Abstract
PDIP38 (polymerase delta interacting protein 38) was originally discovered as a protein that interacts with DNA polymerase δ and PCNA. PDIP38 is present in multiple intracellular locations and is a multifunctional protein that has been implicated in several diverse cellular functions. We investigated the nuclear localization of PDIP38 in order to gain insights to its response to UV damage. PDIP38 was found to form distinct nuclear foci in response to UV irradiation in several cell lines, including HeLa S3 and A549 cells. However, these foci were not those associated with UV repair foci. Using various markers for different nuclear subcompartments, the UV-induced PDIP38 foci were identified as spliceosomes/nuclear speckles, the storage and assembly sites for mRNA splicing factors. To assess the role of PDIP38 in the regulation of splicing events, the effects of PDIP38 depletion on the UV-induced alternate splicing of MDM2 transcripts were examined by nested RT-PCR. Alternatively spliced MDM2 products were induced by UV treatment but were greatly reduced in cells expressing shRNA targeting PDIP38. These findings indicate that upon UV-induced DNA damage, PDIP38 is translocated to spliceosomes and contributes to the UV-induced alternative splicing of MDM2 transcripts. Similar results were obtained when cells were subjected to transcriptional stresses with actinomycin D or α-amanitin. Taken together, these studies show that PDIP38 is a protein regulated in a dynamic manner in response to genotoxic stress, as evidenced by its translocation to the spliceosomes. Moreover, PDIP38 is required for the induction of the alternative splicing of MDM2 in response to UV irradiation.
Introduction
Genomic stability depends on the faithful duplication and maintenance of the integrity of DNA and is essential for the survival of all living organisms. DNA polymerase δ (Pol δ) is a key enzyme that participates in the replication of the eukaryotic genome.Citation1-Citation3 The Pol δ holoenzyme (Pol δ4) is a 4-subunit complex consisting of the p125, p50, p68, and p12 subunits in mammalian cells.Citation1,Citation4 Pol δ is regulated in response to UV damage by the degradation of the p12 subunit, which results in the conversion of Pol δ4 to the trimer, Pol δ3 (reviewed in Lee et al.Citation1). The function of Pol δ is facilitated by the DNA sliding clamp, PCNA, which acts as a processivity factor.Citation5 PCNA also coordinates the interaction of Pol δ with flap endonuclease 1 (FEN1) and DNA ligase I proteins during Okazaki fragment processing.Citation6,Citation7 The functions of Pol δ and PCNA are not exclusive to DNA replication; these 2 proteins also participate in gap filling during DNA repair processes, e.g., in nucleotide excision repair (NER)Citation8-Citation12 and in D-loop extension during homologous recombination.Citation13
PCNA also plays a key role in the regulation of the DNA damage tolerance pathway.Citation14,Citation15 UV irradiation introduces bulky lesions, including cyclopyrimidine butane dimers (CPDs), which create barriers for DNA polymerases that can result in replication fork arrest and cell death. To avoid cell death, the DNA tolerance pathway is activated, in which specialized translesion synthesis (TLS) polymerases such as Pol η are transiently switched for the replicative polymerase. The TLS polymerases bypass the DNA lesions and allow replication to continue. The process of translesion synthesis requires the mono-ubiquitination of PCNA. These repair and TLS processes take place at DNA repair foci at sites of UV-damaged DNA. PCNA, Pol δ, and other repair proteins are characteristically recruited to these DNA repair foci, which are readily detected by fluorescence microscopy.Citation10
PDIP38 was discovered by a yeast 2-hybrid screen and was found to interact with the p50 subunit of Pol δ and with PCNA.Citation16 PDIP38 has been found in multiple sites within the cell. It was initially reported to be localized mainly to the mitochondria, as well as to the nucleus, and possesses a mitochondrial peptidase cleavage site.Citation17 PDIP38 was found to associate with mitochondrial single-stranded binding protein.Citation18 PDIP38 was also shown to shuttle between the cytosol and the nucleus, and also to be recruited to the plasma membrane under the influence of CEACAM1.Citation19 The latter is a cell adhesion molecule that is involved in the control of proliferation in hematopoietic and endothelial cells.Citation20 PDIP38 was also shown to be associated with the mitotic spindle, and PDIP38 depletion was associated with aberrant spindle formation, chromosome segregation, as well as multinucleation.Citation21 PDIP38 interacts with several translesion polymerases, Pol η, Rev1, and Rev7, suggesting a role in TLS.Citation22 While these studies broadly support a role in DNA metabolism, PDIP38 was also reported to regulate Nox4 (NADPH oxidase 4) and proposed to have a role in linking focal adhesion turnover and vascular smooth muscle migration to reactive oxygen species production.Citation23 Recently, a PDIP38-deficient mouse model was generated. The PDIP38 homozygous state was embryonically lethal, and the heterozygous mice exhibited defects in vascular functions.Citation24 In correspondence with these findings, the roles of PDIP38 are likely to be complex, and this multifunctional protein appears to participate in multiple and diverse cellular functions.
Numerous proteins participate in the cellular response to DNA damage. Proteins involved in post-transcriptional regulation have emerged as important regulators in the cell cycle progression, DNA damage responses, DNA repair, and apoptosis. These include many RNA binding proteins that influence stability of selective mRNA transcripts or facilitate the splicing of pre-mRNAs encoding proteins involved in the DNA damage response.Citation25 There are reports of numerous genes, including MDM2, that are alternatively spliced in response to genotoxic stress.Citation26 Dysregulation of alternative splicing of numerous genes, including MDM2,Citation29-Citation32 have been associated with cancer.Citation27,Citation28
The goal of this study was to understand the nuclear functions of PDIP38. Initially this quest was directed toward understanding the response of PDIP38 to genotoxic stress induced by DNA damaging agents, and how this contributes to the DNA damage tolerance pathway, based on recent findings of its association with TLS (translesion synthesis) polymerases.Citation22 Unexpectedly, however, we found that PDIP38 is not recruited to sites of DNA damaged by UV. Instead, we found PDIP38 is translocated to spliceosomes/nuclear speckles in response to UV irradiation. Directed by this observation, we identified a novel function of PDIP38, viz., its involvement in pre-mRNA processing, specifically in alternative splicing of MDM2 induced by UV irradiation.
Results
Indirect immunofluorescence localization of PDIP38
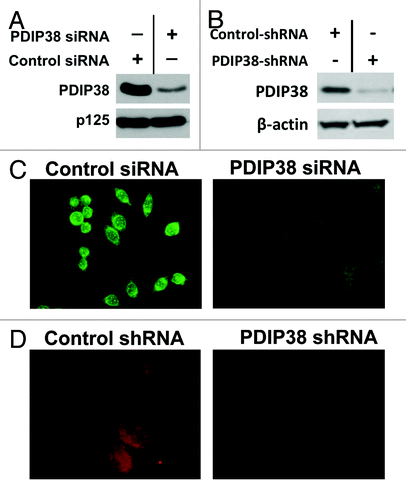
An anti-PDIP38 antibody was raised in rabbits (Materials and Methods), and its use in fluorescence microscopy was optimized. siRNA and shRNA depletion of PDIP38 were utilized to establish the specificity of the antibody. siRNA depletion of PDIP38 was performed in HeLa S3 cervical carcinoma cells. PDIP38-knockdown cells were generated by the use of PDIP38 shRNA in A549 lung adenocarinoma cells (Materials and Methods). PDIP38 was efficiently depleted by both siRNA () and shRNA knockdowns (). PDIP38 immunofluorescence in the control HeLa S3 cells was largely present in the cytoplasm with some nuclear staining but was severely reduced in the PDIP38 siRNA-treated cells (). PDIP38 immunofluorescence was essentially absent in A549 cell lines stably expressing shRNA targeted against PDIP38 as compared with the control cell line (). These experiments validated the specificity of the antibody, and eliminated the possibility that observations of multiple subcellular distributions of immunofluorescence are due to the detection of artifacts produced by binding of nonspecific antigen components in cells subjected to DNA damage.
