Abstract
DNA double-strand breaks are caused by both intracellular physiological processes and environmental stress. In this study, we used laser microbeam cut (abbreviated microcut or cut), which allows specific DNA damage in the pronucleus of a fertilized egg and in individual blastomere(s) of an early embryo, to investigate the response of early embryos to DNA double-strand breaks. Line type γH2AX foci were detected in the cut region, while Chk2 phosphorylation staining was observed in the whole nuclear region of the cut pronuclei or blastomeres. Zygotes with cut male or female pronucleus showed poor developmental capability: the percentage of cleavage embryos was significantly decreased, and the embryos failed to complete further development to blastocysts. The cut blastomeres in 2-cell, 4-cell, and 8-cell embryos ceased cleavage, and they failed to incorporate into compacted morulae, but instead underwent apoptosis and cell death at the blastocyst stage; the uncut part of embryos could develop to blastocysts, with a reduced percentage or decreased cell number. When both blastomeres of the 2-cell embryos were cut by laser microbeam, cell death occurred 24 h earlier, suggesting important functions of the uncut blastomere in delaying cell death of the cut blastomere. Taken together, we conclude that microbeam-induced DNA damage in early embryos causes compromised development, and that embryos may have their own mechanisms to exclude DNA-damaged blastomeres from participating in further development.
Introduction
Genomic stability is critically important for normal cell growth and proliferation in various types of somatic cells. DNA strand breaks can be caused by normal physiological process, such as mismatches during DNA replication or incorrect activity of topoisomerase I and II. It can also be caused by harmful factors or components in the environment, such as chemical agents,Citation1 heavy metals,Citation2 UV light,Citation3 and ionizing radiation.Citation4 If DNA damage is not repaired or incorrectly repaired, it may cause large scope aberrations in the genome and lead to cell cycle arrest or to cell apoptosis. DNA damage response induced by chemicals, drugs, and ionizing radiation in oocytes,Citation5-Citation8 spermatozoa,Citation9 and pre-implantation embryosCitation10,Citation11 has been reported. However, mitochondrial DNA and active proteins in the cytoplasm may also become injured by such treatments, and these methods cannot be used to experimentally damage only one pronucleus in a fertilized egg or single blastomeres in an early embryo.
Laser microbeam can cut the cell nuclei precisely in distinct areas, and it is regarded as an effective method to induce DNA damage.Citation12,Citation13 UVA (wavelength: 315–400 nm) has been extensively used as laser microbeam to induce double-strand DNA damage in various kinds of somatic cells.Citation14-Citation16 Cells need to be pre-sensitized with nucleotide analogs such as BrdU and 50-iodo-2-deoxyuridine (IdU)Citation17 when a lower dose of UVA is used. However, a high dose of UVA can induce DNA damage without pre-sensitization treatment of the cells.Citation14 In the present study, we used UVA laser as a microcut tool to induce DNA DSB (double-strand breaks) in fertilized eggs and early cleavage embryos. Specifically, we investigated the effect of DNA damage on cell division and embryo development when one pronucleus of a zygote or one blastomere of a cleavage stage embryo was cut by laser microbeam.
Results
Establishment of a microbeam-induced DNA damage model in mouse oocytes and embryos
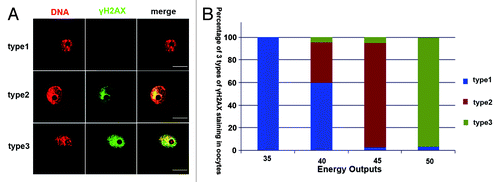
Germinal vesicle stage oocytes were cut by microbeam using different energy outputs. DNA damage of GV oocytes were detected in the cut region by γH2AX (phosphorylates the histone variant H2AX at serine 139) foci. Two types of γ-H2AX foci could be detected in the cut region. If the cut energy was lower than 40 energy output, γH2AX foci could barely be detected in oocytes. When the germinal vesicle was cut using 45 energy output, we could observe γH2AX staining in most oocytes just in the region that was cut in a line style. However, if the energy was increased to 50 energy output, γH2AX staining was extended to the whole germinal vesicle (). In order to avoid other effects (more than DNA damage) induced by the microbeam cut, we used a relatively low energy, and we defined the 45 energy output as the most suitable energy level. Developmental capacity of oocytes cut by microbeam was recently evaluated by our lab.Citation7 We showed that 45 energy output could also induce blastomere DNA damage in the line type in early embryos.
Figure 1. Correlation between DNA damage extent and laser energy output in oocytes and embryos. (A) Microbeam cut oocytes were classified into 3 types. Type 1, oocytes without γH2AX foci; type 2, oocytes with line style γH2AX foci; type 3, γH2AX foci distributed in the whole nucleus. (B) Proportions of 3 types of γH2AX foci after microbeam cut with different energy outputs ranging from 35 to 50. There were no γH2AX foci in the oocytes when energy output was 35; when the energy output was increased to 40, small proportions of oocytes showed γH2AX foci in type 2 or 3; when the energy was increased to 45 nearly all oocytes showed type 2 γH2AX foci; most of the oocytes cut by 50 energy output showed type 3 γH2AX foci (bar = 50 μm).
