Abstract
Epigenetic silencing of tumor suppressor genes frequently occurs and may account for their inactivation in cancer cells. We previously demonstrated that miR-29b is a tumor suppressor microRNA (miRNA) that targets de novo DNA methyltransferases and reduces the global DNA methylation of multiple myeloma (MM) cells. Here, we provide evidence that epigenetic activity of miR-29b leads to promoter demethylation of suppressor of cytokine signaling-1 (SOCS-1), a hypermethylated tumor suppressor gene. Enforced expression of synthetic miR-29b mimics in MM cell lines resulted in SOCS-1 gene promoter demethylation, as assessed by Sequenom MassARRAY EpiTYPER analysis, and SOCS-1 protein upregulation. miR-29b-induced SOCS-1 demethylation was associated with reduced STAT3 phosphorylation and impaired NFκB activity. Downregulation of VEGF-A and IL-8 mRNAs could be detected in MM cells transfected with miR-29b mimics as well as in endothelial (HUVEC) or stromal (HS-5) cells treated with conditioned medium from miR-29b-transfected MM cells. Notably, enforced expression of miR-29b mimics increased adhesion of MM cells to HS-5 and reduced migration of both MM and HUVEC cells. These findings suggest that miR-29b is a negative regulator of either MM or endothelial cell migration. Finally, the proteasome inhibitor bortezomib, which induces the expression of miR-29b, decreased global DNA methylation by a miR-29b-dependent mechanism and induced SOCS-1 promoter demethylation and protein upregulation. In conclusion, our data indicate that miR-29b is endowed with epigenetic activity and mediates previously unknown functions of bortezomib in MM cells.
Introduction
Multiple myeloma (MM) is a hematologic malignancy characterized by abnormal proliferation of bone marrow plasma cells (PCs). Despite the recent huge progress in the understanding of MM biologyCitation1,Citation2 and the availability of novel experimental platformsCitation3-Citation9 and agentsCitation10,Citation11 that have significantly increased survival of MM patients,Citation12 MM is still an incurable disease, and additional therapeutic strategies are urgently awaited. Pathogenesis of MM is a multistep process in which PCs undergo series of molecular and cellular changes within the human bone marrow (huBM) milieu, which supports growth, survival, and drug resistance of tumor cells. Constitutive genetic alterations of MM cells are relevant determinants of their biological behavior within the huBMCitation11,Citation13-Citation16; on the other hand, epigenetic changes might also play an important role in MM pathogenesis and may drive initial steps in neoplastic transformation by reducing genome stability and regulating gene expression.Citation17 The biochemical modifications that govern epigenetics are DNA methylation and post-translational modifications of histone proteins.Citation18,Citation19 About 80% of CpG sites in mammalian cells are methylated, but both the CpG sites and their degree of methylation are unevenly distributed in the genome.Citation20-Citation22 CpG dinucleotides are mostly concentrated in small regions termed “CpG islands”, which are found in about 55% of human gene promoters.Citation22,Citation23 Aberrant methylation of cytosine residues of tumor suppressor genes is the most studied epigenetic modification: in fact, it is well recognized that increased methylation of CpG islands of tumor suppressor genes is associated with transcriptional silencing.Citation24 Aberrant patterns of hypermethylation are also detected in MM, where epigenetic inactivation of tumor suppressor genes is generally associated with an unfavorable prognosis; moreover, drug response and microenvironment interaction pathways are affected by epigenetic inactivation, linking tumor biology to prognosis.Citation25
A better understanding of methylation events occurring in MM appears therefore of great clinical relevance, since it might contribute to early detection, prognostic assessment, and prediction of therapeutic response;Citation26-Citation28 furthermore, the intrinsic characteristic of reversibility of DNA methylation makes it an attractive therapeutic target.
