Abstract
TP53 protects cells from transformation by responding to stresses including aneuploidy and DNA double-strand breaks (DSBs). TP53 induces apoptosis of lymphocytes with persistent DSBs at antigen receptor loci and other genomic loci to prevent these lesions from generating oncogenic translocations. Despite this critical function of TP53, germline Tp53−/− mice succumb to immature T-cell (thymic) lymphomas that exhibit aneuploidy and lack clonal translocations. However, Tp53−/− mice occasionally develop B lineage lymphomas and Tp53 deletion in pro-B cells causes lymphomas with oncogenic immunoglobulin (Ig) locus translocations. In addition, human lymphoid cancers with somatic TP53 inactivation often harbor oncogenic IG or T-cell receptor (TCR) locus translocations. To determine whether somatic Tp53 inactivation unmasks translocations or alters the frequency of B lineage tumors in mice, we generated and analyzed mice with conditional Tp53 deletion initiating in hematopoietic stem cells (HSCs) or in lineage-committed thymocytes. Median tumor-free survival of each strain was similar to the lifespan of Tp53−/− mice. Mice with HSC deletion of Tp53 predominantly succumbed to thymic lymphomas with clonal translocations not involving Tcr loci; however, these mice occasionally developed mature B-cell lymphomas that harbored clonal Ig translocations. Deletion of Tp53 in thymocytes caused thymic lymphomas with aneuploidy and/or clonal translocations, including oncogenic Tcr locus translocations. Our data demonstrate that the developmental stage of Tp53 inactivation affects karyotypes of lymphoid malignancies in mice where somatic deletion of Tp53 initiating in thymocytes is sufficient to cause thymic lymphomas with oncogenic translocations.
Keywords: :
Introduction
The TP53 tumor suppressor maintains cellular homeostasis in response to a wide range of stresses, including aneuploidy, DSBs, genomic instability, and oncogene activation.Citation1,Citation2 These stresses stabilize and activate TP53, leading to changes in expression of target genes, including those involved in cell cycle control and apoptosis.Citation1,Citation2 TP53 is the most frequently inactivated tumor suppressor gene, with mutation or deletion occurring in over 60% of all human cancers,Citation1,Citation3 indicating that TP53 prevents malignant transformation of multiple cell types. Although TP53 inactivation occurs less frequently in lymphoid malignancies than in solid tumors, TP53 loss is more common in aggressive lymphoma subtypes and correlates with increased tumor grade, treatment resistance, and poor patient survival.Citation4-Citation6
Lymphocyte development involves cellular proliferation and antigen receptor gene assembly. Bone marrow HSCs differentiate into early progenitor B cells that remain in the bone marrow or into early thymic progenitors that migrate to the thymus. These cells proliferate and differentiate into pro-B or pro-T cells, respectively, which induce expression of the RAG1/RAG2 (RAG) endonuclease.Citation7,Citation8 RAG catalyzes the assembly of TCR and Ig genes in G1 phase cells through induction of DSBs at variable (V), diversity (D), and joining (J) gene segments.Citation9,Citation10 Non-homologous end-joining (NHEJ) factors repair these DSBs to generate V(D)J coding joins that encode the first exons of TCR and Ig genes.Citation10,Citation11 Assembly of TCRβ, TCRγ, and TCRδ genes occurs in CD4−CD8− “double-negative” (DN) pro-T cells.Citation12,Citation13 Expression of functional TCRγ and TCRδ genes signals differentiation into mature γδ T cells.Citation14 In contrast, expression of functional TCRβ genes triggers proliferation as cells differentiate into CD4+CD8+ “double positive” (DP) thymocytes.Citation15 In DP thymocytes, TCRα gene assembly followed by αβ TCR selection permit differentiation into CD4+ or CD8+ “single positive” (SP) thymocytes that exit the thymus as naive αβ T cells.Citation12,Citation16 Assembly and expression of IgH genes in pro-B cells drives proliferation as cells differentiate into pre-B cells, which must recombine either Igκ or Igλ genes to differentiate into immature B cells that exit the bone marrow and migrate to the spleen as they mature.Citation13,Citation17,Citation18 In response to antigen, mature B cells proliferate and undergo IgH class switch recombination (CSR) through DSB intermediates.Citation19-Citation22 In addition to programmed DSBs in antigen receptor loci, lymphocytes experience spontaneous DSBs that arise from errors in DNA replication during periods of proliferation.Citation23
TP53 inactivation occurs in human B and T lineage lymphomas containing aneuploidy as well as in those exhibiting genomic instability,Citation3,Citation24 suggesting that functions of TP53 in response to chromosome missegregation and DSBs are each important to suppress transformation of differentiating lymphocytes. Aberrant segregation of chromosomes during cellular division leads to TP53-dependent apoptosis of ensuing aneuploid daughter cells.Citation25 Induction of DSBs stabilizes and activates TP53, which promotes temporary cell cycle arrest to provide cells time to repair these lesions or induce apoptosis if they cannot be repaired.Citation2 Germline Tp53 inactivation in mice leads to aneuploidy and genomic instability in differentiating and mature lymphocytesCitation26 and enables pro-T and pro-B cells with un-repaired RAG-induced Tcrδ and IgH locus DSBs to survive, progress into S phase, and generate translocations.Citation27-Citation31 Despite roles of TP53 in response to both chromosome missegregation and DSBs, most Tp53−/− mice succumb to aneuploid TCRβ+ thymic lymphomas that lack Tcr translocations, though a small fraction succumb to B-cell lymphomas that have not been assayed for translocations.Citation32-Citation37 However, mice with combined germline inactivation of Tp53 and NHEJ factors reproducibly succumb to pro-B cell lymphomas with RAG-dependent IgH translocations that amplify the c-Myc oncogeneCitation28,Citation38,Citation39 and occasionally develop TCRβ− thymic lymphomas with Tcrδ translocations.Citation40 In addition, Tp53−/− mice with germline inactivation of the H2ax DSB repair factor predominantly succumb to TCRβ− thymic lymphomas with clonal translocations not involving Tcr loci, but occasionally develop TCRβ− lymphomas with Tcrα/δ translocations or pro-B cell lymphomas with Igh;c-myc translocations.Citation21,Citation36 Furthermore, on a genetic background with a block in αβ T-cell development at the DN stage, germline Tp53 deficiency causes TCRβ− thymic lymphomas with RAG-dependent translocations that may involve Tcrδ or IgH loci.Citation41 Together, these studies showed that TP53 functions in response to chromosome missegregation and DSBs are critical to prevent transformation of differentiating lymphocytes. However, they concluded that Tp53 only suppresses oncogenic translocations in cells with DSB repair or differentiation impaired.