Figure 1. Validation of the anti-PDIP38 antibody using siRNA and shRNA depletion of PDIP38. (A) HeLa S3 cells were transfected with siRNA against PDIP38 and harvested 72 h post transfection (Materials and Methods). Lysates were subjected to 10% SDS-PAGE and immunoblotted with antibodies against PDIP38. Blots for Pol δ p125 were used as a loading control. (B) Lysates of A549 cells in which PDIP38 was knocked down with PDIP38 shRNA were immunoblotted with antibodies against PDIP38; β-actin was used as a loading control (C). HeLa S3 cells transfected with control and PDIP38 siRNA were stained for PDIP38 (green immunofluorescence). (D) A549 control cells and A549 cells in which PDIP38 was knocked down by stable expression of PDIP38 shRNA (Materials and Methods) were fixed and stained for PDIP38 (red immunofluorescence). Images were taken with a Zeiss AxioVision at 40× (C) or 100× magnification (D).
Identification of the foci to which PDIP38 is translocated in response to UV irradiation
PDIP38 interacts with Pol η, Rev1, and Rev7, suggesting that it has a role in TLS.Citation22 Several studies have demonstrated the recruitment of repair proteins, including NER factors, Pol δ subunits, Pol η, and PCNA, to sites of localized UV-induced DNA damage.Citation10 Since PDIP38 associates with Pol δ, PCNA,Citation16 and Pol η,Citation22 we performed experiments to determine if PDIP38 is also recruited to sites of UV damage. We used the technique of irradiating cells through UV-opaque polycarbonate filters containing pores (5 or 10 μm) as a facile methodCitation10 for determining if PDIP38 is recruited to sites of local DNA lesions.
Localized DNA lesions in HeLa S3 cells were induced by UV irradiation through filters at a dose of 75 J/m2, and Pol η and PCNA were used as markers for the sites of UV-induced DNA lesions.Citation10 Pol η and PCNA were readily recruited to local UV-induced DNA damage areas, as shown by their co-localization (). Co-staining for PCNA and PDIP38 demonstrated that PDIP38 was not recruited to the sites of UV-induced DNA damage (). Similar results were obtained with A549 cells (data not shown). Thus, we conclude that PDIP38 is not recruited to sites of UV-damaged DNA, i.e., DNA repair foci. These results suggest that PDIP38 is redistributed elsewhere, perhaps to a unique nuclear compartment, and may participate in processes other than the direct repair of UV-induced DNA damage.
Figure 2. PDIP38 is not recruited to sites of UV-induced DNA damage. (A) Recruitment of PCNA and Pol η to sites of local UV damage. HeLa S3 cells were irradiated with UV (75 J/m2) through Millipore polycarbonate filters with 10 μm pores. Cells were fixed 3 h post-treatment and co-stained for PCNA (red immunofluorescence) and Pol η (green immunofluorescence [“Materials and Methods”]). Merged images are shown in the right panels. Areas of local irradiation are marked by the blue arrows. (B) PDIP38 is not recruited to sites of local UV-induced damage. HeLa S3 cells were subjected to local UV irradiation as described above and co-stained for PCNA (red immunofluorescence) and PDIP38 (green immunofluorescence). Areas of local irradiation are marked by the blue arrows. Images were taken at 40× (A) and 100× magnification (B).
![Figure 2. PDIP38 is not recruited to sites of UV-induced DNA damage. (A) Recruitment of PCNA and Pol η to sites of local UV damage. HeLa S3 cells were irradiated with UV (75 J/m2) through Millipore polycarbonate filters with 10 μm pores. Cells were fixed 3 h post-treatment and co-stained for PCNA (red immunofluorescence) and Pol η (green immunofluorescence [“Materials and Methods”]). Merged images are shown in the right panels. Areas of local irradiation are marked by the blue arrows. (B) PDIP38 is not recruited to sites of local UV-induced damage. HeLa S3 cells were subjected to local UV irradiation as described above and co-stained for PCNA (red immunofluorescence) and PDIP38 (green immunofluorescence). Areas of local irradiation are marked by the blue arrows. Images were taken at 40× (A) and 100× magnification (B).](/cms/asset/4824132a-d5d3-4171-bb96-09e0eb5c3bd3/kccy_a_10926221_f0002.gif)
During the DNA damage response, the orchestration and recruitment of proteins to sites of DNA damage is dynamic and occurs rapidly. ATM and ATR are recruited and activated at sites of DNA damage within minutes.Citation33,Citation34 Many of their downstream substrates activated at sites of the damage are mobilized to other sites to exert and coordinate DNA repair and activate cell cycle checkpoint processes. Since the DNA damage response is very dynamic, we examined PDIP38 localization at a shorter time interval (5 min) after UV irradiation. PDIP38 was not transiently recruited to sites of damaged DNA as shown by co-staining for PDIP38 and PCNA (Fig. S1A) and for PDIP38 and γ-H2AX (Fig. S1B).
PDIP38 is recruited to nuclear speckles in response to UV irradiation
Since we had demonstrated that PDIP38 is not being recruited to the newly organized DNA damage foci, we investigated whether it was being translocated to one of the known subnuclear compartments, as we noted its presence in distinct foci in unstressed cells. Several nuclear bodies are dynamically altered during cellular stress responses and can be suspected to be sites of PDIP38 recruitment. One nuclear compartment subjected to dynamic changes during stress is the promyelocytic leukemia (PML) body.Citation35 The nucleolus is another nuclear subcompartment that exhibits changes during cellular damage.Citation36,Citation37 The trafficking of proteins between these compartments and the nucleoplasm can form regulatory circuits that contribute to cellular responses to genotoxic stress.Citation35,Citation37 Nuclear spliceosomes, also known as nuclear speckles, are compartments enriched with splicing factors and function as storage and assembly sites for splicing factors.Citation38,Citation39 During stress conditions, nuclear speckles undergo morphological changes, and many splicing factors are redistributed.Citation40
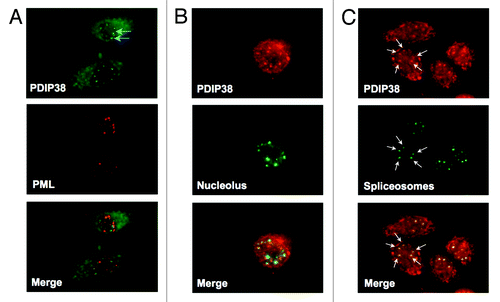
We therefore examined the co-localization of PDIP38 after UV irradiation with proteins that are known markers for these subcompartments. The markers used were C23 for the nucleolus, PML proteinCitation35 for the PML bodies, and splicing regulator protein SC35Citation41 for the spliceosomes. A549 cells were globally irradiated with UV (60 J/m2) and analyzed 4 h after irradiation. Cells were fixed and co-stained for PDIP38, in turn, with each of the 3 marker proteins. We observed that PDIP38 was neither recruited to the PML bodies (), nor to nucleolus () but was distinctly appearing within the spliceosomes ().