MicroRNAs (miRNAs) recently emerged as important regulators of gene expressionCitation29 through their ability to regulate multiple pathways involved in MM pathogenesisCitation30 and MM-related diseases,Citation31-33 as well as for their potential to modulate the immunologic microenvironment of MM cells.Citation34 Increasing evidence demonstrates that multiple genetic and epigenetic mechanisms account for the altered expression of a number of miRNAs in cancer cells,Citation35-Citation37 including MM cells.Citation38-Citation41 Of note, manipulation of aberrantly expressed miRNAs indeed produces profound effects in MM cell proliferation, survival, and drug-resistance and therefore represents a novel strategy for the personalized treatment of MM.Citation10,Citation42
An important class of miRNAs, named epi-miRNAs, is emerging for its ability to regulate gene expression through the downregulation of key enzymes of the epigenetic machinery (such as DNA methyltransferases, histone deacetylases, and polycomb repressive complex genes) and indirectly affects the levels of tumor suppressor genes, whose expression is, in turn, controlled by epigenetic factors.Citation43
We have recently demonstrated that miR-29b targets de novo DNA methyltransferases (DNMTs) and reduces the global DNA methylation of MM cells.Citation31 On the basis of our and others’ findings, it can be thus speculated that miR-29b could act either as tumor suppressor miRNA, via canonical 3′UTR regulation of its anti-apoptotic or pro-survival targets,Citation44 or as epi-miRNA by inducing the reactivation of hypermethylated tumor suppressor genes through negative regulation of DNMTs. Consistently, miR-29b leads to demethylation and re-activation of relevant tumor suppressor genes in lung cancerCitation45 and acute myeloid leukemia cells.Citation46
One important hypermethylated target in MM is the suppressor of cytokine signaling-1 (SOCS-1) gene, a tumor suppressor that negatively regulates several cytokine pathways inducing downregulation of the Janus kinase (JAK)/signal transducer pathway.Citation47-Citation50 In MM, SOCS-1 promoter was found hypermethylated in 62.9% of patients, and its silencing was associated with greater responsiveness to cytokines, thus supporting proliferation and survival of MM cells.Citation51 Moreover, hypermethylation of SOCS-1 was also reported in other hematologic malignanciesCitation52,Citation53 or solid tumors.Citation54-Citation56
In the present study, we investigated whether miR-29b could revert the aberrant hypermethylated status of SOCS-1 gene promoter and affect adhesion and migratory capacities of MM and endothelial cells. Moreover, the effects of the proteasome inhibitor bortezomib, a known miR-29b-inducing agent,Citation40 on the global DNA methylation profile and SOCS-1 expression, were analyzed. All together these findings provide a molecular rationale for developing miR-29b-based epigenetic therapies for MM.
Results
SOCS-1 mRNA levels are downregulated in MM cells
Since it has been reported that SOCS-1 is frequently downregulated in human malignancies,Citation52,Citation54 first we sought to analyze SOCS-1 mRNA espression in primary cells from 55 MM patient and 29 plasma cell leukemia (PCL) patients, which were classified according to the presence of the recurrent IGH chromosomal translocations and cyclin D expression (TC classification)Citation57 as compared with normal PCs from 4 different healthy donors. We found that SOCS-1 mRNA was indeed steadily downregulated in TC(1–4) patient groups and in PCL patients ().
Figure 1. SOCS-1 mRNA expression and its correlation with miR-29b levels in primary MM and PCL samples.(A). Differential expression of SOCS-1 mRNA in immunoselected CD138+ cells from 4 healthy donors (N), 55 MM classified according to the presence of the recurrent IGH chromosomal translocations and cyclin D expression (TC1: n = 12; TC2: n = 13; TC3: n = 12; TC4: n = 12; TC5: n = 6), 21 primary PCL (pPCL), and 8 secondary PCL (sPCL), as obtained by cDNA microarray; raw expression values are reported. Difference between normal and TC classes was assessed by t test using Welch correction or Wilcoxon rank sum test. SOCS-1 was significantly downregulated in all patient groups, except TC5, as compared with N samples (N vs TC1: P = 0.031; N vs TC2: P = 0.023; N vs TC3: P = 0.004; N vs TC4: P = 0.023; N vs TC5: P = 0.11; N vs pPCL: P = 0.03; N vs sPCL: P = 0.008).(B) Correlation analysis between endogenous miR-29b levels and SOCS-1 mRNA levels, determined by high density microarray of mRNA and miRNA expression in a panel of 34 MM (TC1:n = 6; TC2:n = 10; TC3:n = 9; TC4:n = 6; TC5:n = 3) and 18 pPCL samples (P = 0,09). Log values of raw data are reported in graph. Pearson test was used to evaluate the occurrence of correlation.