Early embryonic cleavage and development is disturbed by microbeam-induced DNA damage in the male or female pronucleus
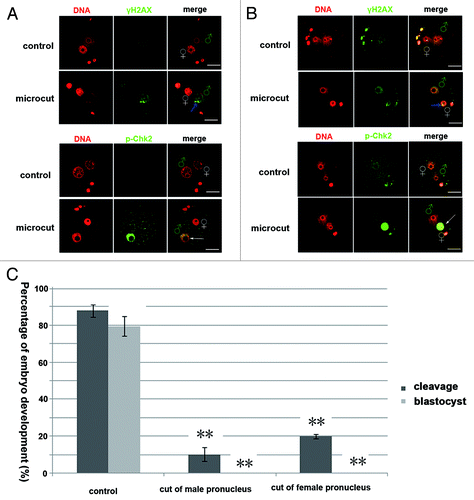
Zygotes with distinguishable male and female pronuclei were cut by microbeam at 45 energy output. The male and female pronuclei could be distinguished by their size (the male pronucleus is larger than the female). γH2AX staining was detected in the cut region of the male or female pronucleus (). However, p-Chk2 staining was found to distribute in the whole pronucleus (either male or female) of the cut zygote (). DNA damage in either male or female pronucleus caused significantly reduced embryo cleavages and subsequent development compared with the control group ().
Figure 2. DNA damage in either male or female pronucleus blocked embryo development. (A) Line style γH2AX foci (marked by blue arrows) was detected in the cut male pronucleus of the zygote, and p-Chk2 staining was detected in the whole male pronucleus. (B) Line style γH2AX foci (marked by blue arrow) was detected in the cut female pronucleus of the zygote, and p-Chk2 staining was detected in the whole female pronucleus. (C) Cleavage percentage (control, 88.02%; cut of male pronucleus, 10.3%; cut of the female pronucleus 19.9%; P < 0.05) and blastocyst percentage (control, 79.60%; cut of male pronucleus, 0%; cut of the female pronucleus 0%; P < 0.05) of the pronuclear cut groups were significantly lower than those in the control group (bar = 50 μm).
DNA damage in one blastomere of 2-cell embryos causes division arrest of the cut blastomere and affects development of the intact blastomere
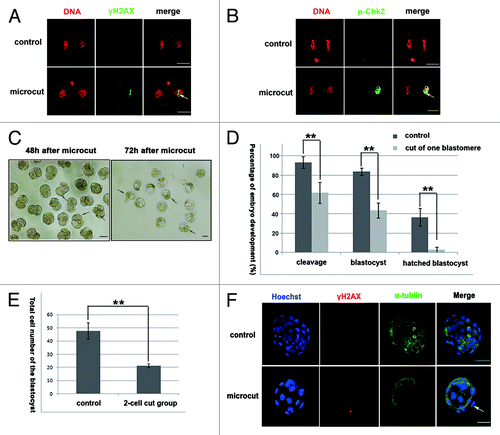
One blastomere of 2-cell stage embryos was cut by a 45 energy output. Phosphorylation of the H2AX and Chk2 could be detected in the nuclear region of the cut blastomere (). Embryos with one cut blastomere were cultured in vitro for another 3 d to analyze their developmental capability. In these embryos, the cut blastomere ceased dividing and was not incorporated into the compacted morula during subsequent development, but the other blastomeres without microbeam cut maintained the capability to divide normally until the blastocoel was observed (, arrows indicating the undivided cut blastomeres). The percentage of cleavage, blastocysts, and hatched blastocysts of the cut embryo groups were significantly lower than those in the control group (cleavage, 62.1% vs. 93.5%; blastocyst, 43.7% vs. 83.9%; hatched blastocyst, 2.9% vs. 36.7%; P < 0.05) (), and the total cell number of blastocysts in the microcut group was significantly lower than that of the control group (21.28 vs. 47.48; P < 0.05) (). Although the developmental capability of the microcut group was significantly decreased, no increased γH2AX staining was detected in the blastocysts of the cut group compared with the control group ().
Figure 3. Microcut induced DNA damage in one blastomere of 2-cell embryos, and decreased embyro developmental capability. (A) γH2AX foci of line style was detected in the cut blastomere in 2-cell embryo. (B) p-Chk2 staining was detected in the whole nucleus in the cut blastomere of 2-cell embryo. (C) The cut blastomere ceased development, while the uncut one developed to the blastocyst stage; the cut blastomere did not participate in the morula compaction of the uncut portion; (D) Cleavage percentage, blastocyst percentage and hatched blastocyst percentage were significantly lower in the cut group compared with the control group (cleavage, 93.5% vs. 62.1%; blastocyst, 83.9% vs. 43.7%, hatched blastocyst, 36.7% vs. 2.9%; P < 0.05); (E) the total cell numbers of the cut group was significantly lower compared with the control group (21.28 vs. 47.48; P < 0.05); (F) γH2AX was not detected when the cut embryo developed to the blastocyst stage; α-tubulin staining was included to indicate the cell boundary and to clearly distinguish the cut blastomere (bar = 50 μm).