Figure 3. PDIP38 is recruited to spliceosomes in response to UV-induced damage. A549 cells were globally irradiated with 60 J/m2 of UV and allowed to recover for 4 h. (A) Cells were fixed and co-stained for PDIP38 (green immunofluorescence) and PML bodies (red immunofluorescence). The merged images show no signs of co-localization. (B) Cells were fixed and co-stained for PDIP38 (red immunofluorescence) and nucleoli (anti-C-23, green immunofluorescence). The merged images show no signs of co-localization. (C) Cells were fixed and co-stained for PDIP38 (red immunofluorescence) and spliceosomes (anti-SC35, green immunofluorescence). Images were captured at 100× magnification. PDIP38 and SC35 are co-localized, as shown for one of the cells by the white arrows.
We examined the dynamics of PDIP38 redistribution to nuclear speckles in response to different UV doses. A549 cells were treated with UV at 0, 10, and 30 J/m2 and the translocation of PDIP38 to the spliceosomes analyzed at 0, 1, 3, 6, 12, and 24 h by co-staining with anti-PDIP38 and anti-SC35. Data for the 10 and 30 J/m2 treatments are shown in Figure S2. No observable changes in distribution of PDIP38 were observed at the lowest dose of 10 J/m2 even up to 24 h post-treatment. PDIP38 was translocated to the spliceosomes at UV doses of 30 J/m2 (Fig. S2). Co-localization was already observed at 1 h, and by 3 h the co-localization of PDIP38 with SC35 was complete and remained so until 12 h.
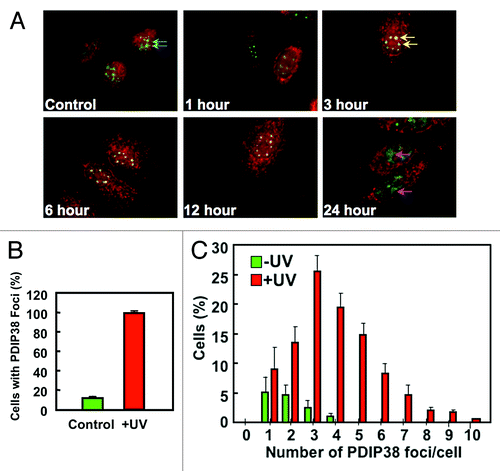
We then examined the co-localization of PDIP38 in more detail, using a UV dose of 60 J/m2. The merged images for the spliceosomes (green SC35 immunofluorescence) and PDIP38 (red immunofluorescence) at various time intervals after UV irradiation showed that translocation of PDIP38 was complete by 3 h (, yellow arrows) and remained so up to 12 h later (). By 24 h the co-localization was lost, and the SC35 immunofluorescence was present in large diffuse areas (, pink arrows), suggesting that apoptosis was taking place. Next, we performed a quantitative analysis of the recruitment of PDIP38 to the spliceosomes for the 3 h time point. Randomly chosen cells were visually scored for spliceosomes containing PDIP38 in the control and UV-treated cells 3 h post-irradiation. In the control cells, 12% of the cells had spliceosomes co-staining with PDIP38. PDIP38 was present in all the spliceosomes in the cells treated with UV (). Next, we determined the frequency of PDIP38 foci (PDIP38 containing spliceosomes) in individual cells, and plotted the number of cells against the number of foci per cell (). The number of PDIP38 foci induced by UV treatment ranged from 1–13 foci per cell, and only those in the range of 1–10 are shown (). The distribution of the number of PDIP38 foci ranged from 1–4 per cell in untreated cells, and the number of spliceosomes (all of which were PDIP38-positive) per cell was increased in the UV-treated cells. These changes in number are consistent with observations that spliceosomes become more numerous after UV irradiation and also undergo alterations of their morphology, viz., condensation and rounding.Citation38
Figure 4. Time course and duration of PDIP38 co-localization with SC35 after UV treatment. (A) A549 cells were globally irradiated with 60 J/m2 of UV and allowed to recover for the indicated times. Cells were fixed and co-stained for PDIP38 (red immunofluorescence) and SC35 (green immunofluorescence). Only the merged images for each time point are shown. Images were captured at 100× magnification. Blue arrows in the control panel indicate the green fluorescence of the spliceosomes. In the 3 h time point panel, the yellow arrows show that most of the spliceosomes exhibit co-localization of PDIP38 and SC35 immunofluorescence. Pink arrows in the 24 h time point panel show the large diffuse areas of SC35 green fluorescence that now do not show association with PDIP38. (B) Untreated A549 cells or globally irradiated (60 J/m2) cells were harvested 3 h post-treatment. Cells were fixed and stained as described above. Quantitation was performed by counting 100 cells in 4 separate experiments and scoring the percentage of cells exhibiting PDIP38 co-localization with SC53 in control (green bars) and UV-treated cells (red bars). Data are plotted as mean ± SD (n = 4). (C) A549 cells were treated with UV as described above. Quantitation was performed by counting the number of PDIP38 foci present in each individual cell. One hundred cells were counted in 4 separate experiments. The percentage of cells was plotted against the number of PDIP38-containing foci per cell (Control, green bars; UV-treated cells, red bars). Data are plotted as mean ± SD (n = 4).
Thus, we have demonstrated that PDIP38 undergoes translocation to the spliceosomes following UV irradiation. This represents a novel cellular response to UV irradiation.