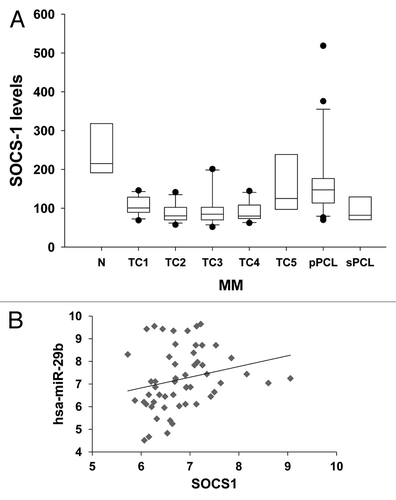
We next attempted to correlate SOCS-1 mRNA and miR-29b levels in our data set (34 MM and 18 pPCL patients), for which both miRNA and gene expression profiling were available. This integrated approach disclosed a positive correlation trend between SOCS-1 mRNA and miR-29b levels among all MM and PCL patients analyzed (); this trend was found statistically significant within the TC2 patients group (n = 10, P = 0.016; not shown), thus suggesting that miR-29b might likely act as a positive regulator of SOCS-1 in the context of a specific MM genetic background. These findings prompted us to investigate the molecular mechanisms by which miR-29b could regulate SOCS-1 expression in MM cells.
Synthetic miR-29b mimics demethylate SOCS-1 in MM cells
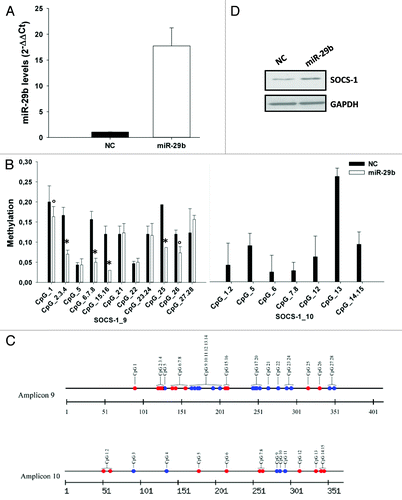
On the basis of our previous findings indicating that miR-29b is endowed with demethylating activity,Citation31 we investigated whether enforcing miR-29b in MM cells could modulate the expression of SOCS-1 through its promoter demethylation. To this aim, NCI-H929 or U266 cells were transfected with synthetic miR-29b mimics or scrambled control oligonucleotides (NC). The levels of miR-29b in transfected cells were assessed by quantitative real-time-PCR (qRT-PCR; ; Fig. S2A). In parallel, genomic DNA was subjected to bisulfite sequencing and Sequenom MassARRAY analysis. The methylation level of each CpG site, in a total of 9 amplicons spanning the SOCS-1 promoter, was evaluated. As shown in and Figure S2B, the methylation levels widely varied, from completely unmethylated up to 100% methylated, across different CpG sites within each amplicon. Notably, miR-29b significantly reduced the methylation percentage of multiple CpG sites, 24 h after transfection, being 6 out of 11 (54.5%) CpGs within amplicon 9, and 7 out of 7 (100%) CpGs within amplicon 10 less or fully demethylated after transfection with miR-29b mimics (). Conversely, methylation of CpGs within amplicons 1 and 12 was not affected by miR-29b, whereas 22% and 44% of CpGs residing in amplicon 16 and 20 were less methylated in the presence of miR-29b (not shown); CpGs within amplicons 2, 5, and 18 were not found methylated at basal level (not shown). miR-29b-induced SOCS-1 promoter demethylation occurred together to increased protein expression, as assessed by western blotting (). Similar results were obtained when U266 cells were transfected with synthetic miR-29b mimics: a significant decrease in methylation of CpGs within amplicon 1, 9, and 10 was indeed observed upon miR-29b transfection (Fig. S2B).
Figure 2. miR-29b demethylates SOCS-1 gene promoter.(A) Quantitative RT-PCR of miR-29b levels in NCI-H929 cells transfected with synthetic miR-29b mimics or scrambled oligonucleotides (NC). Raw Ct values were normalized to RNU44 housekeeping snoRNA and expressed as ΔΔCt values. miR-29b levels in cells transfected with NC were set as internal reference. Data are the average of 2 independent transfection experiments performed in triplicate.(B) Sequenom MassARRAY platform was used for the quantitative methylation analysis. Mean methylation levels of CpG sites in SOCS-1 promoter are reported. Data are expressed as mean ± SD of 2 independent experiments performed in triplicate. CpGs whose methylation was detectable at basal level in NCI-H929 transfected cells, have been analyzed. °P ≤ 0.05; *P ≤ 0.01.(C) Graphic representation of all CpGs within amplicon 9 and 10 of SOCS-1 promoter. Red circles indicate all CpGs found demethylated after miR-29b transfection; blue circles represent CpGs unmethylated at basal level or unaffected by miR-29b.(D) Immunoblot of SOCS-1, 24 h after transfection of NCI-H929 cells with synthetic miR-29b or scrambled oligonucleotides (NC). GAPDH was used as loading control.