Oct4 and Cdx2 are expressed normally in the intact blastomere but not in the cut blastomere of 2-cell embryos
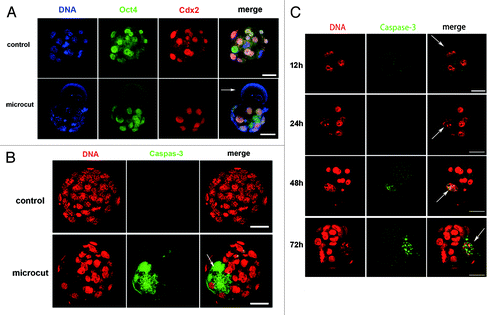
Two-cell embryos had the capability to develop to blastocysts with only one healthy blastomere, and the blastocysts did not show significantly more DNA damage signal (γH2AX foci) than the control group. We thus further investigated why the cut blastomeres lost the developmental capacity, and why the embryos derived from the uncut blastomere showed decreased developmental potential. We used immunofluorescence to determine whether or not these embryos displayed any defect in Oct4 (a marker of the inner cell mass) or Cdx2 (a marker of trophoblast cells) expression when developing to blastocyst stage. In vitro cultured blastomeres of 2-cell embryos were used as controls. Cdx2 and Oct4 staining was detected in the uncut region of these embryos similar to the control, but neither Cdx2 nor Oct4 staining was detected in the cut blastomere ().
Figure 4. Oct4 and Cdx2 expression in developmentally arrested blastomere and blastocyst derived from uncut blastomere (One blastomere of the 2-cell embryo was isolated and cultured as control), and time-dependent caspase-3 staining in early embryos. (A) Oct4 and Cdx2 expression in blastocyst derived from uncut blastomere was similar to that of the control, while developmentally arrested cut blastomere showed faint staining of Oct4 and Cdx2 (arrow); (B) Cleaved caspase-3 staining was detected in the cut blastomere at the blastocyst stage; (C) Caspase-3 staining was evident 48 h after microcut in the cut blastomere (white arrow), and was increasing by 72 h. The caspase-3 staining was located in the nuclear region of the cut blastomere, and later extended to the whole cell as indicated in (bar = 50 μm).
Cleaved caspase-3 is detected in the DNA-damaged blastomeres
Cleaved caspase-3 was used as an apoptosis marker to detect cell apoptosis in the cut embryos. Cut embryos that had intact cell membranes and normal blastocoels were fixed 75–80 h after the microcut. Cleaved caspase-3 staining was detected in the microbeam cut blastomere, and faint cleaved caspase-3 staining was also detected in the embryo derived from the uncut blastomere. However, there was no evident cleaved caspase-3 staining in the control embryo (). We next analyzed 4 time points (12, 24, 48, and 72 h after microbeam cut) to determine the time point when the cleaved caspase-3 became initially increased. The results indicated that cleaved caspase-3 appeared 48 h after the microbeam cut, and was further enhanced by 72 h. The cleaved caspase-3 staining was restricted to the nucleus of the cut blastomere at 48 h and 72 h ().
DNA-damaged blastomere of 4-cell or 8-cell embryo fails to divide and to incorporate into compacted morulae
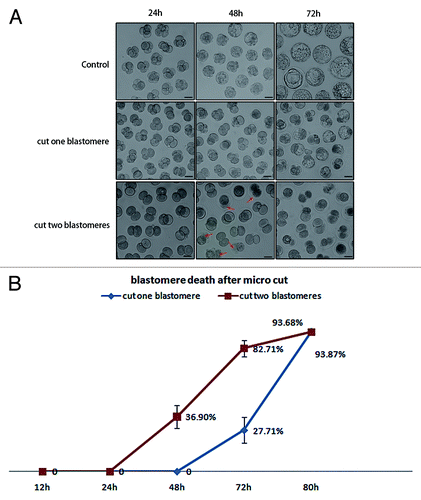
We further cut one blastomere in 4-cell or 8-cell stage embryos by microbeam and observed cell division and embryo development. Consistent with the result of the 2-cell embryo cut experiment, the blastomeres, which were cut at the 4-cell or 8-cell stages, did not divide either, and the uncut portion could develop like normal embryos (). Morula compaction was initiated soon after the embryo developed to the 8-cell stage, but the cut blastomere in the 8-cell stage embryo did not participate in the compaction process. Although 4-cell and 8-cell embryos were cut during later developmental stages compared with the 2-cell embryos, the cut blastomere also died when the embryo developed to the blastocyst stage (about 56 h after microcut in the 4-cell and 32 h after microcut in the 8-cell). As indicated above, the cut blastomere did not die until the blastocyst stage, even if the microcut was performed as early as at the 2-cell stage. PI and Hoechst 33342 double staining was used to determine the integrity of the cell nuclear membrane in the cut blastomere. There was no PI staining before the blastocyst stage, so the permeability of the cell membrane did not change before cell death occurred (). In the two blastomere-cut group, the embryos displayed cell death at 48 h after microcut, 24 h earlier than observed in the one blastomere-cut group (). This time point was consistent with the increase in cleaved caspase-3 in the one blastomere-cut group as indicated in .
