Abstract
BRCA1 and 53BP1 play decisive roles in the choice of DNA double-strand break repair mechanisms. BRCA1 promotes DNA end resection and homologous recombination (HR) during S/G2 phases of the cell cycle, while 53BP1 inhibits end resection and facilitates non-homologous end-joining (NHEJ), primarily during G1. This competitive relationship is critical for genome integrity during cell division. However, their relationship in the many cells in our body that are not cycling is unknown. We discovered profound differences in 53BP1 and BRCA1 regulation between cycling and non-cycling cells. Cellular growth arrest results in transcriptional downregulation of BRCA1 and activation of cathepsin-L (CTSL)-mediated degradation of 53BP1. Accordingly, growth-arrested cells do not form BRCA1 or 53BP1 ionizing radiation-induced foci (IRIF). Interestingly, cell cycle re-entry reverts this scenario, with upregulation of BRCA1, downregulation of CTSL, stabilization of 53BP1, and 53BP1 IRIF formation throughout the cycle, indicating that BRCA1 and 53BP1 are important in replicating cells and dispensable in non-cycling cells. We show that CTSL-mediated degradation of 53BP1, previously associated with aggressive breast cancers, is an endogenous mechanism of non-cycling cells to balance NHEJ (53BP1) and HR (BRCA1). Breast cancer cells exploit this mechanism to ensure genome stability and viability, providing an opportunity for targeted therapy.
Keywords: :
Introduction
The tumor suppressor proteins 53BP1 and BRCA1 have emerged as key regulators of the choice between DNA double-strand break (DSB) repair mechanisms.Citation1-Citation5 Recruitment of BRCA1 to DSBs promotes the DNA end resection that is necessary for homologous recombination (HR),Citation6 a mechanism that repairs DNA with great fidelity using sister chromatids as templates for recombination.Citation7 In contrast, 53BP1 binding to DSBs inhibits end resection and facilitates non-homologous end joining (NHEJ), which is considered an error-prone mechanism of DNA repair.
A number of studies have addressed the functional relationship between 53BP1 and BRCA1 during the cell cycle in an attempt to understand the contribution of HR and NHEJ to the repair of DNA DSBs at different phases of the cell cycle.Citation8-Citation10 Some studies proposed that HR is the preferred mechanism of DSB repair during S and G2 phases of the cell cycle, while NHEJ functions throughout the cell cycle, being dominant in G1. In HR-deficient cells, NHEJ can repair up to 85% of DSBs in G2, supporting the notion that these 2 pathways compete for repair substrate.Citation11-Citation14 Initial studies showed high levels of BRCA1 in exponentially growing cells, which decreased dramatically upon growth factor withdrawal and arrest in G0/G1,Citation15-Citation17 and a progressive upregulation of BRCA1 and formation of BRCA1/CtIP/MRN DNA repair complexes as cells re-entered the cell cycle.Citation18 These data suggested that the regulation of BRCA1 levels during the cell cycle could be responsible for favoring 53BP1-dependent NHEJ during G1 and HR during S and G2 phases of the cell cycle.
However, recent studies using time-lapse microscopy and fluorescent reporters to monitor DSBs in asynchronous cycling cells have challenged this notion. One study confirmed that early in G1, cells repair DSBs exclusively by NHEJ.Citation19 However, cells damaged toward the end of G1 activate HR, which is most active coinciding with the highest levels of replication during S phase. Cells damaged during late S and G2 exhibit both HR and NHEJ. These findings revealed a relationship between the levels of active replication and HR and indicated that NHEJ is the dominant repair mechanism during G1 and G2 phases. Another study has provided mechanistic insights about the functional antagonistic relationship between these 2 DNA repair pathways in cycling cells.Citation20 The model proposed is somewhat different from the previous study, showing that BRCA1 in complex with CtIP promotes end resection of the DSB, while antagonizing the binding of a complex formed by 53BP1 and Rif1 to the break during S and G2 phases of the cell cycle. Inhibition of 53BP1/Rif1 recruitment requires CDK-mediated phosphorylation of CtIP. In turn, recruitment of the 53BP1/Rif1 complex, which requires ATM-dependent phosphorylation of 53BP1 N terminus, inhibits the recruitment of the BRCA1/CtIP complex during G1. This study proposed a cell cycle-regulated circuit whereby the recruitment of BRCA1/CtIP or 53BP1/Rif1 complexes regulate DNA DSB repair pathway choice to ensure predominance of NHEJ during G1 and of HR during S and G2 phases of the cell cycle. Importantly, the latter study reported that the global levels of BRCA1 do not change during the cell cycle.Citation20
Although the specific contributions of BRCA1 and 53BP1 to DNA repair mechanisms during the cell cycle remain controversial, it is well accepted that a balance between BRCA1 and 53BP1 is important for the maintenance of genome integrity. Accordingly, different lines of evidence have associated loss of either of these proteins with accumulation of DNA damage and genomic instability and with increased cancer incidence.Citation21-Citation25 Interestingly, 53BP1 proficiency seems to play a major role in the profound genomic instability that results from BRCA1 deficiency. Recent groundbreaking studies demonstrated that loss of 53BP1 is synthetically viable with the loss of BRCA1.Citation1-Citation5 Loss of 53BP1 in the context of BRCA1 deficiency allows end resection and partially rescues HR defects. Interestingly, loss of 53BP1 rescued embryonic lethality in BRCA1-deficient mice (BRCA1∆11/∆11). Furthermore, reduced levels of 53BP1 were observed in breast cancers with the poorest prognosis, such as BRCA1-mutated and triple-negative (TNBC). Overall, these studies suggest that loss of 53BP1 contributes to tumor progression, especially in those tumors that present HR deficiencies.
The mechanisms responsible for regulating 53BP1 protein levels in normal cells or downregulating 53BP1 in tumor cells remain poorly understood. Our previous studies identified one mechanism modulating the levels of 53BP1 protein in mammalian cells. Initially, we found that depletion of the structural nuclear proteins A-type lamins activate the degradation of 53BP1 by the cysteine protease cathepsin L (CTSL), which translocates into the nucleus in lamins-deficient cells.Citation26,Citation27 Accordingly, depletion or inhibition of CTSL activity rescued the levels of 53BP1, as well as the defects in NHEJ and the genomic instability observed in lamins-deficient cells.Citation28,Citation29 Most recently, we found that CTSL-mediated degradation of 53BP1 is activated upon depletion of BRCA1 in breast cancer cells, which allows BRCA1-deficient cells to rescue HR and overcome genomic instability and growth arrest.Citation30 Interestingly, inhibition of CTSL stabilized 53BP1 protein in the context of BRCA1 deficiency and increased the sensitivity of BRCA1-deficient cells to DNA damaging strategies. Importantly, we also found an inverse linear correlation between nuclear levels of CTSL and 53BP1 in breast tumors from patients with sporadic TNBC and patients carrying germline mutations in the BRCA1 gene. Altogether, these studies demonstrate a key role for CTSL in the regulation of 53BP1 protein levels, thus supporting the notion that inhibition of CTSL-mediated degradation of 53BP1 could represent a new therapeutic strategy for breast cancers with the poorest prognosis, as well as some lamin-related diseases.