Recruitment of PDIP38 to nuclear speckles in response to transcriptional stress
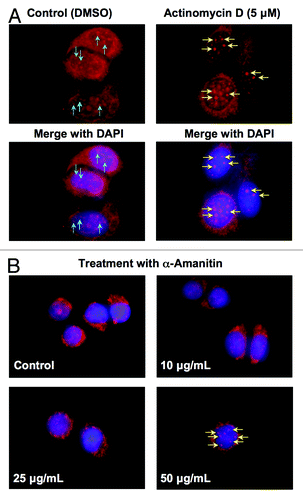
Spliceosomes are dynamic structures within the nucleus and are altered in response to the cellular environment, especially with respect to the transcriptional state of the cell. Many studies have identified changes of nuclear speckles in terms of the size and shape following transcription inhibition.Citation42 Since UV stress is known to cause global transcription inhibition,Citation43-Citation45 we wanted to assess whether transcription inhibition would induce the translocation of PDIP38. The effects of the transcriptional state on PDIP38 localization in the cell was assessed by the use of the transcription inhibitors actinomycin DCitation46 and α-amanitin.Citation47 Images for the localization of PDIP38 in control cells and MCF7 cells treated with 5 μM actinomycin D for 3 h showed that there was weak but visible localization of PDIP38 to the spliceosomes in the control cells (, blue arrows). After actinomycin D treatment, PDIP38 was translocated to the spliceosomes, as shown by strong intensification of the PDIP38 immunofluorescence (, yellow arrows). Since actinomycin D is a DNA intercalator and can potentially elicit the DNA damage response, the effects seen from the use of this compound may not be exclusively due to transcription inhibition. Therefore, we used α-amanitin, a potent inhibitor of RNA polymerase II,Citation47 to inhibit the transcription machinery. A549 cells were treated with various concentrations (10–50 μg/mL) of α-amanitin for 4 h. PDIP38 was recruited to spliceosomes at the highest concentration used (50 μg/mL) but not at lower concentrations (). These findings suggest that PDIP38 is sensitive to changes in transcriptional activity and are consistent with evidence that transcriptional stress can act as a sensor that activates DNA damage signaling pathways.Citation43,Citation44
Figure 5. Transcription inhibition induces translocation of PDIP38 to nuclear spliceosomes. (A) MCF7 cells were treated with 5 μM actinomycin D for 3 h. Top row: cells were fixed and stained for PDIP38 (red immunofluorescence). Bottom row: merged images of the cells co-stained for DNA with DAPI (blue immunofluorescence). Blue arrows show the staining of PDIP38 in the spliceosomes. Yellow arrows show the much more intense staining of the spliceosomes after actinomycin D treatment. (B) A549 cells were treated with α-amanitin (0, 10, 25, and 50 μg/mL) for 4 h. Cells were fixed and stained for PDIP38 (red immunofluorescence) and DAPI (blue immunofluorescence). The merged images are shown. The yellow arrows indicate the increase in intensity of PDIP38 immunofluorescence that occurs at 50 μg/mL of α-amanitin.
PDIP38 contributes to alternative splicing in response to UV-induced DNA damage
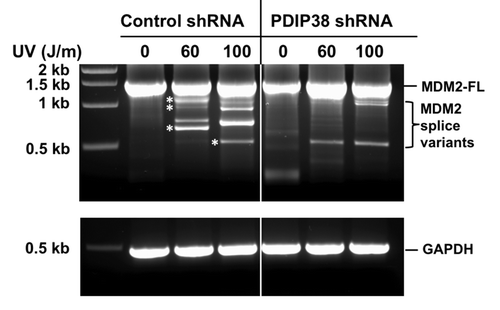
Nuclear speckles are nuclear compartments enriched with splicing factors that function as storage and assembly sites for splicing factors during transcription of pre-mRNAs that can occur along the periphery of nuclear speckles.Citation38,Citation39,Citation42 There are a number of genes whose transcription exhibits changes in alternative splicing in cancer cells and in cells exposed to genotoxic stress.Citation26,Citation48-Citation50 One example of a gene that is alternatively spliced in cancer cells is MDM2, which encodes the E3 ubiquitin ligase that regulates p53.Citation29,Citation31,Citation32 Alternative splicing of MDM2 transcripts is induced in cultured cells by UV and genotoxic stress.Citation26,Citation49,Citation51 The appearance of the splice variants has significance beyond their impact on p53 functions, since it has been shown that the truncated MDM2 proteins may exhibit growth-regulatory properties,Citation29 the ability to promote tumorigenesis in miceCitation52 and to transform cultured cells.Citation32 We therefore evaluated the potential participation of PDIP38 in alternative splicing of MDM2 in response to UV irradiation. A549 control and A549 PDIP38 shRNA-knockdown cells (“Materials and Methods”) were challenged with UV irradiation at 60 and 100 J/m2 and allowed to recover for 24 h post-treatment. RNA was isolated, and nested RT-PCR was used to amplify the MDM2 cDNAs using methods described by Matsumoto et al.Citation31 In control cells, only the full-length 1.5 kb MDM2 transcript was present. Upon UV treatment, MDM2 spliced variants were detected in A549 control cells treated with 60 and 100 J/m2 UV (). MDM2 splice variants represent truncations of the ca. 1.5 kb mature MDM2 transcript by exon skipping, and several of these have been well characterized as Mdm2-a (941 bp), Mdm2-b (707 bp), Mdm2-c (1016 bp), and Mdm2-d (449 bp).Citation31,Citation32 Bands with sizes approximating that of all 4 were observed after UV treatment (). Data shown in are representative of several such experiments. However, in the PDIP38 shRNA-knockdown cells, the appearance and amount of alternative spliced products was severely reduced (). Previous studies have shown that the alternative splicing of MDM2 can be observed at UV doses from 30–50 J/m2 over periods of 6–24 h, with variations in cell type.Citation49,Citation51 Overall, these findings show that PDIP38 is required for the UV induced alternative splicing of MDM2. These findings provide evidence for a novel function of PDIP38 that may be relevant to the molecular mechanisms that may underlie the regulation of alternative splicing of MDM2.
Figure 6. Effect of PDIP38 knockdown on alternative splicing of MDM2 induced by UV irradiation. (A) Control A549 cells (control shRNA) or A549 cells stably expressing shRNA targeted against PDIP38 (PDIP38 shRNA) were treated with 60 and 100 J/m2 of UV. Cells were harvested 24 h post-treatment. Total RNA was isolated and subjected to nested reverse transcription-PCR to detect MDM2 full-length and splice variants, as described in “Material and Methods”. Asterisks denote the bands that corresponded in size to those reported for the MDM2 splice variants, Mdm2-c, Mdm2-a, Mdm2-b, and Mdm2-d.Citation31 (Sizes of the bands were graphically determined from a plot of the marker DNA size against migration.)
Discussion
We have made several novel and unexpected findings regarding the cellular behavior of PDIP38 in response to UV-induced genotoxic stress, stemming from the initial goal of determining if it is involved in DNA repair or translesion synthesis. PDIP38 is not recruited to sites of UV damage in response to UV irradiation. However, PDIP38 was detected in some spliceosomes in unstressed cells, but upon UV irradiation PDIP38 was translocated and was present essentially in all the spliceosomes. This novel DNA damage response suggested that it might be involved in posttranscriptional processing of mRNAs, and we have shown for the first time that PDIP38 is involved in the UV-induced alteration in the posttranscriptional regulation of MDM2, viz., the generation of alternatively spliced MDM2 variants. MDM2 is a well-documented example of a gene whose transcripts are altered in response to genotoxic stress. The induction of MDM2 variants could therefore be considered as a cellular response to DNA damage or transcriptional stress. Moreover, a large number of splice variants of MDM2 have been found to be associated with human cancers. The role of MDM2 splice variants, which are found in cancer tissues and are also generated in response to genotoxic stress, is complex. MDM2 alternatively spliced forms have been proposed to have roles outside of p53 regulatory functionsCitation29 and can take on transforming capabilities that can promote unchecked cell division.Citation26,Citation29,Citation32,Citation48 Our studies therefore point to a role of PDIP38 in the regulation of the alternative splicing of MDM2 in response to genotoxic stress. These splicing variants may transduce cellular processes and reflect adaptations that contribute to the maintenance of genomic stability. However, little is known of how these posttranscriptional events involving MDM2 are mediated, or of the signaling processes that regulate their activation. It should be noted that the cellular functions of the truncated proteins remain an important area of investigation, since these are presumed to be expressed in many cancer cells.Citation29 Further studies are needed to determine the mechanisms of how PDIP38 participates in MDM2 alternative splicing in response to DNA damage, and whether this is specific to MDM2 or involves the splicing of other genes.