Cancers develop through somatic acquisition and selection of mutations such as TP53 inactivation and other oncogenic lesions.Citation42 We previously showed that, while germline H2ax−/−Tp53−/− mice succumb to TCRβ− thymic lymphomas with clonal translocations, conditional deletion of H2ax and Tp53 in mouse DN thymocytes prolongs lifespan and leads to TCRβ+ thymic lymphomas.Citation21,Citation36,Citation43 That somatic inactivation of H2ax and Tp53 causes more mature thymic lymphomas with longer latency compared with germline inactivation indicates that oncogenic lesions prior to T cell commitment drive transformation of thymocytes. We recently showed that conditional deletion of Tp53 in pro-B cells predisposes mice to B lineage lymphomas with oncogenic translocations, including Igh;c-myc and other Ig translocations.Citation44 The tumor-free survival of these mice is similar to that of Tp53−/− mice, suggesting that development of thymic lymphomas from aneuploidy prevents B-cell lymphomas from oncogenic Ig translocations in Tp53−/− mice. To determine whether somatic inactivation of Tp53 unmasks translocations or alters the frequency of B lineage lymphomas in mice, we generated mice with conditional Tp53 deletion initiating in HSCs or DN thymocytes. These strains each succumbed at similar ages to thymic lymphomas, although HSC deletion occasionally caused B lineage lymphomas. HSC deletion of Tp53 led to clonal translocations not involving Tcr loci in thymic lymphomas and Igh translocations in B lymphomas, while DN deletion of Tp53 caused thymic lymphomas with aneuploidy and/or clonal translocations including Tcrα/δ locus translocations. Our data demonstrate that the developmental stage of Tp53 inactivation affects karyotypes of lymphoid cancers in mice, where somatic deletion of Tp53 initiating in thymocytes is sufficient to cause thymic lymphomas with oncogenic translocations.
Results
Conditional deletion of Tp53 in HSCs or thymocytes predisposes mice to thymic lymphomas
To determine whether Tp53 inactivation initiating in HSCs or thymocytes predisposes mice to lymphoma, we established and characterized Vav–cre+/−p53flox/flox (VP) and Lck–cre+/−p53flox/flox (LP) mice. Vav-cre and Lck-cre induce deletion of “floxed” genes in HSCs and DN thymocytes, respectively.Citation45,Citation46 We detected nearly complete deletion of Tp53 in bone marrow cells and thymocytes of VP mice and in thymocytes of LP mice, but no Tp53 deletion in LP bone marrow cells (Fig. S1), confirming the expected developmental timing of Tp53 inactivation in VP and LP mice. We also found grossly normal T and B cell development in VP and LP mice as compared with age-matched, wild-type controls (Fig. S2), consistent with the phenotype of Tp53−/− mice.Citation47 We generated and aged cohorts of 22 VP and 20 LP mice to evaluate their spontaneous predisposition to cancer. We observed that cohort VP mice survived cancer-free between 91–365 d with median age of mortality of 144.5 d, whereas cohort LP mice survived cancer-free between 70–365 d with a median age of mortality of 119 d (). The median ages of cancer-free survival of cohort VP and LP mice were not statistically significant (p = 0.134) and were comparable to the survival of germline Tp53−/− mice.Citation32,Citation33 All VP and LP mice succumbed to lymphoma, except for one LP mouse that was euthanized due to development of a sarcoma and one mouse of each genotype that died without lymphoma ( and ). Most VP mice and LP mice succumbed to lymphomas that were only visible in the thymus ( and ). However, 3 VP mice (nos. 202, 228, and 623) and 2 LP mice (nos. 119 and 983) had lymphoma cells in their spleens, while one VP mouse (no. 602) succumbed to a disseminated lymphoma found in the thymus, spleen, and multiple lymph nodes ( and ). In addition, 2 VP mice (nos. 421 and 426) succumbed to lymphomas that were located in the spleen and multiple lymph nodes but not visible in the thymus. We did not characterize cohorts of Vav–cre+/−, Lck–cre+/−, or p53flox/flox mice, since none of these mice exhibits increased tumor predisposition.Citation45,Citation46,Citation48 Consistent with this notion, none of the Vav–cre+/−, Lck–cre+/−, or p53flox/flox mice we used for breeding until 1 y of age developed cancer.
Figure 1. Mice with conditional inactivation of Tp53 initiating in HSCs or DN thymocytes reproducibly succumb to thymic lymphomas. (A) Kaplan–Meier curves comparing tumor-free survival of 22 VP and 20LP mice. All tumors were thymic lymphomas other than 2 VP mice that succumbed to B lineage lymphomas (indicated by asterisks). Additionally, one LP mouse succumbed to a sarcoma, and one mouse of each cohort did not succumb to any tumors by 1 year of age. Curves were compared using the log-rank (Mantel–Cox) test, P = 0.134 (B) Flow cytometry analyses of VP lymphomas no 118 and 975 and of LP lymphomas no. 740 and 976 showing their surface expression of CD3 and TCRβ or CD4 and CD8. Gates were drawn on normal T cells analyzed in parallel with the lymphomas. The percentages of cells in each gate are indicated. (C) Flow cytometry analyses of VP lymphomas no. 421 and 426 showing their surface expression of B220 and IgM or Igκ and Igλ. Gates were drawn on normal B cells analyzed in parallel with lymphomas. The percentages of tumor cells in each gate are indicated.
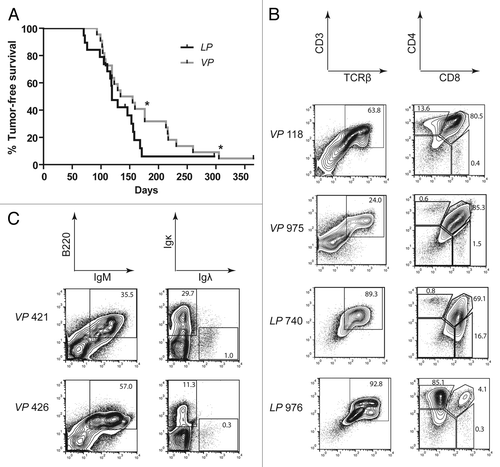
Table 1. Summary of VP tumor cohort
Table 2. Summary of LP tumor cohort
Tp53 inactivation in HSCs or thymocytes causes clonal immature T-cell lymphomas
To determine the lymphocyte lineages and developmental stages to which inactivation of Tp53 in HSCs or DN thymocytes causes cellular transformation, we first analyzed VP and LP lymphomas by flow cytometry using antibodies that recognize cell surface markers of specific lymphocyte lineages and developmental stages. For lymphomas found within the thymus, we assessed cell surface expression of TCRδ, TCRβ, CD4, CD8, and the CD3ε molecule through which αβ TCRs signal.Citation15 For lymphomas found in lymph nodes, we assessed cell surface expression of the B220, IgM, Igκ, and Igλ molecules. Due to the variation in cell staining observed within some tumors, we simplified our classification by denoting a lymphoma as positive for an epitope if more than half of the cells fell within a positive gate established from flow cytometry of non-malignant lymphocytes. Of the 17 VP thymic lymphomas assayed, most were TCRβ+CD3+CD4+CD8+, but a substantial number were also TCRβ−CD3− with or without CD4 and CD8 expression (; ; Fig. S3). Lymphoma cells in the spleens of the 2 VP mice (nos. 421 and 426) that lacked thymic lymphomas were B220+IgM+ (; ; Fig. S3). Lymphoma no. 421 contained Igκ+ and Igκ− cells, while lymphoma no. 426 contained mostly Igκ− cells (; ; Fig. S3). Of the 15 LP thymic lymphomas assayed, 10 were TCRβ+CD3+CD4+CD8+; only one was TCRβ−CD3− ( and ; Fig. S3). Many VP and LP thymic lymphomas displayed subpopulations with different expression patterns of TCRβ, CD3, CD4, and/or CD8, indicating that they represent either oligoclonal lymphomas arising from distinct initiating cells or clonal lymphomas with subpopulations that have differentially silenced and/or re-expressed genes. VP thymic lymphomas more often showed TCRβ, CD4, CD8, and CD3 expression characteristic of immature T-cell developmental stages than LP thymic lymphomas. Collectively, our flow cytometry analysis of VP and LP lymphomas indicates that deletion of Tp53 in mouse HSCs or DN thymocytes causes predominantly immature T-cell lymphomas, with HSC deletion leading to a higher percentage of tumors from an earlier T-cell developmental stage and occasional mature B-cell lymphomas.
