakis PP. Melphalan-induced DNA damage in vitro as a predictor for clinical outcome in multiple myeloma. Haematologica 2007; 92:1505 - 12; http://dx.doi.org/10.3324/haematol.11435; PMID: 18024399
- Vangsted A, Gimsing P, Klausen TW, Nexø BA, Wallin H, Andersen P, Hokland P, Lillevang ST, Vogel U. Polymorphisms in the genes ERCC2, XRCC3 and CD3EAP influence treatment outcome in multiple myeloma patients undergoing autologous bone marrow transplantation. Int J Cancer 2007; 120:1036 - 45; http://dx.doi.org/10.1002/ijc.22411; PMID: 17131345
- Chen Q, Van der Sluis PC, Boulware D, Hazlehurst LA, Dalton WS. The FA/BRCA pathway is involved in melphalan-induced DNA interstrand cross-link repair and accounts for melphalan resistance in multiple myeloma cells. Blood 2005; 106:698 - 705; http://dx.doi.org/10.1182/blood-2004-11-4286; PMID: 15802532
- Yarde DN, Oliveira V, Mathews L, Wang X, Villagra A, Boulware D, Shain KH, Hazlehurst LA, Alsina M, Chen DT, et al. Targeting the Fanconi anemia/BRCA pathway circumvents drug resistance in multiple myeloma. Cancer Res 2009; 69:9367 - 75; http://dx.doi.org/10.1158/0008-5472.CAN-09-2616; PMID: 19934314
- Bharti AC, Donato N, Singh S, Aggarwal BB. Curcumin (diferuloylmethane) down-regulates the constitutive activation of nuclear factor-kappa B and IkappaBalpha kinase in human multiple myeloma cells, leading to suppression of proliferation and induction of apoptosis. Blood 2003; 101:1053 - 62; http://dx.doi.org/10.1182/blood-2002-05-1320; PMID: 12393461
- Xiao H, Xiao Q, Zhang K, Zuo X, Shrestha UK. Reversal of multidrug resistance by curcumin through FA/BRCA pathway in multiple myeloma cell line MOLP-2/R. Ann Hematol 2010; 89:399 - 404; http://dx.doi.org/10.1007/s00277-009-0831-6; PMID: 19756599
- Pan Y, Gao Y, Chen L, Gao G, Dong H, Yang Y, Dong B, Chen X. Targeting autophagy augments in vitro and in vivo antimyeloma activity of DNA-damaging chemotherapy. Clin Cancer Res 2011; 17:3248 - 58; http://dx.doi.org/10.1158/1078-0432.CCR-10-0890; PMID: 21288924
- Katayama M, Kawaguchi T, Berger MS, Pieper RO. DNA damaging agent-induced autophagy produces a cytoprotective adenosine triphosphate surge in malignant glioma cells. Cell Death Differ 2007; 14:548 - 58; http://dx.doi.org/10.1038/sj.cdd.4402030; PMID: 16946731
- Couvé S, Macé-Aimé G, Rosselli F, Saparbaev MK. The human oxidative DNA glycosylase NEIL1 excises psoralen-induced interstrand DNA cross-links in a three-stranded DNA structure. J Biol Chem 2009; 284:11963 - 70; http://dx.doi.org/10.1074/jbc.M900746200; PMID: 19258314
- McNeill DR, Lam W, DeWeese TL, Cheng YC, Wilson DM 3rd. Impairment of APE1 function enhances cellular sensitivity to clinically relevant alkylators and antimetabolites. Mol Cancer Res 2009; 7:897 - 906; http://dx.doi.org/10.1158/1541-7786.MCR-08-0519; PMID: 19470598
- Sousa MML, Zub KA, Aas PA, Hanssen-Bauer A, Demirovic A, Sarno A, Tian E, Liabakk NB, Slupphaug G. An inverse switch in DNA base excision and strand break repair contributes to melphalan resistance in multiple myeloma cells. PLoS One 2013; 8:e55493; http://dx.doi.org/10.1371/journal.pone.0055493; PMID: 23405159