Bortezomib reduces global DNA methylation and SOCS-1 promoter methylation in MM cells
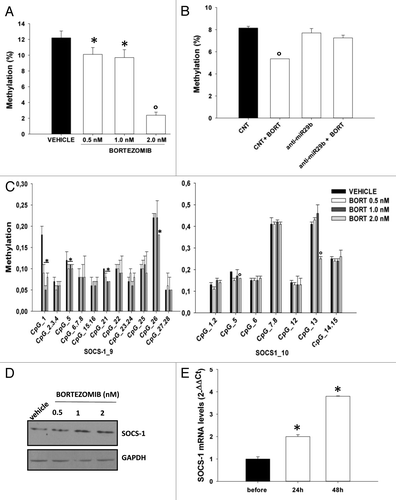
We then investigated whether induction of miR-29b by the proteasome inhibitor bortezomib could mimic the effects induced by enforced expression of synthetic miR-29b oligonucleotides and could thus lead to epigenetic changes in MM cells. In fact, we previously demonstrated that miR-29b is upregulated after treatment with bortezomib and modulates bortezomib responsiveness of MM cells.Citation40 In this light, we first evaluated whether bortezomib could exert demethylating activity in MM cells. NCI-H929 cells were treated for 24 h with bortezomib at concentrations resembling its clinically active doses, and the effects on global as well as SOCS-1 methylation were analyzed. Interestingly, bortezomib decreased global DNA methylation levels in NCI-H929 cells up to low concentrations (0.5 nM; ); of note, this effect was miR-29b-dependent, since it was completely abrogated when miR-29b was stably inhibited by lentiviral transduction of miR-29b inhibitory sequences (). In order to investigate a possible effect of bortezomib on SOCS-1 demethylation, genomic DNA was obtained from bortezomib- or vehicle-treated NCI-H929 cells and subjected to Sequenom MassArray analysis for SOCS-1 promoter. Bortezomib reduced the methylation levels of 4 out of 11 (38%) CpGs within amplicon 9, and 2 out of 7 (28.5%) CpGs located in amplicon 10 (); moreover, bortezomib-induced demethylating effects were paralleled by SOCS-1 protein upregulation (). An additional relevant finding was that primary CD138+ cells immunopurified from PCL patient peripheral blood showed upregulation of SOCS-1 mRNA following bortezomib (1.3 mg/m2) treatment (). Bortezomib-induced SOCS-1 upregulation could not be ascribed to a block in its proteasomal degradation, since no accumulation of ubiquitinated SOCS-1 was evident after bortezomib treatment (Fig. S3A); moreover, inhibition of proteasome activity by MG132 did not affect SOCS-1 protein levels (Fig. S3B), thus ruling out a major role of the proteasome in SOCS-1 upregulation in our cellular model.
Figure 3. Bortezomib reduces global DNA methylation as well as SOCS-1 promoter methylation in MM cells.(A) Global DNA methylation levels were determined, as reported in the “Materials and Methods”, in NCI-H929 cells treated for 24 h with vehicle or bortezomib 0.5, 1.0 and 2.0 nM or(B) in NCI-H929 stably transduced with a control vector or miR-29b inhibitory sequences (anti-miR-29b) and then treated for 24 h with 1 nM bortezomib (BORT).(C) Mean methylation levels of CpG sites in SOCS-1 promoter, as determined by Sequenom MassARRAY analysis, in NCI-H929 cells treated for 24 h with vehicle or bortezomib 0.5, 1.0, or 2.0 nM. Data are expressed as mean ± SD of 2 independent experiments performed in triplicate.(D) Immunoblot of SOCS-1, 24 h after treatment of NCI-H929 cells with vehicle or bortezomib 0.5, 1.0 or 2.0 nM . GAPDH was used as loading control. *P ≤ 0.05; °P ≤ 0.01.(E) Quantitative RT-PCR of miR-29b levels in PCL patient-derived peripheral blood mononuclear cells, 24 and 72 h after systemic bortezomib treatment (1.3 mg/m2). Raw Ct values were normalized to RNU44 housekeeping snoRNA and expressed as ΔΔCt values. *P ≤ 0.01.