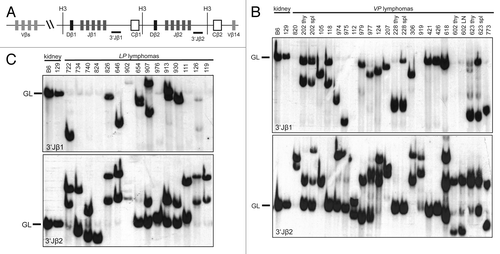
To determine the lymphocyte lineage of the 4 TCRβ−CD3−CD4−CD8− VP thymic lymphomas and to distinguish between oligoclonal and clonal immature T cell lymphomas in VP and LP mice, we next analyzed Tcrβ gene rearrangements in VP and LP lymphomas. For DN thymocytes to survive and differentiate, Tcrβ rearrangements must occur on one allele.Citation49 To characterize Tcrβ rearrangements, we conducted Southern blotting on HindIII-digested genomic DNA of VP and LP lymphomas with 3′Jβ1 and 3′Jβ2 probes (). We isolated DNA from the tumor-containing organ without further purification, under assumption that most cells were malignant. This was confirmed by flow cytometry. Further, this approach does not detect unique rearrangements in single cells, such as those occurring in normal lymphocytes. Thus, only germline configurations in non-lymphoid cells will be detected as background. We detected Tcrβ rearrangements in the TCRβ−CD3−CD4−CD8− VP thymic lymphomas (nos. 228, 602, 618, and 773) (), demonstrating that these malignancies are immature T cell lymphomas. We also found that 16 of 18 VP and all 15 LP thymic lymphomas analyzed contained one or two rearranged Tcrβ alleles and therefore arose from the expansion of a single cancer-initiating cell (). These data indicate that the diverse expression of surface epitopes observed within some VP and LP lymphomas represents tumor subpopulations that have differentially silenced and/or re-expressed these genes. The remaining 2 VP thymic lymphomas (nos. 124 and 618) contained 3 Tcrβ rearrangements (), indicating that these cancers either arose from the expansion of two cancer-initiating cells or one cancer-initiating cell that continued to assemble Tcrβ genes after malignant transformation. We also performed Southern analysis of Tcrβ rearrangements on VP lymphomas found in the spleen or lymph nodes of mice. We found that 3 of these 4 VP lymphomas analyzed contained the same Tcrβ rearrangements as the thymic lymphomas from the same animals (), demonstrating that these mice succumbed to a disseminated clonal immature T cell lymphoma. VP lymphoma (no. 202) displayed an additional Tcrβ rearrangement that was not present in the thymic lymphoma of this mouse (), suggesting that this splenic lymphoma developed from the thymic lymphoma in association with ongoing Tcrβ rearrangement. Our Southern analysis of Tcrβ rearrangements in VP and LP lymphomas indicates that inactivation of Tp53 in mouse HSCs or DN thymocytes causes mainly clonal immature T-cell malignancies.
Figure 2. Mice with conditional inactivation of Tp53 initiating in HSCs or DN thymocytes develop clonal thymic lymphomas. (A) Schematic of the mouse TCRβ locus showing relative locations of representative upstream Vβs, the 2 Dβ-Jβ-Cβ clusters, and the Vβ14 segment. The positions of the HindIII restriction sites (H3) and 3′Jβ1 and 3′Jβ2 probes used for Southern analyses are also shown. (B and C). Southern blot analysis of HindIII-digested DNA isolated from the indicated (B) VP or (C) LP lymphomas or from kidneys of C57BL/6 (B6) or 129/SvEv (129) control mice using the 3′Jβ1 or 3′Jβ2 probe. Germline (GL) bands for each probe are indicated. Membranes were hybridized with 3′Jβ1 probes, stripped, and then hybridized with 3′Jβ2 probes to reveal which lymphomas contained Vβ rearrangements to the Dβ2-Jβ2 cluster on both alleles.
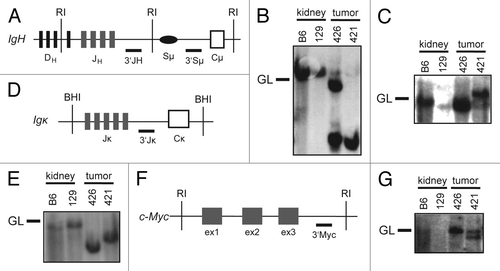
To determine whether the 2 B lineage lymphomas that arose in VP cohort mice were clonal and whether they arose from developing or mature B cells, we analyzed Igh and Igκ rearrangements in these tumors. To characterize Igh rearrangements, we conducted Southern blot analysis of EcoRI-digested genomic DNA of VP lymphomas nos. 421 and 426 with 3′JH and 3′Sμ probes (). We found that these tumors contained one (no. 426) or two (no. 421) JH rearrangements and therefore arose from the clonal expansion of a single cancer-initiating cell (). We also detected Sμ recombination in lymphoma no. 421 (), suggesting that this tumor may have developed from a B cell that had attempted CSR. Southern blot analysis of BamHI-digested genomic DNA from VP lymphomas nos. 421 and 426 with the 3′Jκ probe showed that each of these tumors contained Jκ rearrangements (). This suggests that IgM+Igκ− VP lymphoma no. 426 developed from a B lymphocyte that had developed at least to the pre-B cell stage. Our Southern analysis of these 2 VP B lineage lymphomas indicates that deletion of Tp53 in mouse HSCs can cause clonal B lineage lymphomas.