Unlike most well-defined RNA binding proteins, such as splicing factors, PDIP38 lacks a canonical RNA binding domain, such as the RNA recognition motif (RRM). One possibility is that PDIP38 can interact and mediate its effects on pre-mRNA processing through the existence of non-canonical RNA binding domains. Another possibility is that PDIP38 can influence mRNA processing by serving as a protein scaffold to allow protein–protein interactions that will facilitate splicing events. During splicing reactions, most splicing factors act via protein interactions to promote the stability of splicing complexes during normal and alternative splicing.Citation53,Citation54 In this regard, our findings that PDIP38 is required for alternative splicing of MDM2 suggests that investigation of its potential role in the manifestation of splice variants in cancer could lead to further insights into its functions.
Three other proteins have recently been reported to be recruited to the spliceosomes in response to UV, these being B-Myb, hOGG1, and APE1. B-Myb is a transcription factor involved in cell cycle progression and is recruited to nuclear speckles in response to UVC, concomitant with a switch in its phosphorylation patterns.Citation55 Interestingly, B-Myb has also been reported to be required for S-phase entry and to interact with Poldip1, a Pol δ-interacting protein.Citation56 hOGG1 is a DNA glycosylase involved in the initial step of base excision repair of oxidized purines, and APE1 catalyzes the next step. Both are recruited to nuclear speckles in response to UVA but not by UVC. Their recruitment was found to be dependent on the generation of reactive oxygen species.Citation57
Our studies open yet another window on the potential cellular functions of PDIP38. PDIP38 clearly exhibits a multifunctional behavior; its range of functions and various protein interactions and subcellular localizations points to its role in a gamut of remarkably diverse cellular activities. At the present time, there does not appear to be a single unifying property that can link all of these diverse functions.
One example of PDIP38 functions at the cellular and physiological level that has emerged is its role in vascular function, through its regulation of redox events and H2O2 levels via the NADPH oxidase, Nox4.Citation23,Citation24,Citation58 PDIP38 interacts with p22phox and activates Nox4 and positively regulates reactive oxygen species in vascular smooth muscle cells. These events impact focal adhesion, cytoskeletal remodeling and vascular smooth muscle migration. PDIP38 has also been shown to interact with the cell adhesion receptor CEACAM1, which regulates its shuttling between the cytoplasm and the nucleus.Citation19 PDIP38 was shown to be localized to the mitotic spindle, and PDIP38 loss of function impaired spindle organization and chromosomal segragation.Citation21
The cellular and physiological functions of PDIP38 remain poorly defined in relation to involvement in DNA replication/repair processes. A more direct role for PDIP38 in DNA replication and DNA damage tolerance mediated by TLS polymerases remains to be more fully delineated. The interaction of PDIP38 with Pol η, which suggests a role in translesion synthesis, needs to be re-assessed in the light of our findings that it does not localize to sites of UV damage, as this indicates that it does not directly participate in the process of translesion DNA synthesis. PDIP38 interacts with Pol η by binding to its UBZ (ubiquitin binding zinc finger) domain; this could act to prevent switching of Pol η at replication forks in the absence of damage to prevent its participation in normal DNA replication.Citation22 PDIP38 interaction with the UBZ domain of Pol ηCitation22 could also affect its recruitment and switching with Pol δ at UV damage sites.Citation59 The translocation of PDIP38 could lead to its sequestration from the nucleoplasm, providing a negative regulation of its functions in DNA replication or repair processes. The trafficking of proteins to and from nucleoliCitation36 and PML bodiesCitation35 includes examples of this type of regulation. PDIP38 interacts with Pol δ and PCNA, raising possibilities that it could modify Pol δ functions in DNA replication.Citation16 Much further research is needed in a more detailed investigation of its effects on the various enzymatic reactions involved in DNA replication and repair. In particular, determination of the crystal structure of PDIP38 may provide new insights into its properties and structural domains that could lead to a better understanding of its functions.
While our studies have focused on the effects of UVC on PDIP38, they are also relevant in general to the effects of UV irradiation from other components of the UV spectrum (UVA, UVB) that pose a risk for skin cancer from exposure to natural sunlight. While much of the UVC component of sunlight is blocked before reaching the surface of the earth, all 3 components of the UV spectrum produce damage to DNA through the production of CPDs and other photoproducts as well as the generation of reactive oxygen species.Citation60,Citation61
In conclusion, our studies have revealed a novel response of PDIP38, viz., its translocation to the spliceosomes as a relatively earlier and sustained event, in response to UV-induced genotoxic stress. Furthermore, we have shown that PDIP38 depletion affects the DNA damage-induced alternative splicing of MDM2. Taken together, these findings reveal another function for PDIP38. These new findings indicate that PDIP38 can respond to genotoxic or transcriptional stresses by undergoing translocation to the spliceosomes, where it is a required participant in the regulation of MDM2 alternative splicing. Our studies now open avenues for future work that could yield insights into the posttranscriptional regulation of MDM2 and result in a greater understanding of the functions of its splice variants in the DNA damage response as well as in growth control, which underlie their connection with tumorigenesis.
Materials and Methods
Cell culture
A549, HeLa S3 and MCF7 cells were obtained from ATCC and maintained according to protocols from the supplier. A549 cells are derived from human lung adenocarcinoma. HeLa S3 cells are a clonal HeLa cell line. MCF7 is a human breast adenocarcinoma cell line.
Treatments with DNA damaging agents and transcription inhibitors
UV radiation was delivered through a UVLMS-38EL series 3 UV lamp with a fluence rate of 2 J/m2/s. For global UV irradiation, cells were exposed to UVC (254 nM) at a range of doses. Cell culture media was removed, and cells were rinsed with PBS prior to UV treatment. For local UV irradiation, polycarbonate filters containing multiple 5–10 μm pores (Millipore) were placed in direct contact with cells after the media was removed and subjected to various doses of UVC irradiation.Citation10 Cells were replenished with fresh media immediately after treatment, and cells were allowed to recover between 1–24 h, depending on the experiment, to assess the dynamics of PDIP38 recruitment.
Indirect immunofluorescence microscopy
Indirect immunofluorescence microscopy was performed as previously described.Citation10 One day prior to treatment, cells were seeded onto 2-chamber slides (BD Falcon) at approximately 5 × 104 cells (60% confluent by the time of treatment) and incubated in a 37 °C incubator. After treatments as described above, cells were either fixed with 90% methanol overnight or with 4% paraformaldehyde for 15 min at room temperature. Cells were rinsed with PBS and post-fixed in 70% ethanol overnight at −20 °C. Cells were then permeabilized with 0.1% Triton X-100 in PBS for 10 min at room temperature and blocked with 2% bovine serum albumin (BSA) in PBS for 1 h. Primary antibodies were incubated overnight in 4 °C. Cells were washed 3 times with 2% BSA and incubated with secondary antibody conjugated with a fluorescent dye (Life Technologies) at a 1:200 dilution for 1 h at room temperature. AlexaFluor488 conjugated anti-mouse or anti-rabbit IgG and Rhodamine Red conjugated anti-mouse or anti-rabbit IgG were used for secondary antibodies. Slides were washed with 2% BSA and counterstained with 4’,6-diamidino-2-phenylindole (DAPI) for 10 min to stain the nucleus. Slides were visualized with a Zeiss Axiovert 200M microscope, and images were captured with a black and white CCD AxioCam and pseudocolored using AxioVision software (Release version 4.7).