Despite these recent findings, many questions still remain about the regulation of 53BP1 levels and the functional relationship between 53BP1 and BRCA1 during the cell cycle. In addition, the regulation of these proteins in the many cells in the body that are not cycling remains obscure. Here, we monitored the regulation of 53BP1 levels during exit and re-entry into the cell cycle. We find changes in global levels of 53BP1 protein during growth arrest that mirror BRCA1 expression, and that are regulated by CTSL. Importantly, our study reveals profound differences in the regulation of BRCA1 and 53BP1 proteins between cycling and non-cycling cells.
Results
Inverse correlation between CTSL and levels of 53BP1 and BRCA1 proteins in growth-arrested human fibroblasts
Previous studies showed that BRCA1 expression increases as cells progress through the G1 phase of the cell cycle, peak during S phase, and remain elevated at the G2/M transition.Citation15-Citation17 In contrast, a recent study showed that the levels of BRCA1 do not change during the cell cycle.Citation20 Thus, whether or not BRCA1 levels are regulated during the cell cycle remains controversial. Similarly, how the levels of 53BP1 are regulated during the cell cycle or during growth arrest is not known. Given our previous finding that CTSL regulates 53BP1 stability,Citation27-Citation29 we investigated a possible relationship between CTSL and 53BP1 levels in growth-arrested and cycling cells by using human fibroblasts immortalized with telomerase (BJ+hTert), which have intact cell cycle regulation and growth arrest by contact inhibition. BJ+hTert fibroblasts were plated at high confluency and maintained confluent for 72 h to induce growth arrest. At this point, cells were either collected, or replated at low density to allow re-entry into the cell cycle. Samples of cells were collected at different times after re-entry into the cell cycle. We monitored the cell cycle distribution profile at each condition via propidium iodide content. As shown in , asynchronously growing cells show a normal cell cycle profile, while contact inhibited cells accumulate at G0/G1 phase. To determine if confluency arrests cells in G0 or G1, we monitored the presence of Ki67, a marker that is absent in cells arrested in G0. As shown in Figure S1, most of the cells arrested by confluency are positive for Ki67, indicating that cells arrest primarily in G1. also shows that approximately 24 h after re-plating, cells had re-entered the cell cycle and showed a normal cell cycle distribution profile.
Figure 1. Differences in the levels of DNA repair factors between growth-arrested and cycling human fibroblasts. (A) Cell cycle profile of human fibroblasts expressing telomerase (BJ+hTERT) either growing asynchronously, growth arrested by contact inhibition, or released from the growth arrest. Cells were fixed and labeled with propidium iodide and analyzed for fluorescent DNA content using the Nexcelom Cellometer. The percentage of cells in G0/G1, S, and G2/M phases of the cell cycle is shown under each cell cycle profile. (B) Western blots to monitor the levels of the protease cathepsin L (CTSL), DNA repair factors (53BP1, BRCA1, RPA), cell cycle regulators (Cyclin A, p107), and loading control Lamin A/C in growth-arrested and cycling fibroblasts treated with cathepsin inhibitor E64 or vehicle control. Note the marked increase in CTSL and the decrease in 53BP1 and BRCA1 levels in growth-arrested cells and how E64 treatment prevents 53BP1 decrease. (C) Cell cycle profile monitored as in (A) but in cells incubated with 10 μM E64 throughout the time course of the experiment. (D) Western blots in cells treated with E64 to monitor the same proteins as in (B). Note how inhibition of cathepsin activity with E64 prevents the decrease in 53BP1 protein levels. Representative experiments of at least 3 biological repeats are shown.
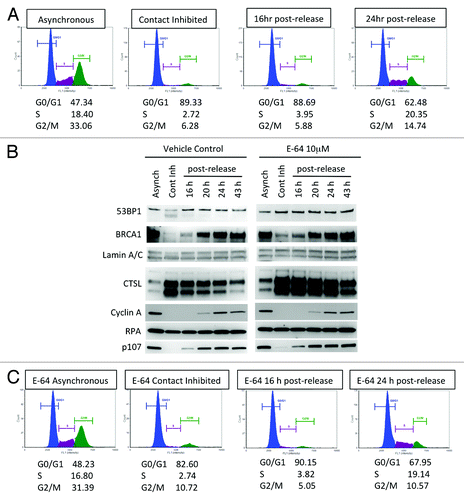
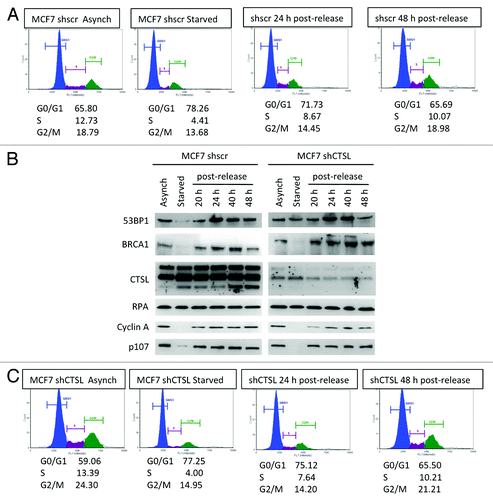
Next, we monitored the levels of BRCA1, CTSL, and 53BP1 during growth arrest and at different times after re-entry into the cell cycle and compared the levels of these proteins to those of asynchronously growing cells (, left panels). Arrest in G1 is accompanied by a profound increase in the levels of CTSL and a decrease in 53BP1 protein compared with asynchronously growing cells. We also observed decreased BRCA1 levels in growth-arrested cells, confirming previous results. As cells re-entered the cell cycle, a reduction in CTSL correlated with increased 53BP1 and BRCA1 protein levels. Consistent with the decrease in CTSL levels upon re-entry into the cell cycle, cathepsin L activity was reduced as early as 16 h post-release from growth arrest (Fig. S2). In contrast, we found that the levels of the single-strand DNA-binding protein RPA remain constant during exit and re-entry into the cell cycle. In addition, we show that the Rb family member p107 also mirror 53BP1 and BRCA1 expression, with low levels in growth-arrested cells, which increase as cells re-enter the cell cycle. Changes in the levels of Cyclin A throughout the course of the experiment were monitored as an additional indicator of cell cycle progression, as Cyclin A is markedly reduced in G1 phase. We monitored levels of A-type lamins (Lamin A/C) as loading control. The data demonstrate that 53BP1 protein levels fluctuate during exit and re-entry into the cell cycle, mirroring the levels of BRCA1, and that CTSL inversely correlates with the levels of both proteins.
To determine whether CTSL contributes to 53BP1 downregulation in growth-arrested cells, we inhibited CTSL activity in BJ+hTert fibroblasts via treatment with the cathepsin inhibitor E64 (10 μM) throughout the time course of the experiment. As shown in , treatment with E64 did not modify the cell cycle profile at any of the conditions tested. Interestingly, we found that E64 treatment prevented the decrease of 53BP1 in contact-inhibited cells (, right panels). In contrast, E64 had no effect on the levels of 53BP1 or any of the other proteins tested in asynchronously growing cells or in cells that re-entered the cell cycle. These results indicate that CTSL participates in the regulation of 53BP1 levels specifically in fibroblasts arrested in G1 by contact inhibition.