Figure 3. Mice with conditional inactivation of Tp53 initiating in HSCs also develop clonal B lineage lymphomas. (A) Schematic of the mouse IgH locus showing relative locations of representative DH segments, the 4 JH segments, the Sμ region, and the first CH exon, Cμ. The positions of the EcoRI restriction sites (RI) and 3′JH and 3′Sμ probes used for Southern blots are also shown. (B and C) Southern blot analysis of EcoRI-digested DNA isolated from VP lymphomas no. 421 or 426 or from kidneys of C57BL/6 (B6) or 129/SvEv (129) control mice using the (B) 3′JH and (C) 3′Sμ probes. Germline (GL) bands for each probe are indicated. (D) Schematic of the mouse Igκ locus showing relative locations of the 5 Jκ segments and the Cκ exon. The positions of the BamHI restriction sites (BHI) and 3′Jκ probe used for Southern blots are also shown. (E) Southern blot analysis of BamHI-digested DNA isolated from VP lymphomas no. 421 or 426 or from kidneys of C57BL/6 (B6) or 129/SvEv (129) control mice using the 3′Jκ probe. Germline (GL) band for the 3′Jκ probe is indicated. (F) Schematic of the mouse c-Myc locus showing relative locations of the three c-Myc exons, and of the EcoRI restriction sites (RI) and 3′Myc probe used for Southern blots. (G) Southern blot analysis of EcoRI-digested DNA isolated from VP lymphomas no. 421 or 426 or from kidneys of C57BL/6 (B6) or 129/SvEv (129) control mice using the 3′Myc probe. Germline (GL) band for the 3′Myc probe is indicated.
Conditional inactivation of Tp53 in HSCs or thymocytes causes lymphomas with clonal translocations
To determine whether conditional deletion of Tp53 initiating in HSCs or DN thymocytes causes lymphomas with chromosomal translocations, we conducted spectral karyotyping (SKY) on 7 VP and eight LP lymphomas. SKY is a molecular cytogenetic approach to visualize all chromosomes in a single metaphase spread to identify translocations and fusions between chromosomes.Citation50 We defined clonal translocations as those found in greater than half of the metaphases analyzed for a given tumor. SKY revealed that 2 of the 5 VP thymic lymphomas (nos. 118 and 820) analyzed had clonal translocations, with lymphoma no. 773 containing 3 different clonal chromosome fusions (; ; Fig. S4). One VP thymic lymphomas (no. 975) contained two non-clonal chromosome fusions (; Fig. S4). The remaining VP thymic lymphoma (no. 207) lacked translocations but exhibited aneuploidy (; Fig. S4). None of the clonal translocations in VP thymic lymphomas involved chromosomes on which Tcr or Ig loci reside. In contrast, SKY revealed that both VP B lymphomas contained clonal translocations involving chromosomes on which the Igh (chromosome 12 in no. 421) or Igκ (chromosome 6 in no. 426) locus resides (; ; Fig. S4). SKY also demonstrated that 3 of the 8 LP thymic lymphomas (nos. 826, 902, and 976) analyzed harbored clonal translocations, with lymphoma no. 902 also containing a clonal chromosome fusion (; ; Fig. S4). Notably, 2 of these tumors (no. 902 and 976) contained clonal translocations involving chromosome 14, which carries the Tcrα/δ locus. The other 5 LP thymic lymphomas lacked translocations but exhibited aneuploidy (). Our SKY analysis shows that conditional deletion of Tp53 deletion in HSCs or thymocytes causes lymphomas with clonal translocations, with thymocyte deletion also causing aneuploid lymphomas.
Figure 4. Mice with conditional inactivation of Tp53 initiating in HSCs or DN thymocytes develop lymphomas with oncogenic antigen receptor locus translocations. (A) Cytogenetic analysis of a metaphase from VP lymphoma no. 820 with the clonal t(2;17) translocation circled or isolated. (A, i) Spectral image. (A, ii) DAPI image. (A, iii) Karyotype table. (B) Cytogenetic analyses of a metaphase or chromosome from VP lymphoma no. 421 with the clonal t(12;15) translocation circled. (B, i) SKY image. (B, ii) DAPI image. (B, iii) Karyotype table. (B, iv). SKY (left) or FISH image (right) of the t(12;15) translocation hybridized with 3′IgH (green) and c-Myc (red) probes. (C) Cytogenetic analyses of a metaphase or chromosome from LP lymphoma no. 902 with the clonal t(4;15;14) translocation circled or isolated. (B, i) SKY image. (B, ii) DAPI image. (B, iii) Karyotype table. (B, iv) SKY (top) or FISH images of the t(4;15;14) translocation. (middle) hybridized with TCRVδ3/Vα6 (green) and TCRCα (red) probes. (bottom) hybridized with c-Myc (green) probe.
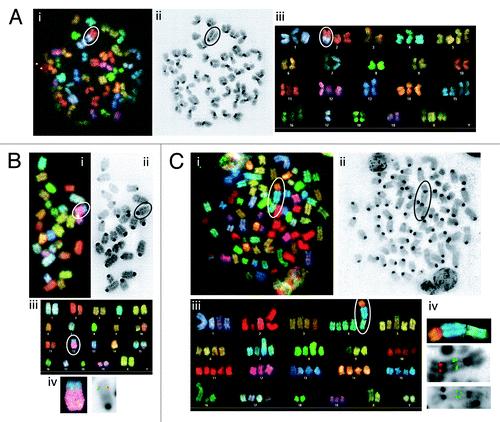
To determine whether conditional deletion of Tp53 initiating in HSCs or DN thymocytes causes lymphomas with clonal Ig or Tcr translocations, respectively, we conducted FISH on the 4 tumors with potential clonal Igh, Igκ, or Tcrα/δ translocations. For this purpose, we hybridized 5′ and 3′ Ig or Tcr locus probes and identified Ig or Tcr translocations by detection of probe signals on different chromosomes (). Since the c-myc oncogene on chromosome 15 is activated by Igh or Tcrα/δ translocations in mouse lymphomas,Citation40,Citation44 we also used a c-myc probe to identify potential Igh;c-myc and Tcrα/δ;c-myc translocations in VP lymphoma no. 421 and LP lymphoma no. 902. FISH revealed co-localization of 3′Igh and c-myc probe signals on one chromosome derivative in VP B lineage lymphoma no. 421 (), indicating that the clonal t(12;15) translocation in this tumor is an Igh;c-myc translocation. Unfortunately, we were unable to determine potential involvement of the Igκ locus in the clonal t(6;4) translocation of VP B lineage lymphoma no. 426 due to insufficient numbers of metaphases from this tumor. FISH revealed splitting of 5′ and 3′ probes on the clonal t(16;14) translocation in metaphases from LP thymic lymphoma no. 976 (Fig. S4 and not shown), indicating that this translocation tumor involves the Tcrα/δ locus. Finally, FISH showed co-localization of multiple copies each of 5′Tcrα/δ and c-myc probe signals on one chromosome derivative in metaphases from LP thymic lymphoma no. 902 ( and not shown), indicating that the clonal t(14;15;4) translocation in this tumor is a Tcrα/δ;c-myc translocation with amplification of the c-myc oncogene and Tcrα sequences. Our FISH analysis of metaphases from VP and LP lymphomas demonstrates that deletion of Tp53 initiating in HSCs or DN thymocytes can cause, respectively, B lineage lymphomas with oncogenic Igh translocations or thymic lymphomas with oncogenic Tcrα/δ translocations.