miR-29b reduces STAT3 phosphorylation and downmodulates STAT-3 targets
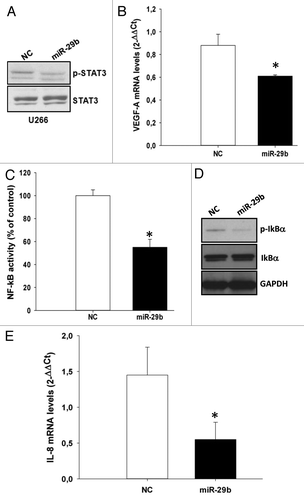
SOCS-1 negatively regulates cell proliferation, migration, and adhesion of cancer cells by inhibiting the JAK/STAT signaling.Citation58 Hence, we asked whether miR-29b-induced SOCS-1 upregulation was able to dampen STAT3 signaling. To address this issue, we used as a model the U266 cell line, characterized by high levels of phosphorylated STAT3 triggered by the autocrine secreted IL-6.Citation59 Cells were transfected with miR-29b mimics, and the levels of unphosphorylated STAT3 or its phosphorylated active form were analyzed by western blotting 24 h after transfection. Importantly, miR-29b was able to reduced STAT3 phosphorylation () even in the presence of BMSCs (Fig. S4A). Moreover, miR-29b was able to reduce the expression of canonical STAT3-transcriptional targets like VEGF-A (). Since STAT3 has been demonstrated to maintain constitutive NF-κB activity in cancer cells,Citation60 we also evaluated the capability of miR-29b to inhibit NF-κB transcriptional activity by transfecting MM cells with synthetic miR-29b mimics or scrambled controls, together with a vector carrying NF-κB-responsive elements cloned upstream of a reporter gene. This assay indeed demonstrated that miR-29b significantly reduced NF-κB transcriptional activity in MM cells (); accordingly, western blot analysis demonstrated reduced IκB-α phosphorylation in miR-29b-transfected U266 cells (). Downregulation of STAT3 and NF-κB signaling might contribute to explain the well-established pro-apoptotic effects exerted by miR-29b mimics. Notably, this effect was demonstrated also in the presence of BMSCs, whose pro-survival activity was indeed overcome by exogenous miR-29b (Fig. S4B). Other important STAT3/NF-κB targets are represented by cytokines and chemokines.Citation58 To evaluate whether miR-29b could influence the levels of IL-6 and IL-8, we performed qRT-PCR in miR-29b transfected cells: interestingly, miR-29b significantly downregulated IL-8 mRNA (), whereas IL-6 mRNA levels did not significantly change (data not shown).
Figure 4. miR-29b inhibits STAT3 and NF-κB signaling in MM cells.(A) Immunoblot of STAT3 phosphorylated at Y705 or unphosphorylated, 24 h after transfection of U266 cells with synthetic miR-29b or scrambled oligonucleotides (NC).(B) Quantitative RT-PCR of VEGF-A 24 h after transfection of U266 cells with synthetic miR-29b or scrambled oligonucleotides (NC). The results are shown as average mRNA expression, in 3 independent experiments, after normalization with GAPDH and ΔΔCt calculations *P < 0.05;(C) Dual luciferase assay was performed in U266 cells transfected with miR-29b or scrambled oligonucleotides (NC), together with a firefly luciferase constructs containing the NFκB-responsive elements and the pRL-TK vector for normalization. The firefly luciferase activity was normalized to renilla luciferase activity. The data are shown as relative luciferase activity of miR-29b-transfected cells as compared with the control (NC), in a total of six experiments from 3 independent transfections. *P < 0.05(D) Immunoblot of IκB-α phosphorylated at Ser32 or unphosphorylated, 24 h after transfection of U266 cells with synthetic miR-29b or scrambled oligonucleotides (NC). GAPDH was used as loading control.(E) Quantitative RT-PCR of IL-8, 24 h after transfection of U266 cells with synthetic miR-29b or scrambled oligonucleotides (NC). The results are shown as average mRNA expression, in 3 independent experiments, after normalization with GAPDH and ΔΔCt calculations. *P < 0.05.