Transcriptional regulation of CTSL, 53BP1, and BRCA1 in growth-arrested fibroblasts
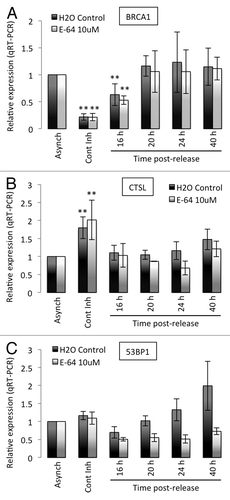
The above results show profound changes in the levels of CTSL, 53BP1, and BRCA1 proteins during exit and re-entry into the cell cycle and demonstrate a role for CTSL in the regulation of 53BP1 levels. Here, we determined whether any of these changes are due to transcriptional regulation of the genes. We performed qRT-PCR in BJ+hTert fibroblasts growing asynchronously, growth arrested by contact inhibition, or allowed to re-enter the cell cycle. We compared the levels of CTSL, 53BP1, and BRCA1 transcripts between cells treated with vehicle control (H2O), or with E64 to inhibit cathepsin activity. As shown in , the levels of BRCA1 transcripts are markedly decreased in contact-inhibited cells and progressively increase as cells re-enter the cell cycle, consistent with previous reports. Importantly, we found that the levels of BRCA1 transcripts are not affected by inhibition of CTSL activity at any condition tested. In contrast, we observed a significant upregulation of CTSL transcripts in growth-arrested cells (). Treatment with E64 did not have a profound effect in the levels of CTSL transcripts throughout the time course of the experiment. Thus, the levels of BRCA1 and CTSL transcripts correlate with proteins levels, indicating that both proteins are regulated transcriptionally during growth arrest. Interestingly, we found that the levels of 53BP1 transcripts remain constant upon contact inhibition (), despite the marked decrease in 53BP1 protein (, left panels). Furthermore, treatment with E64 does not alter the levels of 53BP1 transcripts during growth arrest, although it stabilizes the levels of 53BP1 protein (, right panels). These results indicate that 53BP1 protein stability is regulated during growth arrest, and that CTSL plays a major role in the degradation of 53BP1. Intriguingly, we also found that inhibition of CTSL by E64 treatment results in a consistent downregulation of 53BP1 transcripts levels as cells re-enter the cell cycle. However, these changes in transcription do not translate into decreased 53BP1 protein levels. These results suggest that CTSL impacts on 53BP1 expression at different levels. On one hand, CTSL activity mediates degradation of 53BP1 protein in growth-arrested cells while maintaining 53BP1 transcripts levels in cycling cells.
Figure 2. Transcripts levels of BRCA1, CTSL, and 53BP1 in growth-arrested and cycling human fibroblasts. (A) Quantitation of BRCA1 transcripts levels by qRT-PCR in human fibroblasts expressing telomerase (BJ+hTERT) either growing asynchronously, growth arrested by contact inhibition, or released from the growth arrest. Relative expression is compared in all conditions between cells incubated in water as control and cells incubated with 10 μM E64. (B) Quantitation of CTSL transcripts levels as in (A). (C) Quantitation of 53BP1 transcripts levels as in (A). **Represents P value of statistical significance (P ≤ 0.05) of that sample compared with all the other samples in the experiment. The complete statistical analysis of all the samples under the different conditions is shown in supplemental information.
Formation of 53BP1 and BRCA1 IRIF in growth-arrested and cycling fibroblasts
The decrease in global levels of 53BP1 and BRCA1 shown above suggests that growth-arrested fibroblasts could be compromised in their ability to accumulate these DNA repair factors at sites of DNA damage. To test this hypothesis, we performed immunofluorescence (IF) with 53BP1 and BRCA1 antibodies in cells contact-inhibited and cells growing asynchronously that are irradiated with 8 Gy or mock irradiated. Upon irradiation, 53BP1 and BRCA1 proteins are recruited to sites of DNA damage, forming ionizing radiation-induced foci (IRIF). shows representative images of BRCA1 content in growth-arrested and asynchronous fibroblasts in the absence of ionizing radiation. In growth-arrested cells, BRCA1-positive cells are rarely observed. In contrast, cells positive for BRCA1 are readily observed in asynchronously growing cells, as expected. Interestingly, the few cells positive for BRCA1 consistently showed higher label intensity with DAPI, indicating that these cells have higher DNA content, thus representing a post-replication stage of the cell cycle (S or G2 phases). The graph in shows the quantitation of global BRCA1 fluorescence intensity in asynchronous and growth-arrested cells. We found a statistically significant decrease in BRCA1 intensity in arrested cells. Next, we compared the formation of BRCA1 IRIF between growth-arrested and asynchronously growing cells (). We found that a high percentage of asynchronously growing cells present with BRCA1 IRIF. In contrast, only a few growth-arrrested cells present with BRCA1 IRIF, consistent with the global decrease of BRCA1 protein levels in these cells. As a consequence, the global intensity of BRCA1 fluorescence is markedly decreased in irradiated arrested fibroblasts. Thus, non-cycling fibroblasts are unable to form BRCA1 foci after IR.
Figure 3. Levels of BRCA1 and 53BP1 and formation of IRIF in growth-arrested and cycling cells. (A) Immunofluorescence with BRCA1 antibody was performed in asynchronous and contact-inhibited cells that were irradiated with 8 Gy or mock irradiated. DAPI staining was used to identify all nuclei. Graph shows quantitation of the intensity of fluorescence (relative fluorescence units) in 200 cells per condition. (B) IF with BRCA1 antibody in cells of the same growth conditions as (A) after irradiation with 8 Gy.Note the decrease in BRCA1 intensity of fluorescence, both in control and irradiated growth-arrested cells. (C) The same experiments as in (A) were performed with 53BP1 antibody. (D) The same experiments as in (B) were performed with 53BP1 antibody.Note how 53BP1 levels mirror BRCA1 in all conditions tested, with low levels of 53BP1 in growth-arrested cells irradiated and control. *Represents P value of statistical significance (P ≤ 0.05).
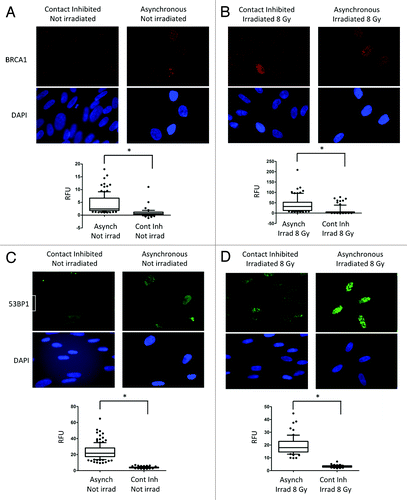
In addition, we compared the content of 53BP1 between growth-arrested and asynchronously growing fibroblasts by IF in the absence of IR. As shown in , growth-arrested fibroblasts show a marked decrease in the intensity of labeling with 53BP1 antibody, consistent with the decrease in the global levels of the protein. Similarly, we found a striking difference in the intensity of 53BP1 IRIF between growth-arrested and asynchronously growing cells (). We confirmed the specificity of the antibody by performing immunofluorescence and immunohistochemistry in cells depleted of 53BP1 via lentiviral transduction with shRNA (Fig. S3). Overall, the data demonstrate that non-cycling cells do not accumulate 53BP1 at DNA repair foci.