Discussion
Our study demonstrates that the context of Tp53 inactivation influences lymphoma predisposition, consistent with diverse roles of Tp53 throughout lymphocyte development and its interactions with known oncogenes. For example, Tp53 is important for termination of activated T-cell responses through induction of the pro-apoptotic SAP protein,Citation51 and inactivation of one or both copies of Tp53 synergizes with C-myc and MafB oncogenes in mouse models of mature B cell lymphomas.Citation52-Citation54 However, our data adds to our understanding of how Tp53 suppresses lymphoid malignancies by demonstrating that developmental timing of Tp53 inactivation in the absence of enforced oncogene expression can alter characteristics of resulting lymphomas. We generated mice with conditional deletion of Tp53 initiating in HSCs or in DN thymocytes and compared their tumor predisposition to the well-characterized cancer phenotype of germline Tp53-deficient mice. We found that HSC initiation of Tp53 inactivation predisposed mice to predominantly thymic lymphomas with clonal translocations not involving antigen receptor loci and occasionally to peripheral B cell lymphomas with clonal Ig translocations. Inactivation of Tp53 in DN thymocytes predisposed mice to thymic lymphomas that exhibited aneuploidy or contained clonal translocations frequently involving Tcrα/δ loci. In contrast, germline inactivation of Tp53 predisposes mice to aneuploid thymic lymphomas lacking clonal translocations.Citation32,Citation33,Citation36 Other than the timing of Tp53 inactivation, the only difference between VP and LP mice and Tp53−/− mice is constitutive expression of Cre. Constitutive Cre expression causes genomic instability, at least in mouse embryonic cells cultured in vitro,Citation55,Citation56 suggesting that the translocations found in VP and LP lymphomas could be Cre-induced lesions. Since the lymphoma predisposition of Vav–cre:Tp53−/− mice has not been reported, we cannot conclude whether the clonal translocations and chromosome fusions found in VP thymic lymphomas arise independently of Vav–cre expression. Yet, considering that Vav–cre mice are not predisposed to cancer,Citation46 our findings demonstrate that Tp53 serves important functions in HSCs and/or thymocytes to suppress malignant transformation in association with genomic instability. Notably, constitutive Cre expression from Lck–cre initiating in DN thymocytes of Tp53−/− mice does not alter onset or karyotype of thymic lymphomas that arise in these mice.Citation57 Therefore, we conclude from the cancer predisposition of LP mice that Tp53 serves critical functions in suppressing generation and/or oncogenic potential of Tcrα/δ translocations during αβ T cell development.
The objective of our study was to determine whether oncogenic lesions arising during embryogenesis and/or in HSCs preclude development of thymic lymphomas with clonal translocations including Tcr translocations. VP and LP mice succumb to tumors at similar ages as Tp53−/−, Lck–cre:Tp53−/− mice, and Mb1–cre:Tp53flox/flox mice.Citation32,Citation33,Citation36,Citation44 In contrast to the aneuploid thymic lymphomas that arise in Tp53−/− mice, we found that VP and LP mice developed lymphomas with aneuploidy or clonal translocations, including Ig or Tcrα/δ translocations. The distinct cancer phenotypes of these mice indicate that loss of Tp53 during embryogenesis, in cells before lymphocyte commitment, and/or in thymocytes masks development of lymphomas with oncogenic translocations in germline Tp53-deficient mice. In addition, the development of LP thymic lymphomas with aneuploidy or clonal translocations indicates that functions of Tp53 in response to both chromosome missegregation and DSBs are each critical for preventing malignant transformation of thymocytes.
T-cell acute lymphoblastic leukemia (T-ALL) remains a significant cause of cancer morbidity and mortality in both children and adults.Citation58,Citation59 Advances have been made in treatment of patients with T-ALL; however, drug-resistance and relapse are common causes of treatment failure, and most patients with tumors that fail treatment do not survive.Citation58 T-ALL is typically treated using genotoxic drugs that can cause serious health issues through effects on normal cells, demonstrating a need to develop more specific and less toxic therapies.Citation60 T-ALLs have heterogeneous karyotypes, with about half being aneuploid and the remainder containing translocations including oncogenic Tcrα/δ translocations.Citation61-Citation63 Although inactivating TP53 mutations are not common in T-ALL, these genetic lesions are often associated with drug resistance, rapid disease progression, and poor survival.Citation3 Therefore, LP mice may provide a useful pre-clinical model to evaluate the potential efficacy of more specific and less toxic treatments for T-ALL with TP53 inactivation.
Materials and Methods
Mice
Vav–cre,Citation46Lck–cre,Citation45 and p53flox/floxCitation48 mice were bred to generate the animals in this study. The genetic background strain of these mice was mixed 129SvEv and C57BL/6, with the 129SvEv strain predominant. We bred Vav–cre+/− and Lck–cre+/− males with p53flox/flox females to generate Vav–cre+/−p53flox/WT and Lck–cre+/−p53flox/WT males. These males were bred with p53flox/flox females to generate cohort Vav–cre+/−p53flox/flox and Lck–cre+/−p53flox/flox mice of either sex. Cohort mice were aged, monitored regularly, and euthanized upon showing visible signs of distress. This study was performed in accordance with national guidelines, and was approved by the Institutional Animal Care and Use Committee of the Children's Hospital of Philadelphia.
Kaplan–Meier analysis
Kaplan–Meier curves were generated in Graphpad Prism 5 (Graphpad Software Inc) and compared using the log-rank (Mantel-Cox) test.
Southern blotting
Genomic DNA (~20 μg) from lymphomas, non-transformed lymphocytes, or kidneys was digested with 100 units of indicated restriction enzymes (New England Biolabs), separated on 0.8% TAE agarose gels, transferred onto Zeta-probe membrane (BioRad), and hybridized with 32P-labeled Tcrβ, Igh, Igκ, or c-myc DNA probes as described.Citation21,Citation43,Citation64
Flow cytometry
Single-cell suspensions of mouse lymphomas or thymuses and spleens of 4–6-week-old control mice were stained in PBS with 3% FBS using antibodies from BD Pharmingen. For analyses of thymic lymphomas and non-malignant T cells, APC-conjugated anti-TCRβ, FITC-conjugated anti-CD8, PE-conjugated anti-CD4, PE-conjugated anti-TCRδ, and FITC-conjugated anti-CD3ε antibodies were used. For analyses of B lymphomas and non-malignant B cells, PE-Cy7-conjugated anti-B220, FITC-conjugated anti-CD43, APC-conjugated anti-IgM, PE-conjugated anti-Igκ, and FITC-conjugated anti-Igλ antibodies were used. Data were collected using a FACSCalibur (BD Biosciences) and CellQuest software (BD Biosciences) and analyzed using FlowJo software (Tree Star).