Figure 5. Cell fate and embryo development after one blastomere was cut by laser microbeam. (A) Developmental pattern of the 2-cell, 4-cell, and 8-cell embryos, when one blastomere was cut by microbeam. The cut blastomere of 2-cell embryos underwent cell death at 80 h after microcut; the 4-cell group underwent cell death at 56 h after microcut; and the 8-cell group underwent cell death at 32 h after microcut. (B) The intact cell membrane was detected by PI and Hoechst 33342 staining, the nucleus which just displayed Hoechst 33342 staining (48 h and 72 h after microcut) indicated intact cell membrane, while the nucleus which had both Hoechst 33342 and PI staining (80 h after microcut) indicated high permeability of the cell membrane and cell death (red arrows indicate the cut blastomeres, bar = 50μm)
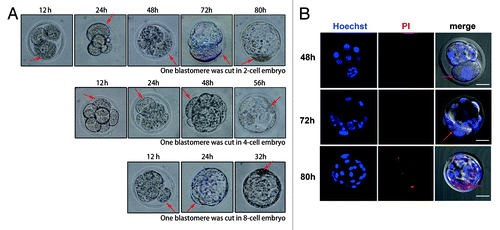
Figure 6. Cell fate and embryo development when one or two blastomeres were exposed to laser microbeam. (A) Dead blastomeres in control group, one blastomere-cut group, and two blastomere-cut group 24 h, 48 h, 72 h after microcut. The two blastomere-cut group displays cell death at 48 h after microcut (red arrows indicate dead cells), while there was no blastomere death at 48 h after microcut (B) Cell death related to the microcut time. Cell death emerged in the two blastomere-cut group at 48 h after microcut (36.90%), while at this time no cell death could be detected in the one blastomere-cut group. The two blastomere-cut group showed cell death 24 h earlier than the one blastomere-cut goup. At 72 h of microcut, 82.71% cell death occurred in the two blasomere-cut group, while only 27.71% cell death was observed in one blastomere-cut group (P < 0.05). At 80 h of microcut, cell death was detected in nearly all embryos (93.68% in one blastomere-cut group and 93.87% in two blastomere-cut group).
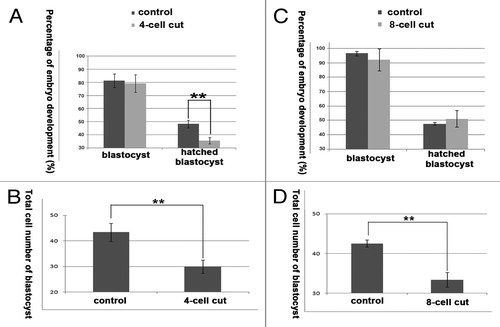
The developmental capability of the cut embryos was further tested. The percentage of blastocyst development was not significantly different between the 4-cell cut group and the control group (79.04% vs. 81.25%), but the percentage of hatched blastocysts and the total cell numbers of the blastocysts were significantly lower in the cut group compared with the control group (hatched blastocyst, 35.57% vs. 48.14%; total cell number of blastocyst, 29.90 vs. 43.33; P < 0.05) (). The percentage of blastocysts and the hatched blastocysts were not significantly different between the 8-cell cut group and control group (blastocyst, 91.97% vs. 96.47%; hatched blastocyst, 51.15% vs. 47.64%), but the total cell numbers were significantly lower in the 8-cell cut group compared with the control group (33.39 vs. 42.50; P < 0.05) ().
Figure 7. The developmental capability of the 4-cell, and 8-cell cut embryos. (A) The percentage of blastocyst development was not significantly different between the 4-cell cut group and control group (79.04% vs. 81.25%), and the percentage of hatched blastocysts was significantly lower in the 4-cell cut group compared with the control group (35.57% vs. 48.14; P < 0.05); (B) The total cell numbers were less in the 4-cell cut group compared with the control group (29.90 vs. 43.33; P < 0.05); (C) The percentages of blastocysts and hatched blastocysts were not significantly different between the 8-cell cut group and control group (blastocyst, 91.97% vs. 96.47%; hatched blastocyst, 51.15% vs. 47.67%); (D) the total cell numbers were significantly lower in the 8-cell cut group compared with the control group (33.39 vs. 42.50; P < 0.05).
Discussions
In this study we investigated the effect of laser microbeam-induced DNA damage in pronuclei of zygotes, and in one blastomere of 2-cell, 4-cell, and 8-cell stage embryos on cell division and embryo development. On one hand, the results indicate that microcut induces DNA damage in distinct regions of embryos, elicits DNA damage response, causes cell cycle arrest, and leads to cell death at the blastocyst stage. Precise regulatory mechanisms exist in the embryos to control the time for cell death in the cut embryos. As a result, the cut blastomeres die at the blastocyst stage regardless of when the microcut was performed. On the other hand, our results suggest that there is a 2-way communication between the cut blastomere and the uncut blastomere. The uncut blastomeres have some beneficial effect on the cut blastomere, as they delay the death of the cut blastomere. Importantly, the embryo may have its own machinery to prevent the DNA-damaged blastomere from participating in development, since even when the blastomere was damaged in the 8-cell embryo before compaction, it will not be incorporated into the compacted morula.