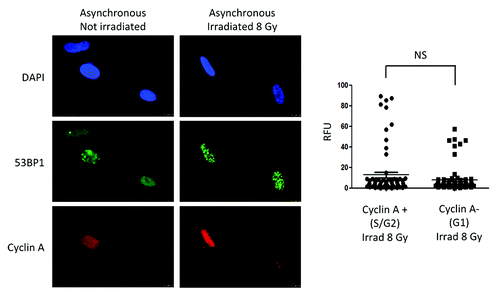
Next, we tested whether activation of CTSL-mediated degradation of 53BP1 is specific of G1-arrested cells, or if it is also a characteristic of the G1 phase in cycling cells. We took advantage of the fact that cyclin A is absent in this phase of the cell cycle. Thus, we performed dual immunofluorescence with cyclin A and 53BP1 antibodies in asynchronous cells control or irradiated with 8 Gy. As shown in , cells positive and negative for cyclin A present with similar global levels of 53BP1. In addition, cells in the G1 phase of the cell cycle (cyclin A-negative) were able to form 53BP1 IRIF with the same intensity as cells in S/G2 phases of the cell cycle (cyclin A-positive), as assessed by quantitation of intensity of fluorescence (graph). These results are consistent with previous studies showing that 53BP1 IRIF can form in all phases of the cell cycle.Citation19,Citation20,Citation31 However, one study in hTert-immortalized retinal pigment epithelial cells (RPE1), showed expression of 53BP1 in cells growth arrested by serum deprivation.Citation32 This discrepancy suggests that different cell lines might regulate 53BP1 in a distinct manner. Whether or not growth-arrested hTert-RPE1 cells can activate CTSL remains unknown. Overall, our data demonstrate profound differences in the regulation of 53BP1 between cycling and non-cycling human fibroblasts. Non-cycling fibroblasts activate CTSL-mediated degradation of 53BP1, while cycling fibroblasts maintain constant levels of 53BP1 throughout the cell cycle.
Figure 4. Levels of 53BP1 and formation of IRIF in cycling cells. Immunofluorescence with 53BP1 and Cyclin A antibodies was performed in asynchronously growing cells that were irradiated with 8 Gy or mock irradiated. DAPI staining was used to identify all nuclei. Cyclin A positive cells represent cells in S/G2 phases of the cell cycle, while Cyclin A-negative cells represent cells in G1. Graphs show quantitation of the intensity of fluorescence (relative fluorescence units) in 200 cells per condition. We compared intensity of fluorescence of 53BP1 between Cyclin A-positive (S/G2) and Cyclin A-negative (G1) cells that were irradiated with 8 Gy. Statistically significant differences were not observed (NS).
Inverse correlation between CTSL and levels of 53BP1 and BRCA1 proteins in serum-starved MCF7 cells
Our results indicate that BJ+hTert fibroblasts growth arrested in G1 by contact inhibition transcriptionally downregulate BRCA1 and activate CTSL-mediated degradation of 53BP1. Our previous studies had shown that breast cancer cells (MCF7) growth arrested by serum deprivation exhibit high levels of CTSL and low levels of BRCA1 and 53BP1.Citation30 Here, we determined whether CTSL is responsible for the decrease of 53BP1 in these cells. MCF7 cells were lentivirally transduced with shRNA specific for depletion of CTSL (shCTSL) or shRNA control (shscr). Both cell lines were grown in complete media or media with 0.1% serum (starved) for 48 h and either collected immediately, or re-plated in complete media containing 10% serum to allow re-entry into the cell cycle. shows the cell cycle profile of cells subjected to the different treatments assessed by propidium iodide staining. Serum deprivation led to growth arrest of MCF7 cells, and approximately 24 h post-release, cells exhibit a normal cell cycle profile. Subsequently, we monitored the levels of 53BP1, BRCA1, and CTSL proteins throughout the time course of the experiment. As shown in (left panels), growth arrest by serum starvation of MCF7 cells proficient in CTSL leads to a marked decrease in the levels of BRCA1 and 53BP1 proteins, concomitant with an increase in CTSL protein, as previously reported.Citation30 In addition, an increase in 53BP1 and BRCA1 protein levels are observed upon re-entry of MCF7 cells into the cell cycle. Thus, exit and re-entry into the cell cycle of MCF7 cells recapitulates the phenotype observed in BJ+hTert fibroblasts, indicating that this is a phenomenon induced by different types of growth arrest and in different cell types. Interestingly, we find that depletion of CTSL prevents the decrease of 53BP1 in serum-starved MCF7 cells (, right panels), indicating that CTSL is responsible for the decrease in 53BP1 protein in this context. Importantly, depletion of CTSL does not affect the cell cycle profile at any of the conditions tested (). In contrast, the levels of BRCA1 were unaffected by the loss of CTSL, showing a decrease in growth-arrested cells. Consistent with observations in BJ+hTert fibroblasts, the levels of RPA were constant throughout the cell cycle, and the levels of Cyclin A and p107 were downregulated in growth-arrested cells. Altogether, our results demonstrate that CTSL plays a role in the regulation of 53BP1 protein levels in growth-arrested cells, allowing 53BP1 protein levels to mirror levels of BRCA1 throughout the cell cycle.
Figure 5. Differences in the levels of DNA repair factors between growth-arrested and cycling breast cancer cells. (A) Cell cycle profile of breast cancer cells (MCF7) either growing asynchronously, growth arrested by serum deprivation (starved), or released from the growth arrest by serum supplementation. Cells were fixed and labeled with propidium iodide and analyzed for fluorescent DNA content using the Nexcelom Cellometer. The percentage of cells in G0/G1, S, and G2/M phases of the cell cycle is shown under each cell cycle profile. (B) Western blots to monitor the levels of the protease cathepsin L (CTSL), DNA repair factors (53BP1, BRCA1, and RPA), and cell cycle regulators (Cyclin A, p107) in MCF7 cells proficient or deficient in CTSL (shscr and shCTSL, respectively). Note the increase in CTSL and the marked decrease in 53BP1 and BRCA1 levels in growth-arrested cells. (C) Cell cycle profile monitored as in (A), but in MCF7 cells depleted of CTSL via lentiviral transduction with a specific shRNA (shCTSL). The blots shown in (B) represent the same experiment and thus levels of all proteins can be compared. Note how depletion of cathepsin L prevents the decrease in 53BP1 protein levels. Representative experiments of at least 3 biological repeats are shown.