Cytogenetics
Metaphase spreads were prepared as previously described.Citation21 Spectral karyotyping and fluorescence in situ hybridization (FISH) were performed according to manufacturers' instructions (Applied Spectral Imaging). FISH probes were labeled with digoxigenin or biotin according to manufacturer’s instructions (Roche). The TCRCα-232F19, TCRVδ3/Vα6–46G9, Igh CH BAC199, and c-myc-454G15 FISH probes have been described previously.Citation43,Citation44 Slides were examined at room temperature under a BX61 microscope (magnification: 600×) from Olympus, controlled by a LAMBDA 10-B Smart Shutter from Sutter Instrument (Novato). Images were captured using a LAMBDA LS light source from Sutter Instrument, and a COOL-1300QS camera ASI, then analyzed through Case Data Manager Version 5.5 configured by Applied Spectral Imaging.
| Abbreviations: | ||
| DSBs | = | DNA double-stranded breaks |
| Tcr | = | T-cell receptor |
| Ig | = | immunoglobulin |
| HSCs | = | hematopoietic stem cells |
| VP | = | Vav-cre+/−p53flox/flox |
| LP | = | Lck-cre+/−p53flox/flox |
Additional material
Download Zip (2.3 MB)Acknowledgments
This research was supported by Training Grant TG GM-07229 of the University of Pennsylvania (AD); and the Department of Pathology and Laboratory Medicine and the Center for Childhood Cancer Research of the Children's Hospital of Philadelphia Research Institute, the Abramson Family Cancer Research Institute of the University of Pennsylvania Perelman School of Medicine, a grant from the Pennsylvania Department of Health, a Leukemia and Lymphoma Society Scholar Award, and the National Institutes of Health R01 Grants CA125195 and CA136470 (CHB).
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26299
Related Research Data
References
- Meek DW. Tumour suppression by p53: a role for the DNA damage response?. Nat Rev Cancer 2009; 9:714 - 23; PMID: 19730431
- Reinhardt HC, Schumacher B. The p53 network: cellular and systemic DNA damage responses in aging and cancer. Trends Genet 2012; 28:128 - 36; http://dx.doi.org/10.1016/j.tig.2011.12.002; PMID: 22265392
- Cheung KJ, Horsman DE, Gascoyne RD. The significance of TP53 in lymphoid malignancies: mutation prevalence, regulation, prognostic impact and potential as a therapeutic target. Br J Haematol 2009; 146:257 - 69; http://dx.doi.org/10.1111/j.1365-2141.2009.07739.x; PMID: 19500100
- Stilgenbauer S, Bullinger L, Lichter P, Döhner H, German CLL Study Group (GCLLSG). Chronic lymphocytic leukemia. Genetics of chronic lymphocytic leukemia: genomic aberrations and V(H) gene mutation status in pathogenesis and clinical course. Leukemia 2002; 16:993 - 1007; http://dx.doi.org/10.1038/sj.leu.2402537; PMID: 12040431
- Bhatia KG, Gutiérrez MI, Huppi K, Siwarski D, Magrath IT. The pattern of p53 mutations in Burkitt’s lymphoma differs from that of solid tumors. Cancer Res 1992; 52:4273 - 6; PMID: 1638540
- Hof J, Krentz S, van Schewick C, Körner G, Shalapour S, Rhein P, Karawajew L, Ludwig WD, Seeger K, Henze G, et al. Mutations and deletions of the TP53 gene predict nonresponse to treatment and poor outcome in first relapse of childhood acute lymphoblastic leukemia. J Clin Oncol 2011; 29:3185 - 93; http://dx.doi.org/10.1200/JCO.2011.34.8144; PMID: 21747090
- Ramírez J, Lukin K, Hagman J. From hematopoietic progenitors to B cells: mechanisms of lineage restriction and commitment. Curr Opin Immunol 2010; 22:177 - 84; http://dx.doi.org/10.1016/j.coi.2010.02.003; PMID: 20207529
- Chi AW, Bell JJ, Zlotoff DA, Bhandoola A. Untangling the T branch of the hematopoiesis tree. Curr Opin Immunol 2009; 21:121 - 6; http://dx.doi.org/10.1016/j.coi.2009.01.012; PMID: 19269149
- Schatz DG, Ji Y. Recombination centres and the orchestration of V(D)J recombination. Nat Rev Immunol 2011; 11:251 - 63; http://dx.doi.org/10.1038/nri2941; PMID: 21394103
- Alt FW, Zhang Y, Meng FL, Guo C, Schwer B. Mechanisms of programmed DNA lesions and genomic instability in the immune system. Cell 2013; 152:417 - 29; http://dx.doi.org/10.1016/j.cell.2013.01.007; PMID: 23374339
- Lieber MR. The mechanism of double-strand DNA break repair by the nonhomologous DNA end-joining pathway. Annu Rev Biochem 2010; 79:181 - 211; http://dx.doi.org/10.1146/annurev.biochem.052308.093131; PMID: 20192759
- Krangel MS, Carabana J, Abbarategui I, Schlimgen R, Hawwari A. Enforcing order within a complex locus: current perspectives on the control of V(D)J recombination at the murine T-cell receptor alpha/delta locus. Immunol Rev 2004; 200:224 - 32; http://dx.doi.org/10.1111/j.0105-2896.2004.00155.x; PMID: 15242408
- Jung D, Giallourakis C, Mostoslavsky R, Alt FW. Mechanism and control of V(D)J recombination at the immunoglobulin heavy chain locus. Annu Rev Immunol 2006; 24:541 - 70; http://dx.doi.org/10.1146/annurev.immunol.23.021704.115830; PMID: 16551259
- Xiong N, Raulet DH. Development and selection of gammadelta T cells. Immunol Rev 2007; 215:15 - 31; http://dx.doi.org/10.1111/j.1600-065X.2006.00478.x; PMID: 17291276
- Bell JJ, Bhandoola A. Putting ThPOK in place. Nat Immunol 2008; 9:1095 - 7; http://dx.doi.org/10.1038/ni1008-1095; PMID: 18800158
- von Boehmer H, Melchers F. Checkpoints in lymphocyte development and autoimmune disease. Nat Immunol 2010; 11:14 - 20; http://dx.doi.org/10.1038/ni.1794; PMID: 20016505
- Kuo TC, Schlissel MS. Mechanisms controlling expression of the RAG locus during lymphocyte development. Curr Opin Immunol 2009; 21:173 - 8; http://dx.doi.org/10.1016/j.coi.2009.03.008; PMID: 19359154
- Nemazee D. Receptor editing in lymphocyte development and central tolerance. Nat Rev Immunol 2006; 6:728 - 40; http://dx.doi.org/10.1038/nri1939; PMID: 16998507