PDIP38 polyclonal antibodies were raised in rabbits using recombinant full-length GST-PDIP38 (Proteintech). Mouse monoclonal antibodies against PCNA (74B1) were as previously described,Citation16 or PC10 from Santa Cruz Biotech. Antibodies for Pol η, p53, MDM2 were from Santa Cruz Biotech. Mouse anti-SC35 (S4045) was obtained from Sigma-Aldritch. Antibodies for p125 subunit of Pol δ were as previously described.Citation10 AlexaFlour488 and Rhodamine Red conjugated secondary antibodies were obtained from Life Technologies. Antibodies against PML protein and C23 were obtained from Santa Cruz Biotech.
Depletion and knockdown of PDIP38
Control and PDIP38 siRNAs were obtained from Invitrogen. Transient transfections were performed using Oligofectamine (Invtrogen). Cells were analyzed after 72 h. Cells with stable knockdown of PDIP38 were generated by transfecting A549 cells with a vector containing lentiviral-expressed short hairpin RNA targeting PDIP38 (PDIP38 shRNA). PDIP38 shRNA and control-shRNA were obtained from Open Biosystems. The shRNAs were packaged into lentivirus by co-transfection with the packaging plasmids Δ8.9 and VSVG into HEK293T cells. Forty-eight hours after transfection, the supernatant was filtered through a sterile 0.45 μm sterile filter and used to infect A549 cells. Following infection, knockdown cells were selected by the addition of 2.5 g/ml of puromycin for 1 wk. The cells were maintained in 0.5 μg/ml puromycin for successful expression of the shRNA. Cells were grown in DMEM medium supplemented with 10% fetal bovine serum and antibiotic/antimycotic mixture and maintained in a humidified 37 °C incubator with 5% CO2.
RNA isolation and cDNA synthesis
Total RNA was extracted from untreated and UV-treated cells using RNeasy mini-kits (Qiagen). Cells were directly lysed with Qiazol reagent, and homogenates were stored in −80°C. Total RNA was measured, and approximately 1 μg of RNA was used as a template for first-strand cDNA synthesis using Revertaid first-strand cDNA synthesis kits (Thermo Scientific) with random hexamers. The reaction mixture was incubated at 25 °C for 5 min. The reverse transcription reaction was at 42 °C for 1 h, and the reaction was terminated by increasing the temperature to 70 °C. cDNA products were immediately used for PCR to analysis of short spliced variants of MDM2 transcripts.
Analysis of MDM2 transcripts
The cDNA obtained by RT-PCR was amplified using the nested PCR primers described by Matsumoto et al.Citation31 The external sense and antisense primers were 5′-CTGGGGAGTC TTGAGGGACC-3′ and 5′-CAGGTTGTCT AAATTCCTAG-3′, respectively. The internal sense and antisense primers were 5′-GCGAAAACCC CGGATGGTGAG-3′ and 5′-CTCTTATAGA CAGGTCAACT AG-3′, respectively. PCR reactions were performed in a total volume of 25 μL containing 1 μL of cDNA from RT-PCR reaction with 1× reaction buffer, 10 pmol of each primer, 0.25 mM dATP, dGTP, dCTP, and dTTP and 0.15 U of Tag DNA polymerase (NEB, Inc). The nested PCR reactions for MDM2 amplification were performed in 2 stages, 1 μL of the first round PCR reaction with the external primers were seeded into the second round reaction with the internal primers. The thermocycler conditions consisted of an initial cycle of 95 °C for 5 min followed by 25 cycles of 95 °C for 30 s, 55 °C for 30 s, and 68 °C for 4.5 min, and a final extension for 8 min at 68 °C. The second PCR was performed the same way except with 35 cycles with internal primers. PCR products were resolved on a 1.5% agarose gel.
| Abbreviations: | ||
| PDIP38 | = | Polymerase delta interacting protein 38 |
| MDM2 | = | mouse double minute 2 homolog |
| Pol δ | = | DNA polymerase δ |
| NER | = | nucleotide excision repair |
| PCNA | = | proliferating cell nuclear antigen |
Additional material
Download Zip (1.4 MB)Acknowledgments
This work was supported by grants from the National Institutes of Health (GM31973 and ES14737) to MYWTL.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26221
Related Research Data
References
- Lee MY, Zhang S, Lin SH, Chea J, Wang X, LeRoy C, Wong A, Zhang Z, Lee EY. Regulation of human DNA polymerase delta in the cellular responses to DNA damage. Environ Mol Mutagen 2012; 53:683 - 98; http://dx.doi.org/10.1002/em.21743; PMID: 23047826
- Garg P, Burgers PM. DNA polymerases that propagate the eukaryotic DNA replication fork. Crit Rev Biochem Mol Biol 2005; 40:115 - 28; http://dx.doi.org/10.1080/10409230590935433; PMID: 15814431
- Kunkel TA, Burgers PM. Dividing the workload at a eukaryotic replication fork. Trends Cell Biol 2008; 18:521 - 7; http://dx.doi.org/10.1016/j.tcb.2008.08.005; PMID: 18824354
- Zhou Y, Meng X, Zhang S, Lee EY, Lee MY. Characterization of human DNA polymerase delta and its subassemblies reconstituted by expression in the MultiBac system. PLoS One 2012; 7:e39156; http://dx.doi.org/10.1371/journal.pone.0039156; PMID: 22723953
- Prelich G, Tan CK, Kostura M, Mathews MB, So AG, Downey KM, Stillman B. Functional identity of proliferating cell nuclear antigen and a DNA polymerase-delta auxiliary protein. Nature 1987; 326:517 - 20; http://dx.doi.org/10.1038/326517a0; PMID: 2882424
- Burgers PM. Polymerase dynamics at the eukaryotic DNA replication fork. J Biol Chem 2009; 284:4041 - 5; http://dx.doi.org/10.1074/jbc.R800062200; PMID: 18835809
- Balakrishnan L, Bambara RA. Okazaki fragment metabolism. Cold Spring Harb Perspect Biol 2013; 5:a10173; http://dx.doi.org/10.1101/cshperspect.a010173; PMID: 23378587
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007; 129:665 - 79; http://dx.doi.org/10.1016/j.cell.2007.05.003; PMID: 17512402
- Mocquet V, Lainé JP, Riedl T, Yajin Z, Lee MY, Egly JM. Sequential recruitment of the repair factors during NER: the role of XPG in initiating the resynthesis step. EMBO J 2008; 27:155 - 67; http://dx.doi.org/10.1038/sj.emboj.7601948; PMID: 18079701