Transcriptional regulation of CTSL, 53BP1, and BRCA1 during growth arrest in MCF7 cells
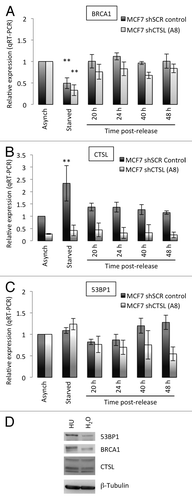
Here, we determined whether the changes in CTSL, 53BP1, and BRCA1 protein levels observed in MCF7 cells during the cell cycle correlate with levels of transcription of the corresponding genes. We also compared transcripts levels between MCF7 cells proficient and deficient in CTSL. We performed qRT-PCR in MCF7 cells growing asynchronously, cells growth arrested by serum starvation, and cells that re-entered the cell cycle. As shown in , serum starvation leads to a statistically significant downregulation of BRCA1 transcripts levels, consistent with the decrease in BRCA1 protein, and recapitulating the phenotype of fibroblasts growth arrested by contact inhibition. In addition, we found an upregulation of BRCA1 transcripts as MCF7 cells re-entered the cell cycle, also consistent with the results in fibroblasts. Importantly, depletion of CTSL did not affect the levels of BRCA1 transcripts at any of the conditions tested, indicating that CTSL does not impact on BRCA1 expression during the cell cycle. In we show the marked depletion of CTSL transcripts achieved by transduction with the specific shRNA. Importantly, serum starvation leads to upregulation of CTSL transcripts, which are downregulated upon re-entry of the cells into the cell cycle. This is also consistent with the results obtained in fibroblasts. Lastly, we find that growth arrest of MCF7 cells by serum starvation does not alter the levels of 53BP1 transcripts despite the marked downregulation of 53BP1 protein. These results are consistent with activation of CTSL-mediated degradation of 53BP1 protein upon growth arrest in different cell lines and by different means. Importantly, treatment of MCF7 cells with hydroxyurea (HU) to inhibit DNA replication and arrest cells in S phase does not lead to downregulation of BRCA1 or activation of CTSL-mediated degradation of 53BP1 (). Rather, increased levels of 53BP1 and BRCA1 proteins are observed in HU-treated MCF7 cells. These results suggest that the decrease of 53BP1 and BRCA1 proteins is specific to growth arrest prior to replication.
Figure 6. Transcripts levels of BRCA1, CTSL, and 53BP1 in growth-arrested and cycling breast cancer cells. (A) Quantitation of BRCA1 transcripts levels by qRT-PCR in breast cancer cells (MCF7) either growing asynchronously, growth arrested by serum deprivation (starved), or released from the growth arrest by serum supplementation. Relative expression is compared in all conditions between CTSL-proficient (shSCR) and CTSL-deficient (shCTSL) cells. (B) Quantitation of CTSL transcripts levels as in (A). (C) Quantitation of 53BP1 transcripts levels as in (A). **Represents P value of statistical significance (P ≤ 0.05) of that sample compared with all the other samples in the experiment. The complete statistical analysis of all the samples under the different conditions is shown in supplemental information. (D) MCF7 cells growing asynchronously were incubated in the presence of hydroxyurea (HU 1mM) or vehicle control (H2O) overnight and processed for western blot to monitor the levels of CTSL, 53BP1, and BRCA1. Note how HU treatment slightly increases the levels of 53BP1 and BRCA1.
Discussion
There is substantial evidence that NHEJ is active in all phases of the cell cycle, while HR is particularly important in S/G2 phases.Citation12 Some studies have shown that BRCA1 levels fluctuate during the cell cycle and markedly increase during S/G2 phases,Citation16,Citation17 while others have reported constant levels of BRCA1 during the cell cycle.Citation20 Recent elegant studies in asynchronously growing cells proposed the notion of a cell cycle-regulated circuit, whereby the recruitment of BRCA1/CtIP or 53BP1/Rif1 complexes regulate the choice of DNA DSB repair pathway during the cell cycle.Citation20 In this model, the choice of mechanism of DNA repair is regulated by recruitment, not by changes in the levels of the proteins/complexes in cycling cells. The present study investigated whether a similar functional relationship exists in non-cycling cells, which is a state of many cells in the body that are not dividing, but that are poised to respond to proliferation stimuli. We find profound differences in the regulation of 53BP1 and BRCA1 between cycling and non-cycling cells. Growth-arrested cells exhibit a marked downregulation of BRCA1 transcripts and protein levels as well as upregulation of CTSL and activation of CTSL-mediated degradation of 53BP1. Importantly, cells that are allowed to re-enter the cell cycle revert to a cycling phenotype, with upregulation of BRCA1 and cessation of CTSL-mediated degradation of 53BP1. These results suggest that BRCA1 and 53BP1 play an important role in replicating cells, but seem to be dispensable in non-dividing cells.
An important question is why growth-arrested cells activate CTSL-mediated degradation of 53BP1. Based on previous studies showing that 53BP1 plays a major role in the genomic instability that characterizes BRCA1-deficient cells,Citation1-Citation5 it is tempting to speculate that a scenario in which BRCA1 is downregulated, keeping high levels of 53BP1, would be detrimental for the cells. In fact, MCF7 cells deficient in both BRCA1 and 53BP1 do not present profound genomic instability.Citation30 However, stabilization of 53BP1 in BRCA1-deficient cells induces a marked increase in genomic instability in response to IR or treatment with PARP inhibitors. These studies suggest that a balance between BRCA1 and 53BP1 is essential for the maintenance of genomic stability. As such, maintenance of 53BP1 levels in the context of BRCA1 deficiency, or maintenance of BRCA1 levels in the context of 53BP1 deficiency are associated with defects in DNA repair, genomic instability, and increased cancer incidence.Citation21-Citation25
We envision that a sensor mechanism could potentially activate CTSL-mediated degradation of 53BP1 to mirror the low levels of BRCA1 in non-cycling cells. Once BRCA1 is upregulated by re-entry into the cell cycle, cells stabilize 53BP1 protein levels. Thus, it is possible that the downregulation of BRCA1 in non-cycling cells is triggering the activation of CTSL-mediated degradation of 53BP1. Although we do not know the nature of such a “sensing” mechanism, this scenario resembles other observations made by us in cells subjected to different stresses. For example, we previously found that depletion of BRCA1 in breast cancer cells activates CTSL-mediated degradation of 53BP1, which, in turn, allows these cells to overcome genomic instability and growth arrest.Citation30 In addition, we demonstrated that CTSL-mediated degradation of 53BP1 is also activated upon depletion of the structural nuclear proteins A-type lamins.Citation28 Interestingly, lamins-deficient cells also downregulate BRCA1 transcripts and protein levels, thus providing another scenario in which loss of BRCA1 coincides with degradation of 53BP1 by CTSL.Citation29 Other reports have also shown activation of CTSL as well as its accumulation in the nucleus in response to expression of oncogenic Ras, although the degradation of 53BP1 in this context was not tested.Citation33,Citation34 Intriguingly, expression of Ras inactivates the BRCA1 DNA repair function by dissociating BRCA1 from chromatin.Citation35
Altogether, the data suggests that this 53BP1 degradation mechanism is activated under different stress conditions, and that in many cases the process is associated with loss of BRCA1 function. Elucidating the molecular mechanisms responsible for activating CTSL-mediated degradation of 53BP1 could reveal new information linking this process with the loss of BRCA1 function. We envision that future studies need to address whether cancer-related mutations in BRCA1 activate this degradation pathway,Citation36 which could have important consequences for tumorigenesis. Furthermore, given that levels of 53BP1 have been linked to increased species lifespan,Citation37 understanding the regulation of 53BP1 levels will be relevant for aging. Overall, our findings support an important role for CTSL not only in the regulation of 53BP1 stability in tumor cells, but also as a regulator of protein stability during growth arrest in both normal and tumor cells. In addition, our studies identify CTSL-mediated degradation of 53BP1 as a mechanism activated in non-cycling cells, which interestingly is exploited by BRCA1-deficient tumor cells to reduce genomic instability and ensure proliferation and viability.Citation30 Furthermore, levels of CTSL inversely correlate with levels of 53BP1 in breast cancers with the poorest prognosis, such as triple-negative and BRCA1-mutated. Therefore, this mechanism of 53BP1 loss provides a new target for the development of therapeutic strategies for specific types of cancers.