In somatic cells, DNA damage response leads to activation of the checkpoint kinase Chk2 through phosphorylation of threonine 68Citation18 by ATM. Then Cdc25A is phosphorylated on serine 123 and degenerated by p-Chk2. Cdk2, which is important for DNA synthesis, cannot be activated due to the degeneration of Chk2.Citation19 As a result, the cell cycle is arrested at the G2 stage. H2AX phosphorylation in DNA-damaged cells provides a platform for recruitment of the necessary checkpoint and repair factors to the DNA damage region.Citation16 DNA damage in the sperm genome caused by chemicals or radiation has been studied by several groupsCitation20-Citation23; γH2AX foci can also be detected in the zygote’s male pronucleus after injection of DNA-damaged spermatozoon into an oocyte. Different treatments of spermatozoa can lead to defects in the embryo’s DNA replication, cell cycle arrest in zygotes, and apoptosis after the first cleavages.Citation24 However, there has been no report on the effect of DNA damage in the female or male pronucleus due to the limitation of treatment methods (chemical or radiation). Microcut is a powerful tool to investigate DNA damage in the zygote’s pronuclei, as it can cause DNA damage restricted to a specified region (just one pronucleus). In our experiment, microcut-caused DNA damage in the zygotes can lead to H2AX and Chk2 phosphorylation as in somatic cellsCitation12,Citation25 The H2AX phosphorylation staining was distinctly localized to the area of the cut region in the line style, and Chk2 phosphorylation can be detected in the entire nuclear region. Microcut-caused DNA damage in either male or female pronucleus leads to failure of cleavage and to the embryo’s developmental arrest. This might have two reasons: one is that the microcut induces more intensive DNA damage than the other methods; and the other is that DNA damage response is more sensitive at the pronuclear stage.
Mouse preimplantation embryos are sensitive to radiation-induced DNA damage.Citation26 DNA damage caused by drug treatment,Citation27,Citation28 hydrogen peroxide,Citation23 heat stress,Citation29 low-frequency electromagnetic fields,Citation30,Citation31 and ionizing radiationCitation10,Citation32 in preimplantation embryos have been investigated in previous studies. However, to our knowledge there is no study on the effects on embryo development when DNA damage takes place in one blastomere. One blastomere of a 2-cell embryo can normally develop to term, creating fertile animals.Citation33 A single blastomere of a mouse 4-cell stage embryo can develop to the blastocyst stage, but it cannot develop into a live mouse, which might be the result of insufficient cell numbers of its inner cell mass.Citation34 One blastomere of rabbitCitation35 and sheepCitation36 4-cell embryos occasionally even 8-cell embryos with more cell numbers when reaching blastocysts are able to develop into an adult animal. In our experiment, 2-cell embryos with one DNA-damaged blastomere can develop to the blastocyst stage, and the uncut blastomere-derived blasotocyst showed a normal Oct4 and Cdx2 expression pattern. However, the developmental capabilities of the cut embryos are significantly lower compared with the control group. This is similar to the phenotype of embryos with fragmentation, which also have low developmental capabilities.Citation37
A major consequence of DNA damage is cell apoptosis or death. Apoptosis can be detected in mouse pre-implantation embryos when they are derived from DNA-damaged sperm.Citation38 We used cleaved caspase-3 as a marker to detect apoptosis in the cut embryos. Caspase-3 is an effectors’ caspase that is activated by upstream initiator caspases (like caspase-6, 8, and 9), and it will lead to the degradation of cellular substrates, which are important for cell survival.Citation39 So it is the last step of the apoptotic cascade, and is expressed just before cell death. Cleaved caspase-3 can be detected in the cut 2-cell embryos located in the nucleus of the cut blastomere 48 h after microcut. However, cell death of cut embryos takes place when the embryos develop to the blastocyst stage. This result is similar to the TUNEL assay of apoptosis as revealed in the mouse and pig pre-implantation embryos in previous studies.Citation40,Citation41 Apoptosis seldom occurs in embryo cells before the blastocyst stage. However, if embryos are treated with specific chemicals or drugs like Staurosporine, apoptosis can be detected in the embryo cells before the 4-cell stage.Citation42 Pre-implantation embryos undergo distinct developmental stages and resist manual intervention. It is well known that blastomeres isolated from pre-implantation embryos undergo morula compaction and blastocoel cavity formation independent of the cell numbers of the blastomeres in the developing embryos, and the timing for development strictly coincides with the timing of the embryo from which the blastomere was derived. The death of the cut blastomere, which takes place at the blastocyst stage, is related to intrinsic regulatory functions of the pre-implantation embryos. However, if both blastomeres of the 2-cell embryos were cut by microbeam, the time for cell death differed from the original pattern.
Although the laser microbeam cut blastomeres survive to the blastocyst stage, they lose the division capability, suggesting a self-protective function of the early embryos to avoid DNA-damaged cells to participate in development. Another important phenomenon is that the laser cut blastomere is not incorporated into the compacted morula, even if the damage occurs at the 8-cell stage, when compaction is about to begin. These results suggest that the embryo has its own machinery to prevent DNA-damaged cells from participating in development.
Another striking finding is that although 4-cell or 8-cell embryos can develop to blastocyst stages, the percentage of the hatching blastocysts decreases when one blastomere of 4-cell embryos is DNA-damaged, and the cell numbers of blastocysts derived from 4-cell cut and 8-cell cut embryos also significantly decreases, indicating reduced embryo quality. In clinical IVF manipulations, individual blastomeres are aspirated for genetic screening when necessary, and such manipulations may cause similar changes. Therefore, care and long-term effect investigations are needed to analyze the effects of such manipulations.
In conclusion, by using a laser microbeam cut technology that allows DNA damage in the pronucleus of a fertilized egg and individual blastomeres of an early embryo, we have shown that DNA damage causes embryonic cell cycle arrest and finally cell death at a particular time point, and that the embryo has its own mechanisms to exclude DNA-damaged cells.