- Chea J, Zhang S, Zhao H, Zhang Z, Lee EY, Darzynkiewicz Z, Lee MY. Spatiotemporal recruitment of human DNA polymerase delta to sites of UV damage. Cell Cycle 2012; 11:2885 - 95; http://dx.doi.org/10.4161/cc.21280; PMID: 22801543
- Ogi T, Limsirichaikul S, Overmeer RM, Volker M, Takenaka K, Cloney R, Nakazawa Y, Niimi A, Miki Y, Jaspers NG, et al. Three DNA polymerases, recruited by different mechanisms, carry out NER repair synthesis in human cells. Mol Cell 2010; 37:714 - 27; http://dx.doi.org/10.1016/j.molcel.2010.02.009; PMID: 20227374
- Overmeer RM, Gourdin AM, Giglia-Mari A, Kool H, Houtsmuller AB, Siegal G, Fousteri MI, Mullenders LH, Vermeulen W. Replication factor C recruits DNA polymerase delta to sites of nucleotide excision repair but is not required for PCNA recruitment. Mol Cell Biol 2010; 30:4828 - 39; http://dx.doi.org/10.1128/MCB.00285-10; PMID: 20713449
- Sebesta M, Burkovics P, Juhasz S, Zhang S, Szabo JE, Lee MY, Haracska L, Krejci L. Role of PCNA and TLS polymerases in D-loop extension during homologous recombination in humans. DNA Repair (Amst) 2013; 12:691 - 8; http://dx.doi.org/10.1016/j.dnarep.2013.05.001; PMID: 23731732
- Zhang W, Qin Z, Zhang X, Xiao W. Roles of sequential ubiquitination of PCNA in DNA-damage tolerance. FEBS Lett 2011; 585:2786 - 94; http://dx.doi.org/10.1016/j.febslet.2011.04.044; PMID: 21536034
- Sale JE, Lehmann AR, Woodgate R. Y-family DNA polymerases and their role in tolerance of cellular DNA damage. Nat Rev Mol Cell Biol 2012; 13:141 - 52; http://dx.doi.org/10.1038/nrm3289; PMID: 22358330
- Liu L, Rodriguez-Belmonte EM, Mazloum N, Xie B, Lee MY. Identification of a novel protein, PDIP38, that interacts with the p50 subunit of DNA polymerase delta and proliferating cell nuclear antigen. J Biol Chem 2003; 278:10041 - 7; http://dx.doi.org/10.1074/jbc.M208694200; PMID: 12522211
- Xie B, Li H, Wang Q, Xie S, Rahmeh A, Dai W, Lee MY. Further characterization of human DNA polymerase delta interacting protein 38. J Biol Chem 2005; 280:22375 - 84; http://dx.doi.org/10.1074/jbc.M414597200; PMID: 15811854
- Cheng X, Kanki T, Fukuoh A, Ohgaki K, Takeya R, Aoki Y, Hamasaki N, Kang D. PDIP38 associates with proteins constituting the mitochondrial DNA nucleoid. J Biochem 2005; 138:673 - 8; http://dx.doi.org/10.1093/jb/mvi169; PMID: 16428295
- Klaile E, Müller MM, Kannicht C, Otto W, Singer BB, Reutter W, Obrink B, Lucka L. The cell adhesion receptor carcinoembryonic antigen-related cell adhesion molecule 1 regulates nucleocytoplasmic trafficking of DNA polymerase delta-interacting protein 38. J Biol Chem 2007; 282:26629 - 40; http://dx.doi.org/10.1074/jbc.M701807200; PMID: 17623671
- Singer BB, Scheffrahn I, Obrink B. The tumor growth-inhibiting cell adhesion molecule CEACAM1 (C-CAM) is differently expressed in proliferating and quiescent epithelial cells and regulates cell proliferation. Cancer Res 2000; 60:1236 - 44; PMID: 10728682
- Klaile E, Kukalev A, Obrink B, Müller MM. PDIP38 is a novel mitotic spindle-associated protein that affects spindle organization and chromosome segregation. Cell Cycle 2008; 7:3180 - 6; http://dx.doi.org/10.4161/cc.7.20.6813; PMID: 18843206
- Tissier A, Janel-Bintz R, Coulon S, Klaile E, Kannouche P, Fuchs RP, Cordonnier AM. Crosstalk between replicative and translesional DNA polymerases: PDIP38 interacts directly with Poleta. DNA Repair (Amst) 2010; 9:922 - 8; http://dx.doi.org/10.1016/j.dnarep.2010.04.010; PMID: 20554254
- Lyle AN, Deshpande NN, Taniyama Y, Seidel-Rogol B, Pounkova L, Du P, Papaharalambus C, Lassègue B, Griendling KK. Poldip2, a novel regulator of Nox4 and cytoskeletal integrity in vascular smooth muscle cells. Circ Res 2009; 105:249 - 59; http://dx.doi.org/10.1161/CIRCRESAHA.109.193722; PMID: 19574552
- Sutliff RL, Hilenski LL, Amanso AM, Parastatidis I, Dikalova AE, Hansen L, Datla SR, Long JS, El-Ali AM, Joseph G, et al. Polymerase Delta Interacting Protein 2 Sustains Vascular Structure and Function. [Epub ahead of print] Arterioscler Thromb Vasc Biol 2013; 33:2154 - 61; http://dx.doi.org/10.1161/ATVBAHA.113.301913; PMID: 23825363
- Boucas J, Riabinska A, Jokic M, Herter-Sprie GS, Chen S, Höpker K, Reinhardt HC. Posttranscriptional regulation of gene expression-adding another layer of complexity to the DNA damage response. Front Genet 2012; 3:159; http://dx.doi.org/10.3389/fgene.2012.00159; PMID: 22936947
- Dutertre M, Sanchez G, Barbier J, Corcos L, Auboeuf D. The emerging role of pre-messenger RNA splicing in stress responses: sending alternative messages and silent messengers. RNA Biol 2011; 8:740 - 7; http://dx.doi.org/10.4161/rna.8.5.16016; PMID: 21712650
- David CJ, Manley JL. Alternative pre-mRNA splicing regulation in cancer: pathways and programs unhinged. Genes Dev 2010; 24:2343 - 64; http://dx.doi.org/10.1101/gad.1973010; PMID: 21041405
- Venables JP. Unbalanced alternative splicing and its significance in cancer. Bioessays 2006; 28:378 - 86; http://dx.doi.org/10.1002/bies.20390; PMID: 16547952
- Okoro DR, Rosso M, Bargonetti J. Splicing up mdm2 for cancer proteome diversity. Genes Cancer 2012; 3:311 - 9; http://dx.doi.org/10.1177/1947601912455323; PMID: 23150764
- Bartel F, Harris LC, Würl P, Taubert H. MDM2 and its splice variant messenger RNAs: expression in tumors and down-regulation using antisense oligonucleotides. Mol Cancer Res 2004; 2:29 - 35; PMID: 14757843
- Matsumoto R, Tada M, Nozaki M, Zhang CL, Sawamura Y, Abe H. Short alternative splice transcripts of the mdm2 oncogene correlate to malignancy in human astrocytic neoplasms. Cancer Res 1998; 58:609 - 13; PMID: 9485008