Materials and Methods
Cell culture
Human fibroblasts expressing telomerase (BJ+hTERT) and MCF7 cells were maintained in DMEM supplemented with 10% FBS, antibiotics, and antimycotics (complete media).
E64 treatment
Cells were incubated with the broad-spectrum cathepsin inhibitor E-64 (E3132, Sigma-Aldrich) diluted in H2O at a concentration of 10 µM for the whole duration of the experiments.
Ionizing radiation
For assaying IRIF formation, cells were irradiated with 8 Gy and fixed and processed for immunofluorescence 1 h post-irradiation.
Contact inhibition of fibroblasts
Eight × 106 BJ+hTERT fibroblasts were plated in 100-mM tissue culture (TC) dishes in complete media. Cells were maintained confluent for 72 h prior to being collected as “contact inhibited” or plated in complete media at 1 × 106 cells in 100 mM TC dishes to allow re-entry into the cell cycle. For time point collection, all media, washes, and trypsinized cells were collected by centrifugation for analysis.
Serum starvation of MCF7 cells
MCF7 cells were plated in DMEM containing 0.1% FBS, antibiotics and antimycotics (starvation media) and maintained in this media for 48 h prior to being collected as “serum starved” or media being refreshed with FBS to a final concentration of 10% to allow re-entry into the cell cycle. For time point collection, all media, washes and trypsinized cells were collected by centrifugation for analysis.
Immunoblotting
Cells were lysed in RIPA buffer (150 mM NaCl, 50 mM Tris-HCl pH 7.4, 1% NP-40, 0.2% SDS, 0.25% sodium deoxycholate, and 1 mM EDTA) supplemented with HALT protease and phosphatase inhibitor cocktail (Thermo Pierce). Lysates were sheared using 10 passes through a 26-gauge needle followed by 10 passes through a 30-gauge needle and sonication for 7 min. Protein concentration was determined using the BioRad Dc Protein Assay, and 60–80 μg of total protein was separated by SDS-PAGE on a 4–15% Criterion TGX Gel (Bio-Rad) and transferred to a nitrocellulose membrane using the Trans-Blot Turbo system (Bio-Rad). Antibodies used include: Lamin A/C (sc-20681, SCBT); 53BP1 (H-300: sc-22760, SCBT); RAD51 (H-92: sc-8349, SCBT); BRCA1 (D-9: sc-6954, SCBT); CTSL (C1391, Leinco); p107 (C-18: sc-318, SCBT); RPA (NA18, Millipore); and Cyclin A (sc-271682, SCBT); donkey anti-rabbit HRP-secondary antibody (ab16284, Abcam); bovine anti-goat HRP-secondary antibody (sc-2352, SCBT); goat anti-mouse HRP-secondary antibody (31430, Thermo Scientific). Nitrocellulose membranes were blocked using 3% milk prepared in PBS+0.1% Tween-20. Antibodies were diluted in 3% milk prepared in PBS+0.1% Tween-20, and membranes were washed 3 times using PBS+0.1% Tween-20 after both primary and secondary antibody incubations. Membranes were developed using Thermo Pierce ECL (Fisher).
Immunofluorescence
Cells growing on coverslips were processed directly for IF, or irradiated with 8 Gy and processed 1 h post-irradiation for 53BP1, or 4 h post irradiation for BRCA1. For immunofluorescence of 53BP1, cells were fixed in 3.7% formaldehyde + 0.2% Triton-X 100 in PBS for 10 min at RT, and blocked for 1 h at 37 °C in 2% BSA/0.1% Triton-X100 in PBS. Incubations with 53BP1 antibody (1:1000, NB100–304, Novus) and Alexa Fluor 488 goat anti-rabbit (1:1000, A11008, Invitrogen) secondary antibody were performed for 1 h at 37 °C. Washes were performed in PBS and slides were counterstained with DAPI in Vectashield (H-1200, Vector Labs). Microscopy and photo capture was performed on a Leica DM5000 B microscope using 63× or 100× oil objective lenses (NA 1.4 and 1.3, respectively) with a Leica DFC350FX digital camera and the Leica Application Suite (Version 4.1.0).
BRCA1 immunofluorescence was performed following a protocol from the Fernandez–Capetillo laboratory. Briefly, cells were washed twice with PBS, and incubated in CSK I buffer (10 mM PIPES pH6.8, 100 mM NaCl, 300 mM sucrose, 3 mM MgCl2, 1 mM EGTA, and 0.5% Triton X-100) for 5 min. Coverslips were washed 5 times with cold PBS and fixed in modified STF buffer (150 mM 2-Bromo-2-nitro-1,3-propanediol, 108 mM diazolidinyl urea, 10 mM Na citrate, and 50 nM EDTA pH 5.7) for 30 min at RT. Coverslips were then washed twice with cold PBS and permeabilized (PB buffer: 100 mM Tris-HCl pH 7.4, 50 mM EDTA pH 8.0, and 0.5% Triton X-100) for 15 min at room temperature followed by 2 washes in PBS. Blocking, antibody staining, and mounting were performed as described above, with BRCA1 antibody (sc-6954, Santa Cruz) diluted at 1:200 and Alexa Fluor 594 goat anti-mouse secondary antibody (A11005, Invitrogen) diluted at 1:1000.
Fluorescence intensity was measured using ImageJ. Measurements were taken individually for each cell, as well as for a small adjacent area to account for background. The corrected total cell fluorescence (CTCF) was obtained by subtracting the product of the cell’s area and the mean fluorescence of the background, from the integrated density of the target cell, following the equation: CTSF = integrated density – (mean background fluorescence × area of cell).
Analysis of cell cycle profile
Cells were collected from asynchronous culture, contact-inhibited culture (fibroblasts), serum-starved culture (MCF7), or after re-entry into the cell cycle at various time points. Cell pellets were washed with PBS once and suspended in 500 μl PBS prior to the dropwise addition of 4.5 ml ice-cold 70% ethanol while being vortexed at 600 rpm to fix. The cell suspension was stored at 4 °C until analysis. Fixed cells were pelleted at 1000 × g, washed with PBS, and suspended in 1 ml propidium iodide staining solution (400 μg /ml). Cells were incubated in the staining solution for 15 min at 37 °C. Stained cells were analyzed for fluorescent DNA content using the Nexcelom Cellometer Vision CBL, and cell cycle profiles were created by the program Fcs Express 4 Research Edition.
Quantitative reverse-transcription PCR
RNA was purified from cell pellets using the Bio-Rad Aurum Kit. cDNA was generated by reverse transcription of 750 ng total RNA using Bio-Rad iScript. qRT-PCR was performed using the 7900HT Fast Real-Time PCR system (Applied Biosystems) with the Taqman® Universal PCR Master Mix and Taqman® probe assays targeting HsBRCA1 (Hs01556193_m1), HsTP53BP1 (Hs00996818_m1), HsCTSL (Hs00377632_m1), and HsPPIB (Hs00168719_m1) according to the manufacturer’s directions. Reactions were performed in triplicate and target gene and endogenous controls were amplified in the same plate. Relative quantitative measurements of target genes were determined by using ΔCt calculations.