- Longerich S, Basu U, Alt F, Storb U. AID in somatic hypermutation and class switch recombination. Curr Opin Immunol 2006; 18:164 - 74; http://dx.doi.org/10.1016/j.coi.2006.01.008; PMID: 16464563
- Keim C, Kazadi D, Rothschild G, Basu U. Regulation of AID, the B-cell genome mutator. Genes Dev 2013; 27:1 - 17; http://dx.doi.org/10.1101/gad.200014.112; PMID: 23307864
- Bassing CH, Suh H, Ferguson DO, Chua KF, Manis J, Eckersdorff M, Gleason M, Bronson R, Lee C, Alt FW. Histone H2AX: a dosage-dependent suppressor of oncogenic translocations and tumors. Cell 2003; 114:359 - 70; http://dx.doi.org/10.1016/S0092-8674(03)00566-X; PMID: 12914700
- Boboila C, Alt FW, Schwer B. Classical and alternative end-joining pathways for repair of lymphocyte-specific and general DNA double-strand breaks. Adv Immunol 2012; 116:1 - 49; http://dx.doi.org/10.1016/B978-0-12-394300-2.00001-6; PMID: 23063072
- Bassing CH, Alt FW. The cellular response to general and programmed DNA double strand breaks. DNA Repair (Amst) 2004; 3:781 - 96; http://dx.doi.org/10.1016/j.dnarep.2004.06.001; PMID: 15279764
- Dicker F, Herholz H, Schnittger S, Nakao A, Patten N, Wu L, Kern W, Haferlach T, Haferlach C. The detection of TP53 mutations in chronic lymphocytic leukemia independently predicts rapid disease progression and is highly correlated with a complex aberrant karyotype. Leukemia 2009; 23:117 - 24; http://dx.doi.org/10.1038/leu.2008.274; PMID: 18843282
- Thompson SL, Compton DA. Proliferation of aneuploid human cells is limited by a p53-dependent mechanism. J Cell Biol 2010; 188:369 - 81; http://dx.doi.org/10.1083/jcb.200905057; PMID: 20123995
- Fukasawa K, Wiener F, Vande Woude GF, Mai S. Genomic instability and apoptosis are frequent in p53 deficient young mice. Oncogene 1997; 15:1295 - 302; http://dx.doi.org/10.1038/sj.onc.1201482; PMID: 9315097
- Dujka ME, Puebla-Osorio N, Tavana O, Sang M, Zhu C. ATM and p53 are essential in the cell-cycle containment of DNA breaks during V(D)J recombination in vivo. Oncogene 2010; 29:957 - 65; http://dx.doi.org/10.1038/onc.2009.394; PMID: 19915617
- Zhu C, Mills KD, Ferguson DO, Lee C, Manis J, Fleming J, Gao Y, Morton CC, Alt FW. Unrepaired DNA breaks in p53-deficient cells lead to oncogenic gene amplification subsequent to translocations. Cell 2002; 109:811 - 21; http://dx.doi.org/10.1016/S0092-8674(02)00770-5; PMID: 12110179
- Nacht M, Strasser A, Chan YR, Harris AW, Schlissel M, Bronson RT, Jacks T. Mutations in the p53 and SCID genes cooperate in tumorigenesis. Genes Dev 1996; 10:2055 - 66; http://dx.doi.org/10.1101/gad.10.16.2055; PMID: 8769648
- Difilippantonio MJ, Petersen S, Chen HT, Johnson R, Jasin M, Kanaar R, Ried T, Nussenzweig A. Evidence for replicative repair of DNA double-strand breaks leading to oncogenic translocation and gene amplification. J Exp Med 2002; 196:469 - 80; http://dx.doi.org/10.1084/jem.20020851; PMID: 12186839
- Gladdy RA, Taylor MD, Williams CJ, Grandal I, Karaskova J, Squire JA, Rutka JT, Guidos CJ, Danska JS. The RAG-1/2 endonuclease causes genomic instability and controls CNS complications of lymphoblastic leukemia in p53/Prkdc-deficient mice. Cancer Cell 2003; 3:37 - 50; http://dx.doi.org/10.1016/S1535-6108(02)00236-2; PMID: 12559174
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215 - 21; http://dx.doi.org/10.1038/356215a0; PMID: 1552940
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1 - 7; http://dx.doi.org/10.1016/S0960-9822(00)00002-6; PMID: 7922305
- Liao MJ, Zhang XX, Hill R, Gao J, Qumsiyeh MB, Nichols W, Van Dyke T. No requirement for V(D)J recombination in p53-deficient thymic lymphoma. Mol Cell Biol 1998; 18:3495 - 501; PMID: 9584189
- Jacobs C, Huang Y, Masud T, Lu W, Westfield G, Giblin W, Sekiguchi JM. A hypomorphic Artemis human disease allele causes aberrant chromosomal rearrangements and tumorigenesis. Hum Mol Genet 2011; 20:806 - 19; http://dx.doi.org/10.1093/hmg/ddq524; PMID: 21147755
- Celeste A, Difilippantonio S, Difilippantonio MJ, Fernandez-Capetillo O, Pilch DR, Sedelnikova OA, Eckhaus M, Ried T, Bonner WM, Nussenzweig A. H2AX haploinsufficiency modifies genomic stability and tumor susceptibility. Cell 2003; 114:371 - 83; http://dx.doi.org/10.1016/S0092-8674(03)00567-1; PMID: 12914701
- Ward JM, Tadesse-Heath L, Perkins SN, Chattopadhyay SK, Hursting SD, Morse HC 3rd. Splenic marginal zone B-cell and thymic T-cell lymphomas in p53-deficient mice. Lab Invest 1999; 79:3 - 14; PMID: 9952106
- Difilippantonio MJ, Zhu J, Chen HT, Meffre E, Nussenzweig MC, Max EE, Ried T, Nussenzweig A. DNA repair protein Ku80 suppresses chromosomal aberrations and malignant transformation. Nature 2000; 404:510 - 4; http://dx.doi.org/10.1038/35006670; PMID: 10761921
- Guidos CJ, Williams CJ, Grandal I, Knowles G, Huang MT, Danska JS. V(D)J recombination activates a p53-dependent DNA damage checkpoint in scid lymphocyte precursors. Genes Dev 1996; 10:2038 - 54; http://dx.doi.org/10.1101/gad.10.16.2038; PMID: 8769647
- Rooney S, Sekiguchi J, Whitlow S, Eckersdorff M, Manis JP, Lee C, Ferguson DO, Alt FW. Artemis and p53 cooperate to suppress oncogenic N-myc amplification in progenitor B cells. Proc Natl Acad Sci U S A 2004; 101:2410 - 5; http://dx.doi.org/10.1073/pnas.0308757101; PMID: 14983023
- Haines BB, Ryu CJ, Chang S, Protopopov A, Luch A, Kang YH, Draganov DD, Fragoso MF, Paik SG, Hong HJ, et al. Block of T cell development in P53-deficient mice accelerates development of lymphomas with characteristic RAG-dependent cytogenetic alterations. Cancer Cell 2006; 9:109 - 20; http://dx.doi.org/10.1016/j.ccr.2006.01.004; PMID: 16473278
- Aparicio S, Caldas C. The implications of clonal genome evolution for cancer medicine. N Engl J Med 2013; 368:842 - 51; http://dx.doi.org/10.1056/NEJMra1204892; PMID: 23445095