- Crawford LJA, Walker B, Ovaa H, Chauhan D, Anderson KC, Morris TCM, Irvine AE. Comparative selectivity and specificity of the proteasome inhibitors BzLLLCOCHO, PS-341, and MG-132. Cancer Res 2006; 66:6379 - 86; http://dx.doi.org/10.1158/0008-5472.CAN-06-0605; PMID: 16778216
- Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2011; 2:209 - 21; PMID: 21444945
- Voorhees PM, Orlowski RZ. The proteasome and proteasome inhibitors in cancer therapy. Annu Rev Pharmacol Toxicol 2006; 46:189 - 213; http://dx.doi.org/10.1146/annurev.pharmtox.46.120604.141300; PMID: 16402903
- Jacquemont C, Taniguchi T. Proteasome function is required for DNA damage response and fanconi anemia pathway activation. Cancer Res 2007; 67:7395 - 405; http://dx.doi.org/10.1158/0008-5472.CAN-07-1015; PMID: 17671210
- Murakawa Y, Sonoda E, Barber LJ, Zeng W, Yokomori K, Kimura H, Niimi A, Lehmann A, Zhao GY, Hochegger H, et al. Inhibitors of the proteasome suppress homologous DNA recombination in mammalian cells. Cancer Res 2007; 67:8536 - 43; http://dx.doi.org/10.1158/0008-5472.CAN-07-1166; PMID: 17875693
- Neri P, Ren L, Gratton K, Stebner E, Johnson J, Klimowicz A, Duggan P, Tassone P, Mansoor A, Stewart DA, et al. Bortezomib-induced “BRCAness” sensitizes multiple myeloma cells to PARP inhibitors. Blood 2011; 118:6368 - 79; http://dx.doi.org/10.1182/blood-2011-06-363911; PMID: 21917757
- Roussel M, Moreau P, Huynh A, Mary J-Y, Danho C, Caillot D, Hulin C, Fruchart C, Marit G, Pégourié B, et al, Intergroupe Francophone du Myélome (IFM). Bortezomib and high-dose melphalan as conditioning regimen before autologous stem cell transplantation in patients with de novo multiple myeloma: a phase 2 study of the Intergroupe Francophone du Myelome (IFM). Blood 2010; 115:32 - 7; http://dx.doi.org/10.1182/blood-2009-06-229658; PMID: 19884643
- Steele JM. Carfilzomib: A new proteasome inhibitor for relapsed or refractory multiple myeloma. J Oncol Pharm Pract 2013; 19:348 - 54; http://dx.doi.org/10.1177/1078155212470388; PMID: 23292972
- Wang Q-E, Wani MA, Chen J, Zhu Q, Wani G, El-Mahdy MA, Wani AA. Cellular ubiquitination and proteasomal functions positively modulate mammalian nucleotide excision repair. Mol Carcinog 2005; 42:53 - 64; http://dx.doi.org/10.1002/mc.20065; PMID: 15547920
- Ito T, Ando H, Suzuki T, Ogura T, Hotta K, Imamura Y, Yamaguchi Y, Handa H. Identification of a primary target of thalidomide teratogenicity. Science 2010; 327:1345 - 50; http://dx.doi.org/10.1126/science.1177319; PMID: 20223979
- Zhu Y-X, Braggio E, Shi C-X, Bruins LA, Schmidt JE, Van Wier S, Chang XB, Bjorklund CC, Fonseca R, Bergsagel PL, et al. Cereblon expression is required for the antimyeloma activity of lenalidomide and pomalidomide. Blood 2011; 118:4771 - 9; http://dx.doi.org/10.1182/blood-2011-05-356063; PMID: 21860026
- Moalli PA, Pillay S, Weiner D, Leikin R, Rosen ST. A mechanism of resistance to glucocorticoids in multiple myeloma: transient expression of a truncated glucocorticoid receptor mRNA. Blood 1992; 79:213 - 22; PMID: 1728309
- Nojima M, Maruyama R, Yasui H, Suzuki H, Maruyama Y, Tarasawa I, Sasaki Y, Asaoku H, Sakai H, Hayashi T, et al. Genomic screening for genes silenced by DNA methylation revealed an association between RASD1 inactivation and dexamethasone resistance in multiple myeloma. Clin Cancer Res 2009; 15:4356 - 64; http://dx.doi.org/10.1158/1078-0432.CCR-08-3336; PMID: 19549772