Viral transduction
Lentiviral transductions were performed as described.Citation27 Briefly, 293T cells were transfected with viral packaging (pHR’8.2ΔR) and envelope plasmids (p-CMV-VSV-G) along with the vector containing the shRNA of interest (shscramble or shSCR and shCTSL). After 48 h, virus-containing media was harvested to infect target cells (MCF7). Following a 4 h infection, cells were allowed to recover for 48 h and selected with puromycin. Viral envelope and packaging plasmids were gifts from Sheila Stewart (Washington University). shRNAs were obtained from Sigma-Aldrich.
Cathepsin L activity
Cathepsin L and total Cathepsin activity was measured by using the fluorogenic substrate Z-Phe-Arg-AMC from the InnoZyme™ Cathepsin L Activity Kit (EMD Chemicals) following the manufacturer’s instructions. Briefly, 106 cells were lysed in Lysis Buffer (400 mM Na phosphate buffer pH 6, 75 mM NaCl, 4 mM EDTA, 0.25% Triton X-100). One hundred µg of protein extract were used per sample. Samples were measured in duplicate. Cleaved Z-Phe-Arg-AMC substrate was detected by fluorescence emission (Exc. max: 360 nm; Emi. max: 460 nm). Cathepsin L activity was determined in the presence of CA 074, a specific Cathepsin B inhibitor. As a negative control, the Cathepsin L inhibitor Z-FY(t-but)-DMK was used. Total cysteine activity was determined as the cleavage of the substrate in the absence of inhibitors.
Statistical analysis
For all the experiments, a “2-tailed” Student t test was used to calculate statistical significance of the observed differences. Microsoft Excel v.2010 was used for the calculations. In all cases, differences were considered statistically significant when P < 0.05. For some figures the 95% confidence interval based on an exact binomial distribution was calculated to determine significant differences among samples.
| Abbreviations: | ||
| CTSL | = | cathepsin L |
| DSBs | = | double-strand breaks |
| HR | = | homologous recombination |
| NHEJ | = | non-homologous end joining |
| IRIF | = | ionizing radiation induced foci |
Additional material
Download Zip (390.5 KB)Acknowledgments
We thank J Caceres and M Carron for technical assistance. Research in the laboratory of SG was supported by NIGMS Grant RO1 GM094513-01, DOD BCRP Idea Award BC110089, RDA from Siteman Cancer Center, and Presidential Research Award from St Louis University. Authors declare no conflict of interest.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Author contributions
MC and MAN performed most of the experiments, and DAG. and SCY contributed with some initial experiments. SG supervised the research and the preparation of the manuscript.
Related Research Data
References
- Bouwman P, Aly A, Escandell JM, Pieterse M, Bartkova J, van der Gulden H, Hiddingh S, Thanasoula M, Kulkarni A, Yang Q, et al. 53BP1 loss rescues BRCA1 deficiency and is associated with triple-negative and BRCA-mutated breast cancers. Nat Struct Mol Biol 2010; 17:688 - 95; http://dx.doi.org/10.1038/nsmb.1831; PMID: 20453858
- Bunting SF, Callén E, Wong N, Chen HT, Polato F, Gunn A, Bothmer A, Feldhahn N, Fernandez-Capetillo O, Cao L, et al. 53BP1 inhibits homologous recombination in Brca1-deficient cells by blocking resection of DNA breaks. Cell 2010; 141:243 - 54; http://dx.doi.org/10.1016/j.cell.2010.03.012; PMID: 20362325
- Cao L, Xu X, Bunting SF, Liu J, Wang RH, Cao LL, Wu JJ, Peng TN, Chen J, Nussenzweig A, et al. A selective requirement for 53BP1 in the biological response to genomic instability induced by Brca1 deficiency. Mol Cell 2009; 35:534 - 41; http://dx.doi.org/10.1016/j.molcel.2009.06.037; PMID: 19716796
- Lowndes NF. The interplay between BRCA1 and 53BP1 influences death, aging, senescence and cancer. DNA Repair (Amst) 2010; 9:1112 - 6; http://dx.doi.org/10.1016/j.dnarep.2010.07.012; PMID: 20724228
- Bothmer A, Robbiani DF, Feldhahn N, Gazumyan A, Nussenzweig A, Nussenzweig MC. 53BP1 regulates DNA resection and the choice between classical and alternative end joining during class switch recombination. J Exp Med 2010; 207:855 - 65; http://dx.doi.org/10.1084/jem.20100244; PMID: 20368578
- Moynahan ME, Chiu JW, Koller BH, Jasin M. Brca1 controls homology-directed DNA repair. Mol Cell 1999; 4:511 - 8; http://dx.doi.org/10.1016/S1097-2765(00)80202-6; PMID: 10549283
- Johnson RD, Jasin M. Sister chromatid gene conversion is a prominent double-strand break repair pathway in mammalian cells. EMBO J 2000; 19:3398 - 407; http://dx.doi.org/10.1093/emboj/19.13.3398; PMID: 10880452
- Schwarz K, Ma Y, Pannicke U, Lieber MR. Human severe combined immune deficiency and DNA repair. Bioessays 2003; 25:1061 - 70; http://dx.doi.org/10.1002/bies.10344; PMID: 14579247
- Shrivastav M, De Haro LP, Nickoloff JA. Regulation of DNA double-strand break repair pathway choice. Cell Res 2008; 18:134 - 47; http://dx.doi.org/10.1038/cr.2007.111; PMID: 18157161
- van Gent DC, Hoeijmakers JH, Kanaar R. Chromosomal stability and the DNA double-stranded break connection. Nat Rev Genet 2001; 2:196 - 206; http://dx.doi.org/10.1038/35056049; PMID: 11256071
- Beucher A, Birraux J, Tchouandong L, Barton O, Shibata A, Conrad S, Goodarzi AA, Krempler A, Jeggo PA, Löbrich M. ATM and Artemis promote homologous recombination of radiation-induced DNA double-strand breaks in G2. EMBO J 2009; 28:3413 - 27; http://dx.doi.org/10.1038/emboj.2009.276; PMID: 19779458
- Rothka mM K, Krüger I, Thompson LH, Löbrich M. Pathways of DNA double-strand break repair during the mammalian cell cycle. Mol Cell Biol 2003; 23:5706 - 15; http://dx.doi.org/10.1128/MCB.23.16.5706-5715.2003; PMID: 12897142
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17:5497 - 508; http://dx.doi.org/10.1093/emboj/17.18.5497; PMID: 9736627
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4:435 - 45; http://dx.doi.org/10.1038/nrm1127; PMID: 12778123