miR-29b impairs MM and endothelial cell migration
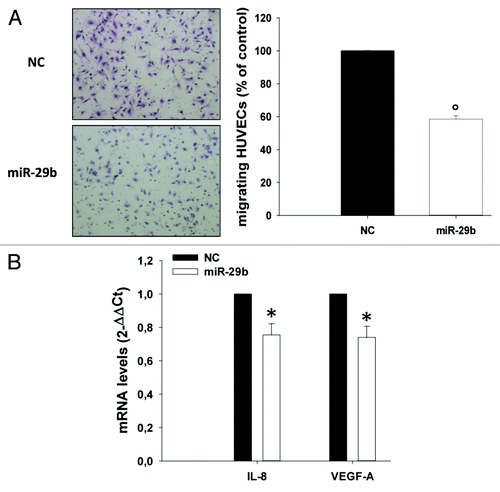
miR-29b-induced downregulation of IL-8, which is known to promote cell migration in different biological contexts,Citation61,Citation62 suggested that miR-29b might modulate the migratory capacity of MM cells. To address this point, MM cells were transfected with miR-29b or scrambled controls; the levels of miR-29b after transfection were determined by qRT-PCR (). Transfected cells were assessed for their capacity to migrate by using transwell migration assays in the presence or absence of IL-8 as chemo-attractant. Notably, synthetic miR-29b mimics were able to decrease both basal and IL-8-induced migration of OPM2 cells (). miR-29b-induced inhibition of cell migration occurred together with downregulation of IL-8, VEGF-A and MMP2 mRNAs (). On the other hand, miR-29b-expressing OPM2 cells were more prone to adhere to HS-5 stromal cells (). Of note, a decrease in IL-8 and VEGF-A mRNAs could be also detected in HS-5 treated with conditioned medium from miR-29b transfected OPM2 cells (). Altogether these findings indicate that miR-29b decreases motility and increases adhesion of MM cells by different mechanisms. Since IL-8 positively regulates migration of endothelial cells,Citation63 we verified whether miR-29b could also impair migration of HUVEC cells. To this aim, transwell migration assays were performed with HUVEC cells using as chemo-attractant conditioned medium from OPM2 cells transfected with miR-29b or scrambled controls. Notably, we observed a marked decrease of HUVEC cell migration when conditioned medium from miR-29b-transfected cells was added (). Most importantly, IL-8 and VEGF-A mRNA levels were significantly downregulated in HUVEC cells cultured with conditioned medium from miR-29b transfected MM cells (), thus suggesting a prominent role of miR-29b in negatively regulating the expression and/or secretion of gene products which promote migration and angiogenesis of the endothelial compartment.
Figure 5. miR-29b inhibits MM cell migration.(A) Quantitative RT-PCR of miR-29b levels in OPM2 cells, 24 h after transfection with synthetic miR-29b mimics or scrambled oligonucleotides (NC). Raw Ct values were normalized to RNU44 housekeeping snoRNA and expressed as ΔΔCt values. miR-29b levels in cells transfected with NC were set as internal reference. Data are the average of 2 independent transfection experiments performed in triplicate.(B) Transwell migration assay of OPM2 cells, 24 h after transfection with miR-29b or scrambled oligonucleotides (NC), in the presence or absence of 10 ng/mL IL-8 added to the lower chamber of the transwell.(C). Quantitative RT-PCR of IL-8, MMP2 and VEGF-A, 24 h after transfection of OPM2 cells with synthetic miR-29b or scrambled oligonucleotides (NC). *P < 0.05, °P < 0.01.(D) Adhesion assay on HS-5 BM stromal cells performed with OPM2 cells, 24 h after transfection of MM cells with miR-29b or scrambled (NC) controls. Cells were stained, photographed under a light microscope equipped with a digital camera (original magnification, 20×) and then counted, as detailed in the “Materials and Methods”.(E) Quantitative RT-PCR of IL-8 and VEGF-A expression in HS-5 cells treated for 24 h with conditioned medium from OPM2 cells (0.5 × 106), collected 24 h after transfection with synthetic miR-29b or scrambled oligonucleotides (NC). *P < 0.01.