- Chauhan D, Li G, Hideshima T, Podar K, Mitsiades C, Mitsiades N, Catley L, Tai YT, Hayashi T, Shringarpure R, et al. Hsp27 inhibits release of mitochondrial protein Smac in multiple myeloma cells and confers dexamethasone resistance. Blood 2003; 102:3379 - 86; http://dx.doi.org/10.1182/blood-2003-05-1417; PMID: 12855565
- Moreaux J, Legouffe E, Jourdan E, Quittet P, Rème T, Lugagne C, Moine P, Rossi JF, Klein B, Tarte K. BAFF and APRIL protect myeloma cells from apoptosis induced by interleukin 6 deprivation and dexamethasone. Blood 2004; 103:3148 - 57; http://dx.doi.org/10.1182/blood-2003-06-1984; PMID: 15070697
- Tai Y-T, Anderson KC. Bruton’s tyrosine kinase: oncotarget in myeloma. Oncotarget 2012; 3:913 - 4; PMID: 22989914
- Cibeira MT, de Larrea CF, Navarro A, Díaz T, Fuster D, Tovar N, Rosiñol L, Monzó M, Bladé J. Impact on response and survival of DNA repair single nucleotide polymorphisms in relapsed or refractory multiple myeloma patients treated with thalidomide. Leuk Res 2011; 35:1178 - 83; http://dx.doi.org/10.1016/j.leukres.2011.02.009; PMID: 21435719
- Burington B, Barlogie B, Zhan F, Crowley J, Shaughnessy JD Jr.. Tumor cell gene expression changes following short-term in vivo exposure to single agent chemotherapeutics are related to survival in multiple myeloma. Clin Cancer Res 2008; 14:4821 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-07-4568; PMID: 18676754
- Barrera LN, Rushworth SA, Bowles KM, MacEwan DJ. Bortezomib induces heme oxygenase-1 expression in multiple myeloma. Cell Cycle 2012; 11:2248 - 52; http://dx.doi.org/10.4161/cc.20343; PMID: 22617388
- Lord CJ, Ashworth A. The DNA damage response and cancer therapy. Nature 2012; 481:287 - 94; http://dx.doi.org/10.1038/nature10760; PMID: 22258607
- Smith J, Tho LM, Xu N, Gillespie DA. Chapter 3 - The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. 1st ed. Elsevier Inc; 2010.
- Pei X-Y, Dai Y, Youssefian LE, Chen S, Bodie WW, Takabatake Y, Felthousen J, Almenara JA, Kramer LB, Dent P, et al. Cytokinetically quiescent (G0/G1) human multiple myeloma cells are susceptible to simultaneous inhibition of Chk1 and MEK1/2. Blood 2011; 118:5189 - 200; http://dx.doi.org/10.1182/blood-2011-02-339432; PMID: 21911831
- Smith J, Tho LM, Xu N, Gillespie DA. Chapter 3 - The ATM-Chk2 and ATR-Chk1 Pathways in DNA Damage Signaling and Cancer. 1st ed. Elsevier Inc; 2010.
- Landau HJ, McNeely SC, Nair JS, Comenzo RL, Asai T, Friedman H, Jhanwar SC, Nimer SD, Schwartz GK. The checkpoint kinase inhibitor AZD7762 potentiates chemotherapy-induced apoptosis of p53-mutated multiple myeloma cells. Mol Cancer Ther 2012; 11:1781 - 8; http://dx.doi.org/10.1158/1535-7163.MCT-11-0949; PMID: 22653969
- Caraux A, Vincent L, Bouhya S, Quittet P, Moreaux J, Requirand G, Veyrune JL, Olivier G, Cartron G, Rossi JF, et al. Residual malignant and normal plasma cells shortly after high dose melphalan and stem cell transplantation. Highlight of a putative therapeutic window in Multiple Myeloma?. Oncotarget 2012; 3:1335 - 47; PMID: 23154454