- Chen Y, Farmer AA, Chen CF, Jones DC, Chen PL, Lee WH. BRCA1 is a 220-kDa nuclear phosphoprotein that is expressed and phosphorylated in a cell cycle-dependent manner. Cancer Res 1996; 56:3168 - 72; PMID: 8764100
- Gudas JM, Li T, Nguyen H, Jensen D, Rauscher FJ 3rd, Cowan KH. Cell cycle regulation of BRCA1 messenger RNA in human breast epithelial cells. Cell Growth Differ 1996; 7:717 - 23; PMID: 8780885
- Vaughn JP, Davis PL, Jarboe MD, Huper G, Evans AC, Wiseman RW, Berchuck A, Iglehart JD, Futreal PA, Marks JR. BRCA1 expression is induced before DNA synthesis in both normal and tumor-derived breast cells. Cell Growth Differ 1996; 7:711 - 5; PMID: 8780884
- Chen L, Nievera CJ, Lee AY, Wu X. Cell cycle-dependent complex formation of BRCA1.CtIP.MRN is important for DNA double-strand break repair. J Biol Chem 2008; 283:7713 - 20; http://dx.doi.org/10.1074/jbc.M710245200; PMID: 18171670
- Karanam K, Kafri R, Loewer A, Lahav G. Quantitative live cell imaging reveals a gradual shift between DNA repair mechanisms and a maximal use of HR in mid S phase. Mol Cell 2012; 47:320 - 9; http://dx.doi.org/10.1016/j.molcel.2012.05.052; PMID: 22841003
- Escribano-Díaz C, Orthwein A, Fradet-Turcotte A, Xing M, Young JT, Tkáč J, Cook MA, Rosebrock AP, Munro M, Canny MD, et al. A cell cycle-dependent regulatory circuit composed of 53BP1-RIF1 and BRCA1-CtIP controls DNA repair pathway choice. Mol Cell 2013; 49:872 - 83; http://dx.doi.org/10.1016/j.molcel.2013.01.001; PMID: 23333306
- Deng CX, Scott F. Role of the tumor suppressor gene Brca1 in genetic stability and mammary gland tumor formation. Oncogene 2000; 19:1059 - 64; http://dx.doi.org/10.1038/sj.onc.1203269; PMID: 10713690
- Morales JC, Xia Z, Lu T, Aldrich MB, Wang B, Rosales C, Kellems RE, Hittelman WN, Elledge SJ, Carpenter PB. Role for the BRCA1 C-terminal repeats (BRCT) protein 53BP1 in maintaining genomic stability. J Biol Chem 2003; 278:14971 - 7; http://dx.doi.org/10.1074/jbc.M212484200; PMID: 12578828
- Nakamura K, Sakai W, Kawamoto T, Bree RT, Lowndes NF, Takeda S, Taniguchi Y. Genetic dissection of vertebrate 53BP1: a major role in non-homologous end joining of DNA double strand breaks. DNA Repair (Amst) 2006; 5:741 - 9; http://dx.doi.org/10.1016/j.dnarep.2006.03.008; PMID: 16644291
- Scully R, Livingston DM. In search of the tumour-suppressor functions of BRCA1 and BRCA2. Nature 2000; 408:429 - 32; http://dx.doi.org/10.1038/35044000; PMID: 11100717
- Ward IM, Minn K, van Deursen J, Chen J. p53 Binding protein 53BP1 is required for DNA damage responses and tumor suppression in mice. Mol Cell Biol 2003; 23:2556 - 63; http://dx.doi.org/10.1128/MCB.23.7.2556-2563.2003; PMID: 12640136
- Gonzalez-Suarez I, Redwood AB, Gonzalo S. Loss of A-type lamins and genomic instability. Cell Cycle 2009; 8:3860 - 5; http://dx.doi.org/10.4161/cc.8.23.10092; PMID: 19901537
- Gonzalez-Suarez I, Redwood AB, Perkins SM, Vermolen B, Lichtensztejin D, Grotsky DA, Morgado-Palacin L, Gapud EJ, Sleckman BP, Sullivan T, et al. Novel roles for A-type lamins in telomere biology and the DNA damage response pathway. EMBO J 2009; 28:2414 - 27; http://dx.doi.org/10.1038/emboj.2009.196; PMID: 19629036
- Gonzalez-Suarez I, Redwood AB, Grotsky DA, Neumann MA, Cheng EH, Stewart CL, Dusso A, Gonzalo S. A new pathway that regulates 53BP1 stability implicates cathepsin L and vitamin D in DNA repair. EMBO J 2011; 30:3383 - 96; http://dx.doi.org/10.1038/emboj.2011.225; PMID: 21750527
- Redwood AB, Perkins SM, Vanderwaal RP, Feng Z, Biehl KJ, Gonzalez-Suarez I, Morgado-Palacin L, Shi W, Sage J, Roti-Roti JL, et al. A dual role for A-type lamins in DNA double-strand break repair. Cell Cycle 2011; 10:2549 - 60; http://dx.doi.org/10.4161/cc.10.15.16531; PMID: 21701264
- Grotsky DA, Gonzalez-Suarez I, Novell A, Neumann MA, Yaddanapudi SC, Croke M, Martinez-Alonso M, Redwood AB, Ortega-Martinez S, Feng Z, et al. BRCA1 loss activates cathepsin L-mediated degradation of 53BP1 in breast cancer cells. J Cell Biol 2013; 200:187 - 202; http://dx.doi.org/10.1083/jcb.201204053; PMID: 23337117
- Sakaue-Sawano A, Kurokawa H, Morimura T, Hanyu A, Hama H, Osawa H, Kashiwagi S, Fukami K, Miyata T, Miyoshi H, et al. Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 2008; 132:487 - 98; http://dx.doi.org/10.1016/j.cell.2007.12.033; PMID: 18267078
- Chapman JR, Sossick AJ, Boulton SJ, Jackson SP. BRCA1-associated exclusion of 53BP1 from DNA damage sites underlies temporal control of DNA repair. J Cell Sci 2012; 125:3529 - 34; http://dx.doi.org/10.1242/jcs.105353; PMID: 22553214
- Collette J, Ulku AS, Der CJ, Jones A, Erickson AH. Enhanced cathepsin L expression is mediated by different Ras effector pathways in fibroblasts and epithelial cells. Int J Cancer 2004; 112:190 - 9; http://dx.doi.org/10.1002/ijc.20398; PMID: 15352030
- Goulet B, Sansregret L, Leduy L, Bogyo M, Weber E, Chauhan SS, Nepveu A. Increased expression and activity of nuclear cathepsin L in cancer cells suggests a novel mechanism of cell transformation. Mol Cancer Res 2007; 5:899 - 907; http://dx.doi.org/10.1158/1541-7786.MCR-07-0160; PMID: 17855659
- Tu Z, Aird KM, Bitler BG, Nicodemus JP, Beeharry N, Xia B, Yen TJ, Zhang R. Oncogenic RAS regulates BRIP1 expression to induce dissociation of BRCA1 from chromatin, inhibit DNA repair, and promote senescence. Dev Cell 2011; 21:1077 - 91; http://dx.doi.org/10.1016/j.devcel.2011.10.010; PMID: 22137763
- Dever SM, Golding SE, Rosenberg E, Adams BR, Idowu MO, Quillin JM, Valerie N, Xu B, Povirk LF, Valerie K. Mutations in the BRCT binding site of BRCA1 result in hyper-recombination. Aging (Albany NY) 2011; 3:515 - 32; PMID: 21666281
- Fink LS, Roell M, Caiazza E, Lerner C, Stamato T, Hrelia S, Lorenzini A, Sell C. 53BP1 contributes to a robust genomic stability in human fibroblasts. Aging (Albany NY) 2011; 3:836 - 45; PMID: 21931182