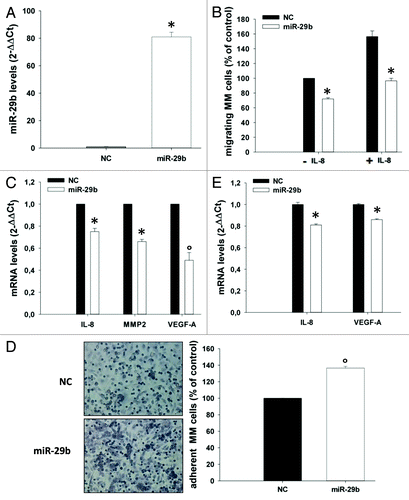
Figure 6. miR-29b inhibits endothelial cell migration.(A) Transwell migration assay of HUVEC cells. HUVECs were incubated for 6 h using as chemo-attractant conditioned medium of OPM2 cells, collected 24 h after transfection with synthetic miR-29b mimics or scrambled oligonucleotides; migrating cells were quantified as indicated in materials and methods. A representative picture of migrating cells is shown (original magnification, 20×).(B) Quantitative RT-PCR of IL-8 and VEGF-A mRNA levels in HUVEC cells treated for 24 h with conditioned medium from OPM2 cells, collected 24 h after transfection with synthetic miR-29b or scrambled oligonucleotides (NC). *P < 0.05, °P < 0.01.
Discussion
A signature of tumor suppressor gene-specific hypermethylation is linked to disease progression and worse prognosis of MM patients.Citation64 In this light, demethylating agents may represent powerful therapeutic tools for reverting aberrant hypermethylation in MM. To date, only 2 azanucleosides (5-azacitidine and decitabine) have been approved by FDA for the treatment of myelodisplastic syndromes (MDS) and are currently in clinical trials for other types of cancers.Citation65 Despite the tremendous advancement in the experimental approaches aimed at analyzing the genome-wide methylation of cancer cells,Citation66 few attempts have been addressed to the design of novel therapeutic strategies interfering with abnormal methylation of cancer cells.Citation67 The discovery of miRNAs has significantly changed the landscape of cancer biologyCitation68 and novel therapeutic modalities based on the re-expression or the inhibition of deregulated miRNAs are emerging as useful tools for the personalized treatment of human cancer.Citation42,Citation69 The term “epi-miRNAs” has been proposed to indicate a specific class of tumor suppressor miRNAs able to downregulate epigenetic enzymes, which, in turn, negatively affect the expression of tumor suppressor genes.Citation43 We have previously reported that miR-29b is a tumor suppressor miRNA that exerts in vitro and in vivo anti-MM activity and reduces global DNA methylation in MM cells through the targeting of DNMTs.Citation31 In the present report, we have investigated SOCS-1 as a putative target of epi-miR-29b. SOCS-1 acts as a classical tumor suppressor gene by inhibiting the signaling mediated by JAK/STAT, an oncogenic pathway which triggers cytokine-driven signals thus supporting cell proliferation, survival, and metastatic behavior of cancer cells.Citation47 By exploiting the Sequenom MassARRAY technology, we performed the first thorough quantitative study of SOCS-1 methylation in MM cell lines and we indeed highlighted its demethylation by miR-29b. In fact, enforced expression of miR-29b in MM cells with hypermethylated SOCS-1 geneCitation70 significantly reduced the methylation status of several CpGs within the SOCS-1 promoter. Interestingly, the proteasome inhibitor bortezomib, a known miR-29b-inducing agent,Citation40 reduced global DNA methylation levels in a miR-29b-dependent fashion, since miR-29b inhibitors were capable to antagonize such effect. Bortezomib is currently used in the treatment of MM with a significant benefit in the relapsed and/or refractory settings.Citation71 Beside its first-discovered and widely studied effects on 26S proteasome inhibition, bortezomib exerts a plethora of intracellular effects,Citation72 in some cases mediated by miRNAs.Citation40,Citation73 Our findings on the epigenetic activity of bortezomib in MM are in agreement with recent emerging evidence indicating that bortezomib is endowed with epigenetic activity in acute myeloid leukemia (AML), where it reduces global DNA methylation by interfering with Sp1/NF-κB-dependent DNMT activity.Citation74 Indeed, our findings add further molecular insights on the demethylating effects triggered by such drug, since we have identified miR-29b as a relevant effector required by bortezomib to elicit its global DNA-demethylating activity. Of note, we found that bortezomib demethylated SOCS-1 promoter, although at a lesser extent than miR-29b, and upregulated SOCS-1 protein levels in vitro by a proteasome-independent mechanism. More importantly, the analysis of SOCS-1 mRNA levels in circulating peripheral blood primary PCL cells exposed to conventional systemic bortezomib regimen demonstrated strong upregulation of SOCS-1 mRNA after bortezomib treatment, thus strenghtening the pivotal role of bortezomib on SOCS-1 re-activation in vivo.