- Yin B, Yang-Iott KS, Chao LH, Bassing CH. Cellular context-dependent effects of H2ax and p53 deletion on the development of thymic lymphoma. Blood 2011; 117:175 - 85; http://dx.doi.org/10.1182/blood-2010-03-273045; PMID: 20947684
- Rowh MA, DeMicco A, Horowitz JE, Yin B, Yang-Iott KS, Fusello AM, Hobeika E, Reth M, Bassing CH. Tp53 deletion in B lineage cells predisposes mice to lymphomas with oncogenic translocations. Oncogene 2011; 30:4757 - 64; http://dx.doi.org/10.1038/onc.2011.191; PMID: 21625223
- Lee PP, Fitzpatrick DR, Beard C, Jessup HK, Lehar S, Makar KW, Pérez-Melgosa M, Sweetser MT, Schlissel MS, Nguyen S, et al. A critical role for Dnmt1 and DNA methylation in T cell development, function, and survival. Immunity 2001; 15:763 - 74; http://dx.doi.org/10.1016/S1074-7613(01)00227-8; PMID: 11728338
- Georgiades P, Ogilvy S, Duval H, Licence DR, Charnock-Jones DS, Smith SK, Print CG. VavCre transgenic mice: a tool for mutagenesis in hematopoietic and endothelial lineages. Genesis 2002; 34:251 - 6; http://dx.doi.org/10.1002/gene.10161; PMID: 12434335
- Lowe SW, Schmitt EM, Smith SW, Osborne BA, Jacks T. p53 is required for radiation-induced apoptosis in mouse thymocytes. Nature 1993; 362:847 - 9; http://dx.doi.org/10.1038/362847a0; PMID: 8479522
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29:418 - 25; http://dx.doi.org/10.1038/ng747; PMID: 11694875
- Jiang D, Lenardo MJ, Zúñiga-Pflücker JC. p53 prevents maturation to the CD4+CD8+ stage of thymocyte differentiation in the absence of T cell receptor rearrangement. J Exp Med 1996; 183:1923 - 8; http://dx.doi.org/10.1084/jem.183.4.1923; PMID: 8666950
- Liyanage M, Coleman A, du Manoir S, Veldman T, McCormack S, Dickson RB, Barlow C, Wynshaw-Boris A, Janz S, Wienberg J, et al. Multicolour spectral karyotyping of mouse chromosomes. Nat Genet 1996; 14:312 - 5; http://dx.doi.org/10.1038/ng1196-312; PMID: 8896561
- Madapura HS, Salamon D, Wiman KG, Lain S, Klein G, Klein E, Nagy N. p53 contributes to T cell homeostasis through the induction of pro-apoptotic SAP. Cell Cycle 2012; 11:4563 - 9; http://dx.doi.org/10.4161/cc.22810; PMID: 23165210
- Fiancette R, Rouaud P, Vincent-Fabert C, Laffleur B, Magnone V, Cogné M, Denizot Y. A p53 defect sensitizes various stages of B cell development to lymphomagenesis in mice carrying an IgH 3′ regulatory region-driven c-myc transgene. J Immunol 2011; 187:5772 - 82; http://dx.doi.org/10.4049/jimmunol.1102059; PMID: 22039300
- Rouaud P, Fiancette R, Vincent-Fabert C, Magnone V, Cogné M, Dubus P, Denizot Y. Mantle cell lymphoma-like lymphomas in c-myc-3’RR/p53+/- mice and c-myc-3’RR/Cdk4R24C mice: differential oncogenic mechanisms but similar cellular origin. Oncotarget 2012; 3:586 - 93; PMID: 22592113
- Vicente-Dueñas C, González-Herrero I, García Cenador MB, García Criado FJ, Sánchez-García I. Loss of p53 exacerbates multiple myeloma phenotype by facilitating the reprogramming of hematopoietic stem/progenitor cells to malignant plasma cells by MafB. Cell Cycle 2012; 11:3896 - 900; http://dx.doi.org/10.4161/cc.22186; PMID: 22983007
- Loonstra A, Vooijs M, Beverloo HB, Allak BA, van Drunen E, Kanaar R, Berns A, Jonkers J. Growth inhibition and DNA damage induced by Cre recombinase in mammalian cells. Proc Natl Acad Sci U S A 2001; 98:9209 - 14; http://dx.doi.org/10.1073/pnas.161269798; PMID: 11481484
- Silver DP, Livingston DM. Self-excising retroviral vectors encoding the Cre recombinase overcome Cre-mediated cellular toxicity. Mol Cell 2001; 8:233 - 43; http://dx.doi.org/10.1016/S1097-2765(01)00295-7; PMID: 11511376
- Cheung AM, Hande MP, Jalali F, Tsao MS, Skinnider B, Hirao A, McPherson JP, Karaskova J, Suzuki A, Wakeham A, et al. Loss of Brca2 and p53 synergistically promotes genomic instability and deregulation of T-cell apoptosis. Cancer Res 2002; 62:6194 - 204; PMID: 12414647
- Smith MA, Seibel NL, Altekruse SF, Ries LA, Melbert DL, O’Leary M, Smith FO, Reaman GH. Outcomes for children and adolescents with cancer: challenges for the twenty-first century. J Clin Oncol 2010; 28:2625 - 34; http://dx.doi.org/10.1200/JCO.2009.27.0421; PMID: 20404250
- Maloney KW, Giller R, Hunger SP. Recent advances in the understanding and treatment of pediatric leukemias. Adv Pediatr 2012; 59:329 - 58; http://dx.doi.org/10.1016/j.yapd.2012.04.010; PMID: 22789585
- Bhatia S. Long-term complications of therapeutic exposures in childhood: lessons learned from childhood cancer survivors. Pediatrics 2012; 130:1141 - 3; http://dx.doi.org/10.1542/peds.2012-2884; PMID: 23166341
- Mrózek K, Harper DP, Aplan PD. Cytogenetics and molecular genetics of acute lymphoblastic leukemia. [v.] Hematol Oncol Clin North Am 2009; 23:991 - 1010, v; http://dx.doi.org/10.1016/j.hoc.2009.07.001; PMID: 19825449
- Le Noir S, Ben Abdelali R, Lelorch M, Bergeron J, Sungalee S, Payet-Bornet D, Villarèse P, Petit A, Callens C, Lhermitte L, et al. Extensive molecular mapping of TCRα/δ- and TCRβ-involved chromosomal translocations reveals distinct mechanisms of oncogene activation in T-ALL. Blood 2012; 120:3298 - 309; http://dx.doi.org/10.1182/blood-2012-04-425488; PMID: 22948044
- Graux C, Cools J, Michaux L, Vandenberghe P, Hagemeijer A. Cytogenetics and molecular genetics of T-cell acute lymphoblastic leukemia: from thymocyte to lymphoblast. Leukemia 2006; 20:1496 - 510; http://dx.doi.org/10.1038/sj.leu.2404302; PMID: 16826225
- Savic V, Yin B, Maas NL, Bredemeyer AL, Carpenter AC, Helmink BA, Yang-Iott KS, Sleckman BP, Bassing CH. Formation of dynamic gamma-H2AX domains along broken DNA strands is distinctly regulated by ATM and MDC1 and dependent upon H2AX densities in chromatin. Mol Cell 2009; 34:298 - 310; http://dx.doi.org/10.1016/j.molcel.2009.04.012; PMID: 19450528