- Sigalas I, Calvert AH, Anderson JJ, Neal DE, Lunec J. Alternatively spliced mdm2 transcripts with loss of p53 binding domain sequences: transforming ability and frequent detection in human cancer. Nat Med 1996; 2:912 - 7; http://dx.doi.org/10.1038/nm0896-912; PMID: 8705862
- Harper JW, Elledge SJ. The DNA damage response: ten years after. Mol Cell 2007; 28:739 - 45; http://dx.doi.org/10.1016/j.molcel.2007.11.015; PMID: 18082599
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542 - 8; http://dx.doi.org/10.1126/science.1083430; PMID: 12791985
- Dellaire G, Bazett-Jones DP. Beyond repair foci: subnuclear domains and the cellular response to DNA damage. Cell Cycle 2007; 6:1864 - 72; http://dx.doi.org/10.4161/cc.6.15.4560; PMID: 17660715
- Emmott E, Hiscox JA. Nucleolar targeting: the hub of the matter. EMBO Rep 2009; 10:231 - 8; http://dx.doi.org/10.1038/embor.2009.14; PMID: 19229283
- Mayer C, Grummt I. Cellular stress and nucleolar function. Cell Cycle 2005; 4:1036 - 8; http://dx.doi.org/10.4161/cc.4.8.1925; PMID: 16205120
- Spector DL, Lamond AI. Nuclear speckles. Cold Spring Harb Perspect Biol 2011; 3; http://dx.doi.org/10.1101/cshperspect.a000646; PMID: 20926517
- Lamond AI, Spector DL. Nuclear speckles: a model for nuclear organelles. Nat Rev Mol Cell Biol 2003; 4:605 - 12; http://dx.doi.org/10.1038/nrm1172; PMID: 12923522
- Sakashita E, Endo H. SR and SR-related proteins redistribute to segregated fibrillar components of nucleoli in a response to DNA damage. Nucleus 2010; 1:367 - 80; http://dx.doi.org/10.4161/nucl.1.4.12683; PMID: 21327085
- Lin S, Xiao R, Sun P, Xu X, Fu XD. Dephosphorylation-dependent sorting of SR splicing factors during mRNP maturation. Mol Cell 2005; 20:413 - 25; http://dx.doi.org/10.1016/j.molcel.2005.09.015; PMID: 16285923
- Misteli T. Cell biology of transcription and pre-mRNA splicing: nuclear architecture meets nuclear function. J Cell Sci 2000; 113:1841 - 9; PMID: 10806095
- Ljungman M. The DNA damage response--repair or despair?. Environ Mol Mutagen 2010; 51:879 - 89; http://dx.doi.org/10.1002/em.20597; PMID: 20818630
- Ljungman M. The transcription stress response. Cell Cycle 2007; 6:2252 - 7; http://dx.doi.org/10.4161/cc.6.18.4751; PMID: 17700065
- Ljungman M, Zhang F, Chen F, Rainbow AJ, McKay BC. Inhibition of RNA polymerase II as a trigger for the p53 response. Oncogene 1999; 18:583 - 92; http://dx.doi.org/10.1038/sj.onc.1202356; PMID: 9989808
- Sobell HM. Actinomycin and DNA transcription. Proc Natl Acad Sci U S A 1985; 82:5328 - 31; http://dx.doi.org/10.1073/pnas.82.16.5328; PMID: 2410919
- Weinmann R, Raskas HJ, Roeder RG. Role of DNA-dependent RNA polymerases II and III in transcription of the adenovirus genome late in productive infection. Proc Natl Acad Sci U S A 1974; 71:3426 - 39; http://dx.doi.org/10.1073/pnas.71.9.3426; PMID: 4530313
- Bartel F, Taubert H, Harris LC. Alternative and aberrant splicing of MDM2 mRNA in human cancer. Cancer Cell 2002; 2:9 - 15; http://dx.doi.org/10.1016/S1535-6108(02)00091-0; PMID: 12150820
- Chandler DS, Singh RK, Caldwell LC, Bitler JL, Lozano G. Genotoxic stress induces coordinately regulated alternative splicing of the p53 modulators MDM2 and MDM4. Cancer Res 2006; 66:9502 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-05-4271; PMID: 17018606
- Jeyaraj S, O’Brien DM, Chandler DS. MDM2 and MDM4 splicing: an integral part of the cancer spliceome. Front Biosci (Landmark Ed) 2009; 14:2647 - 56; http://dx.doi.org/10.2741/3402; PMID: 19273224
- Dias CS, Liu Y, Yau A, Westrick L, Evans SC. Regulation of hdm2 by stress-induced hdm2alt1 in tumor and nontumorigenic cell lines correlating with p53 stability. Cancer Res 2006; 66:9467 - 73; http://dx.doi.org/10.1158/0008-5472.CAN-05-3013; PMID: 17018602
- Jones SN, Hancock AR, Vogel H, Donehower LA, Bradley A. Overexpression of Mdm2 in mice reveals a p53-independent role for Mdm2 in tumorigenesis. Proc Natl Acad Sci U S A 1998; 95:15608 - 12; http://dx.doi.org/10.1073/pnas.95.26.15608; PMID: 9861017
- Manley JL, Tacke R. SR proteins and splicing control. Genes Dev 1996; 10:1569 - 79; http://dx.doi.org/10.1101/gad.10.13.1569; PMID: 8682289
- Shin C, Manley JL. The SR protein SRp38 represses splicing in M phase cells. Cell 2002; 111:407 - 17; http://dx.doi.org/10.1016/S0092-8674(02)01038-3; PMID: 12419250
- Werwein E, Dzuganova M, Usadel C, Klempnauer KH. B-Myb switches from Cyclin/Cdk-dependent to Jnk- and p38 kinase-dependent phosphorylation and associates with SC35 bodies after UV stress. Cell Death Dis 2013; 4:e511; http://dx.doi.org/10.1038/cddis.2013.36; PMID: 23449447
- Werwein E, Schmedt T, Hoffmann H, Usadel C, Obermann N, Singer JD, Klempnauer KH. B-Myb promotes S-phase independently of its sequence-specific DNA binding activity and interacts with polymerase delta-interacting protein 1 (Pdip1). Cell Cycle 2012; 11:4047 - 58; http://dx.doi.org/10.4161/cc.22386; PMID: 23032261
- Campalans A, Amouroux R, Bravard A, Epe B, Radicella JP. UVA irradiation induces relocalisation of the DNA repair protein hOGG1 to nuclear speckles. J Cell Sci 2007; 120:23 - 32; http://dx.doi.org/10.1242/jcs.03312; PMID: 17148573
- Lassègue B, Griendling KK. NADPH oxidases: functions and pathologies in the vasculature. Arterioscler Thromb Vasc Biol 2010; 30:653 - 61; http://dx.doi.org/10.1161/ATVBAHA.108.181610; PMID: 19910640
- Lehmann AR. Replication of damaged DNA by translesion synthesis in human cells. FEBS Lett 2005; 579:873 - 6; http://dx.doi.org/10.1016/j.febslet.2004.11.029; PMID: 15680966
- Sage E, Girard PM, Francesconi S. Unravelling UVA-induced mutagenesis. Photochem Photobiol Sci 2012; 11:74 - 80; http://dx.doi.org/10.1039/c1pp05219e; PMID: 21901217
- Ikehata H, Ono T. The mechanisms of UV mutagenesis. J Radiat Res 2011; 52:115 - 25; http://dx.doi.org/10.1269/jrr.10175; PMID: 21436607