Diffuse large B-cell lymphoma (DLBCL) is the most frequent lymphoma subtype and accounts for approximately 30–40% of all cases in adults.Citation1 DLBCL represents a heterogeneous diagnostic category with respect to morphology, biology, and clinical outcome.Citation1 The use of gene expression profiling significantly improved our understanding of this heterogeneity, as 2 major molecular DLBCL subtypes that arise from different stages of B-cell differentiation were identified by this approach.Citation2 Germinal center B-cell-like (GCB) DLBCLs are derived from germinal center B-cells and, accordingly, express genes such as BCL6 or LMO2 that are detectable in normal germinal center B-cells. In contrast, activated B-cell-like (ABC) DLBCLs originate from activated B-cells that are in the process of differentiation into plasma cells. Intriguingly, these entities not only differ with respect to their gene expression profile, but they are also characterized by significant differences in survival when treated with standard therapy.Citation3 Finally, ABC and GCB DLBCLs are addicted to different oncogenic pathways due to divergent genetic aberrations.
A hallmark of ABC DLBCLs is their dependency to constitutive activity of the oncogenic nuclear factor kappa B (NFκB) pathway that is caused by recurrent mutations in positive and negative regulators of this pathway.Citation4 In contrast, GCB DLBCLs are characterized by different genetic aberrations, such as BCL2 translocations leading to inhibition of apoptosis, or by somatically acquired mutations affecting EZH2 that encode for a histone methyltransferase.Citation5
However, recently we have shown that heterogeneity prevails even within these clearly defined molecular subtypes, when we screened primary DLBCL patient samples for the expression of the tumor suppressor PTEN (phosphatase and tensin homolog, ).Citation6 PTEN is the physiologic antagonist of the oncogenic phosphoinositide 3-kinase (PI3K)/protein kinase B (AKT) pathway.Citation7 Constitutive activation of PI3K/AKT is a hallmark of various different cancer types. The PI3K signaling cascade is initiated with the phosphorylation of phosphatidylinositol 4,5-bisphosphate (PIP2) to phosphatidylinositol 3,4,5-trisphosphate (PIP3). The conversion to PIP3 is tightly regulated by the opposing activities of the lipid phosphatase PTEN and class I PI3K family members. The PI3Ks phosphorylate PIP2 to PIP3, whereas PTEN hydrolyzes the 3-phosphate to generate PIP2. Upon PTEN loss PIP3 accumulates, and AKT and mTOR are activated, promoting cell survival, proliferation, and cell growth.Citation7
Figure 1. Combination of gene expression profiling and immunohistochemical PTEN staining defines a germinal center B-cell-like subtype that is dependent on PI3K/AKT and MYC signaling.
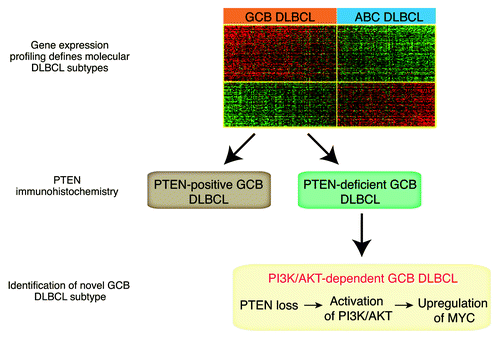
We detected that more than 50% of primary GCB DLBCL patient samples are characterized by loss of PTEN protein expression.Citation6 In contrast, PTEN is expressed in the vast majority of ABC DLBCLs. Loss of PTEN in GCB DLBCLs is inversely correlated with constitutive activation of the PI3K/AKT signaling pathway, and functional analyses demonstrated a dependency on PI3K signaling in these lymphomas. In contrast, PI3K/AKT activation is rarely detectable in PTEN-positive GCB DLBCLs. These results indicate that loss of PTEN is the predominant molecular mechanism of PI3K/AKT activation in GCB DLBCL. Further analyses showed that the addiction to PI3K/AKT signaling in these lymphomas is in part caused by upregulation of the transcription factor MYC. Inhibition of PI3K/AKT either by re-expression of PTEN or by pharmacologic inhibition using a PI3K inhibitor, significantly downregulated MYC protein expression, suggesting that PTEN loss leads to upregulation of MYC via constitutive activation of PI3K/AKT.
The molecular mechanisms that cause PTEN loss in GCB DLBCL remain largely unknown. Deletions of the PTEN locus on chromosome 10q23 as well as somatically acquired PTEN mutations are only detectable in the minority of PTEN-deficient GCB DLBCL cases.Citation6,Citation8 In contrast, in the vast majority of primary GCB DLBCL patient samples, we could not uncover the molecular mode of PTEN silencing. Various microRNAs (miRNAs), such as miR-17–92 or miR-21, have been shown to downregulate PTEN expression.Citation7 It is conceivable that miRNAs play a crucial role in the regulation of PTEN in GCB DLBCL. Data from a recent study using array comparative genomic hybridization (aCGH) to investigate DLBCL patient samples identified recurrent amplifications of the miR-17–92 locus and subsequent overexpression in more than 10% of GCB DLBCLs.Citation8 Intriguingly, these aberrations were not detectable in other molecular DLBCL subtypes,Citation8 supporting the notion that PTEN loss is a specific feature of GCB DLBCLs.
From a clinical point of view, these data might be of major importance. Using a PI3K inhibitor, only PTEN-deficient cell line models responded to PI3K inhibition. In contrast, PTEN-positive models were resistant to inhibitor treatment, indicating that the PTEN protein expression status can be utilized to predict response to PI3K inhibitor treatment (). These results underscore the necessity to stratify patients according to their oncogenic dependencies. To this end, techniques such as gene expression profiling or next generation sequencing need to be implemented in clinical trials to characterize patients upfront of therapy, especially if specific inhibitors are incorporated in these trials. This approach will lead to a better understanding of the responses achieved by novel compounds and will eventually pave the way to more specific and less toxic treatment regimens in DLBCL.
References
- Nogai H, et al. J Clin Oncol 2011; 29:1803 - 11; http://dx.doi.org/10.1200/JCO.2010.33.3252; PMID: 21483013
- Alizadeh AA, et al. Nature 2000; 403:503 - 11; http://dx.doi.org/10.1038/35000501; PMID: 10676951
- Lenz G, et al, Lymphoma/Leukemia Molecular Profiling Project. N Engl J Med 2008; 359:2313 - 23; http://dx.doi.org/10.1056/NEJMoa0802885; PMID: 19038878
- Compagno M, et al. Nature 2009; 459:717 - 21; http://dx.doi.org/10.1038/nature07968; PMID: 19412164
- Morin RD, et al. Nat Genet 2010; 42:181 - 5; http://dx.doi.org/10.1038/ng.518; PMID: 20081860
- Pfeifer M, et al. Proc Natl Acad Sci U S A 2013; 110:12420 - 5; http://dx.doi.org/10.1073/pnas.1305656110; PMID: 23840064
- Song MS, et al. Nat Rev Mol Cell Biol 2012; 13:283 - 96; http://dx.doi.org/10.1038/nrm3330; PMID: 22473468
- Lenz G, et al. Proc Natl Acad Sci U S A 2008; 105:13520 - 5; http://dx.doi.org/10.1073/pnas.0804295105; PMID: 18765795