Materials and Methods
Oocyte collection
Animal care and handling were performed in accordance with Animal Research Committee policies of the Institute of Zoology, Chinese Academy of Sciences. The GV stage oocytes were collected from ovaries of 6–8-wk-old female ICR mice in M2 medium with 2.5 μM milrinone, which was used to keep oocytes at the GV stage. Fully grown oocytes were further used for microbeam cut by power outputs of 35, 40, 45, and 50, respectively. Control oocytes and microbeam cut oocytes (10 min after the cut) were fixed for immunofluorescent staining.
Mouse embryo collection and in vitro culture
Female ICR mice were superovulated by injection of 5 IU of PMSG, followed by 5 IU of hCG administration 48 h later, and then they were mated (males > 10 wk of age). Zygotes, 2-cell embryos, 4-cell embryos, and 8-cell embryos were flushed from oviducts at 20, 40, 60, and 68 h of hCG administration. Two-cell embryos were treated with acidic Tyrode solution for 30 s to remove the zona pellucida (ZP), and then embryos were gently pipetted to obtain blastomeres. Embryos and blastomeres were cultured in vitro in KSOM medium in a humidified incubator of 5% CO2 at 37°C for another 2–3 d depending on the experimental requirements. Cleavage percentage of zygotes was assessed at 48 h of hCG injection, cleavage percentage of the cut 2-cell embryos were assessed at 24 h after microbeam cut. The percentage of blastocyst development was counted at 96 h of hCG administration, and hatched blastocysts were counted at 120 h of hCG administration.
Microbeam cut of oocytes and embryos
For microbeam cut, 10–15 embryos were placed in a drop of medium in a 35 mm dish covered with mineral oil and placed in the dish holder 35 CC of the Palm microlaser workstation (P.A.L.M. Laser Technologies). A pulsed nitrogen laser (30 Hz, 337 nm) attached to the microscope was focused through a LD 60 ×; NA 0.6 Zeiss Achroplan objective to draw a line in size of approximately 10 μm in the nucleus of oocytes or embryos. The parameters used for microbeam cut were speed percent, 5; microbeam cut energy, 35–50; focus, 50; and focus delta, 3. After microbeam cut the embryos were immediately transferred to the incubator for further development, or fixed 10 min later, depending on the experimental requirements. Normal embryos placed in another drop of this dish were used as control.
Antibodies and immunofluorescence
Antibody against γH2AX was purchased from Bioworld. Antibodies for detecting Oct4, Cdx2 were purchased from Santa Cruz Biotechnology Inc. Antibodies for p-Chk2 (Thr68) and cleaved caspase-3 (Asp175) were purchased from Cell Signaling Technology. Immunofluorescent staining was performed using standard protocols. Briefly, all oocytes and embryos were fixed in 4% paraformaldehyde for 30 min, and then treated with 0.5% Triton X-100 for 20 min. After blocking in 1% BSA for 1 h, oocytes and embryos were incubated in the primary antibody (1:200) at 4°C overnight. After 5 washes in PBS with 0.05% Tween 20, the oocytes were incubated with the second antibody (1:200 in PBS with 0.05% Tween 20) for 1 h at room temperature. After 3 washes in PBS with 0.05% Tween 20, the oocytes and embryos were incubated with PI/DAPI (propidium iodide/4, 6-diamino-, 2-phenyl indole) in PBS containing 0.05% Tween-20 for 10 min. Finally, the oocytes or embryos were mounted on glass slides and examined with a laser scanning confocal microscope (Zeiss 710 META). PI and Hoechst 33342 double staining was performed to detect if the cut embryos still displayed intact plasma membranes. Embryos at specific stages were stained in PI and Hoechst33342 mixture without any other treatment for 30 min and then examined using a confocal microscope.
Statistical analysis
All percentage data were subjected to arcsine transformation before statistical analysis. Data were analyzed by analysis of variance (ANOVA) with SPSS. Differences at P < 0.05 were considered significant. Data were from at least 3 replicates, and the numbers of embryos in each group were at least 30.
| Abbreviations: | ||
| GV | = | germinal vesicle |
| UVA | = | Ultraviolet A |
| DSB | = | double-strand breaks |
Acknowledgments
This study was supported by Major Basic Research Program (2012CB944404, 2011CB944501) and National Natural Science Foundation of China (30930065) to QYS.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest are disclosed.
Related Research Data
References
- Ummat A, Rechkoblit O, Jain R, Roy Choudhury J, Johnson RE, Silverstein TD, Buku A, Lone S, Prakash L, Prakash S, et al. Structural basis for cisplatin DNA damage tolerance by human polymerase η during cancer chemotherapy. Nat Struct Mol Biol 2012; 19:628 - 32; http://dx.doi.org/10.1038/nsmb.2295; PMID: 22562137
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160:1 - 40; http://dx.doi.org/10.1016/j.cbi.2005.12.009; PMID: 16430879
- Meyer JN, Bess AS. Involvement of autophagy and mitochondrial dynamics in determining the fate and effects of irreparable mitochondrial DNA damage. Autophagy 2012; 8:1822 - 3; http://dx.doi.org/10.4161/auto.21741; PMID: 22929123
- Ward JF. DNA damage produced by ionizing radiation in mammalian cells: identities, mechanisms of formation, and reparability. Prog Nucleic Acid Res Mol Biol 1988; 35:95 - 125; http://dx.doi.org/10.1016/S0079-6603(08)60611-X; PMID: 3065826
- Marangos P, Carroll J. Oocytes progress beyond prophase in the presence of DNA damage. Curr Biol 2012; 22:989 - 94; http://dx.doi.org/10.1016/j.cub.2012.03.063; PMID: 22578416
- Carroll J, Marangos P. The DNA damage response in mammalian oocytes. Front Genet 2013; 4:117; http://dx.doi.org/10.3389/fgene.2013.00117; PMID: 23805152
- Ma JY, Ou Yang YC, Wang ZW, Wang ZB, Jiang ZZ, Luo SM, Hou Y, Liu ZH, Schatten H, Sun QY. The effects of DNA double-strand breaks on mouse oocyte meiotic maturation. Cell Cycle 2013; 12:1233 - 41; http://dx.doi.org/10.4161/cc.24311; PMID: 23518501
- Yuen WS, Merriman JA, O’Bryan MK, Jones KT. DNA double strand breaks but not interstrand crosslinks prevent progress through meiosis in fully grown mouse oocytes. PLoS One 2012; 7:e43875; http://dx.doi.org/10.1371/journal.pone.0043875; PMID: 22928046
- Dar S, Grover SA, Moskovtsev SI, Swanson S, Baratz A, Librach CL. In vitro fertilization-intracytoplasmic sperm injection outcome in patients with a markedly high DNA fragmentation index (>50%). Fertil Steril 2013; 100:75 - 80; http://dx.doi.org/10.1016/j.fertnstert.2013.03.011; PMID: 23562046
- Yukawa M, Oda S, Mitani H, Nagata M, Aoki F. Deficiency in the response to DNA double-strand breaks in mouse early preimplantation embryos. Biochem Biophys Res Commun 2007; 358:578 - 84; http://dx.doi.org/10.1016/j.bbrc.2007.04.162; PMID: 17498656
- Mu XF, Jin XL, Farnham MM, Li Y, O’Neill C. DNA damage-sensing kinases mediate the mouse 2-cell embryo’s response to genotoxic stress. Biol Reprod 2011; 85:524 - 35; http://dx.doi.org/10.1095/biolreprod.110.089334; PMID: 21593482
- Lukas C, Falck J, Bartkova J, Bartek J, Lukas J. Distinct spatiotemporal dynamics of mammalian checkpoint regulators induced by DNA damage. Nat Cell Biol 2003; 5:255 - 60; http://dx.doi.org/10.1038/ncb945; PMID: 12598907
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, Jackson SP. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37 - 45; http://dx.doi.org/10.1038/ncb1337; PMID: 16327781
- Bekker-Jensen S, Lukas C, Kitagawa R, Melander F, Kastan MB, Bartek J, Lukas J. Spatial organization of the mammalian genome surveillance machinery in response to DNA strand breaks. J Cell Biol 2006; 173:195 - 206; http://dx.doi.org/10.1083/jcb.200510130; PMID: 16618811
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 16; http://dx.doi.org/10.1083/jcb.146.5.905; PMID: 10477747
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886 - 95; http://dx.doi.org/10.1016/S0960-9822(00)00610-2; PMID: 10959836
- Kong X, Mohanty SK, Stephens J, Heale JT, Gomez-Godinez V, Shi LZ, Kim JS, Yokomori K, Berns MW. Comparative analysis of different laser systems to study cellular responses to DNA damage in mammalian cells. Nucleic Acids Res 2009; 37:e68; http://dx.doi.org/10.1093/nar/gkp221; PMID: 19357094
- Matsuoka S, Rotman G, Ogawa A, Shiloh Y, Tamai K, Elledge SJ. Ataxia telangiectasia-mutated phosphorylates Chk2 in vivo and in vitro. Proc Natl Acad Sci U S A 2000; 97:10389 - 94; http://dx.doi.org/10.1073/pnas.190030497; PMID: 10973490
- Falck J, Mailand N, Syljuåsen RG, Bartek J, Lukas J. The ATM-Chk2-Cdc25A checkpoint pathway guards against radioresistant DNA synthesis. Nature 2001; 410:842 - 7; http://dx.doi.org/10.1038/35071124; PMID: 11298456
- Gawecka JE, Marh J, Ortega M, Yamauchi Y, Ward MA, Ward WS. Mouse zygotes respond to severe sperm DNA damage by delaying paternal DNA replication and embryonic development. PLoS One 2013; 8:e56385; http://dx.doi.org/10.1371/journal.pone.0056385; PMID: 23431372
- Barton TS, Robaire B, Hales BF. DNA damage recognition in the rat zygote following chronic paternal cyclophosphamide exposure. Toxicol Sci 2007; 100:495 - 503; http://dx.doi.org/10.1093/toxsci/kfm242; PMID: 17872895
- Derijck AA, van der Heijden GW, Giele M, Philippens ME, van Bavel CC, de Boer P. gammaH2AX signalling during sperm chromatin remodelling in the mouse zygote. DNA Repair (Amst) 2006; 5:959 - 71; http://dx.doi.org/10.1016/j.dnarep.2006.05.043; PMID: 16837249
- Xiao J, Liu Y, Li Z, Zhou Y, Lin H, Wu X, Chen M, Xiao W. Effects of the insemination of hydrogen peroxide-treated epididymal mouse spermatozoa on γH2AX repair and embryo development. PLoS One 2012; 7:e38742; http://dx.doi.org/10.1371/journal.pone.0038742; PMID: 22745678
- Fatehi AN, Bevers MM, Schoevers E, Roelen BA, Colenbrander B, Gadella BM. DNA damage in bovine sperm does not block fertilization and early embryonic development but induces apoptosis after the first cleavages. J Androl 2006; 27:176 - 88; http://dx.doi.org/10.2164/jandrol.04152; PMID: 16304212
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 2012; 46:212 - 25; http://dx.doi.org/10.1016/j.molcel.2012.01.026; PMID: 22424773
- Jacquet P. Sensitivity of germ cells and embryos to ionizing radiation. J Biol Regul Homeost Agents 2004; 18:106 - 14; PMID: 15471212
- Wang QL, Sun SC, Han J, Kwak YC, Kim NH, Cui XS. Doxorubicin induces early embryo apoptosis by inhibiting poly(ADP ribose) polymerase. In Vivo 2012; 26:827 - 34; PMID: 22949597
- Yamauchi H, Katayama K, Ueno M, Kanemitsu H, Nam C, Mikami T, Saito A, Ishida Y, Uetsuka K, Doi K, et al. Etoposide induces TRP53-dependent apoptosis and TRP53-independent cell cycle arrest in trophoblasts of the developing mouse placenta. Biol Reprod 2009; 80:813 - 22; http://dx.doi.org/10.1095/biolreprod.108.069419; PMID: 19109225
- Matsuzuka T, Ozawa M, Nakamura A, Ushitani A, Hirabayashi M, Kanai Y. Effects of heat stress on the redox status in the oviduct and early embryonic development in mice. J Reprod Dev 2005; 51:281 - 7; http://dx.doi.org/10.1262/jrd.16089; PMID: 15699582
- Borhani N, Rajaei F, Salehi Z, Javadi A. Analysis of DNA fragmentation in mouse embryos exposed to an extremely low-frequency electromagnetic field. Electromagn Biol Med 2011; 30:246 - 52; http://dx.doi.org/10.3109/15368378.2011.589556; PMID: 22047462
- Luo Q, Yang J, Zeng QL, Zhu XM, Qian YL, Huang HF. 50-Hertz electromagnetic fields induce gammaH2AX foci formation in mouse preimplantation embryos in vitro. Biol Reprod 2006; 75:673 - 80; http://dx.doi.org/10.1095/biolreprod.106.052241; PMID: 16855210
- Streffer C. Chromosomal damage in preimplantation mouse embryos and its development through the cell cycle. Mutat Res 1993; 299:313 - 5; http://dx.doi.org/10.1016/0165-1218(93)90108-P; PMID: 7683099
- Tarkowski AK. Experiments on the development of isolated blastomers of mouse eggs. Nature 1959; 184:1286 - 7; http://dx.doi.org/10.1038/1841286a0; PMID: 13836947
- Van de Velde H, Cauffman G, Tournaye H, Devroey P, Liebaers I. The four blastomeres of a 4-cell stage human embryo are able to develop individually into blastocysts with inner cell mass and trophectoderm. Hum Reprod 2008; 23:1742 - 7; http://dx.doi.org/10.1093/humrep/den190; PMID: 18503052
- Moore NW, Adams CE, Rowson LE. Developmental potential of single blastomeres of the rabbit egg. J Reprod Fertil 1968; 17:527 - 31; http://dx.doi.org/10.1530/jrf.0.0170527; PMID: 5715682
- Willadsen SM. The development capacity of blastomeres from 4- and 8-cell sheep embryos. J Embryol Exp Morphol 1981; 65:165 - 72; PMID: 6895911
- Chi HJ, Koo JJ, Choi SY, Jeong HJ, Roh SI. Fragmentation of embryos is associated with both necrosis and apoptosis. Fertil Steril 2011; 96:187 - 92; http://dx.doi.org/10.1016/j.fertnstert.2011.04.020; PMID: 21550043
- Toyoshima M. Analysis of p53 dependent damage response in sperm-irradiated mouse embryos. J Radiat Res 2009; 50:11 - 7; http://dx.doi.org/10.1269/jrr.08099; PMID: 19218778
- Cryns V, Yuan J. Proteases to die for. Genes Dev 1998; 12:1551 - 70; http://dx.doi.org/10.1101/gad.12.11.1551; PMID: 9620844
- Kamjoo M, Brison DR, Kimber SJ. Apoptosis in the preimplantation mouse embryo: effect of strain difference and in vitro culture. Mol Reprod Dev 2002; 61:67 - 77; http://dx.doi.org/10.1002/mrd.1132; PMID: 11774377
- Hao Y, Lai L, Mao J, Im GS, Bonk A, Prather RS. Apoptosis and in vitro development of preimplantation porcine embryos derived in vitro or by nuclear transfer. Biol Reprod 2003; 69:501 - 7; http://dx.doi.org/10.1095/biolreprod.103.016170; PMID: 12700186
- Weil M, Jacobson MD, Coles HS, Davies TJ, Gardner RL, Raff KD, Raff MC. Constitutive expression of the machinery for programmed cell death. J Cell Biol 1996; 133:1053 - 9; http://dx.doi.org/10.1083/jcb.133.5.1053; PMID: 8655578