At the functional level, synthetic miR-29b mimicked SOCS-1 activity on STAT3 signaling: transfection of miR-29b reduced STAT3 phosphorylation as well as the activation of STAT3-transcriptional targets like VEGF-ACitation58; moreover, miR-29b also reduced NF-κB activity, which is functionally related to STAT3.Citation60 It has to be pointed out that some STAT3-transcriptional targets and miR-29b validated canonical targets, like MCL-1,Citation40 MMP2, and VEGF-A,Citation75 overlap. Altogether, these findings indicate that miR-29b may interfere with STAT3 pathway in a direct (via canonical 3′UTR-binding of target mRNAs) or indirect (via STAT3 functional downregulation) fashion. In light of several lines of evidence indicating that inhibition of STAT3 indeed has therapeutic value in preclinical models of solid and hematologic cancers,Citation76-Citation78 it can be speculated that miR-29b enforcement, through STAT-3 inhibition, could likely represent a promising strategy in a wide variety of malignancies.
An additional finding highlighted in this report, which increases available information about the biological role of miR-29b in MM, is the ability of this miRNA to impair MM cell migration, an effect which could be likely dependent on downregulation of MMP2, a validated miR-29b-target,Citation75 as well as of IL-8, a chemokine supporting proliferation and migration of cancerCitation61,Citation62 and endothelial cells.Citation63 Importantly, it has been demonstrated that IL-8 production by BMSCs correlates with MM disease activity and with NF-κB function and bone marrow angiogenesis.Citation79 By transwell migration assays, we showed that miR-29b inhibited MM and HUVEC cell migration, and this effect was associated with downregulation of IL-8 in MM cells as well as in stromal and endothelial cells treated with conditioned medium from miR-29b mimics-transfected cells, suggesting a paracrine effect exerted by miR-29b on the BM microenvironment. Of note, miR-29b-induced downregulation of VEGF-A and IL-8 in endothelial cells suggests a potential antagonism of miR-29b on the angiogenetic process. However, these latter observations warrant further investigation in follow-up studies.
In conclusion, our study provides novel insights on the epigenetic activity exerted by miR-29b, thus broadening the toolbox of agents targeting aberrant methylation in cancer cells; moreover, our findings shed light on novel biochemical pathways and phenotypical changes that can be modulated by therapeutic strategies involving restoration of miR-29b function in MM.
Materials and Methods
Primary samples, cell lines, and drugs
Bone marrow mononuclear cells (BMMNCs) and primary MM cells from 55 newly diagnosed untreated MM, 21 primary plasma cell leukemia (PCL), 8 secondary PCL patients, and 4 normal healthy donors were isolated by Ficoll–Hypaque density gradient sedimentation and obtained during standard diagnostic procedures after the patient had provided informed consent in accordance with institutional guidelines and University Magna Graecia IRB approval. MM patient cells were separated from BM samples by antibody-mediated positive selection using anti-CD138 magnetic activated cell separation microbeads (Miltenyi Biotech). The purity of the positively selected PCs was assessed by means of morphology and flow cytometry and was ≥90% in all cases. NCI-H929 and U266 MM cell lines were cultured in RPMI 1640 (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (Lonza Group Ltd). HS-5 BM-stromal cells were purchased from ATCC and cultured in DMEM (Gibco, Life Technologies) supplemented with 10% fetal bovine serum (Lonza Group Ltd). HUVEC were obtained from Lonza and grown in endothelial growth medium (EGM) according to supplier’s information (Clonetics). Clinical-grade bortezomib was purchased from Millennium Pharmaceuticals (Takeda) and dissolved in sodium chloride saline solution at 1 mg/ml.
Generation of MM cells stably expressing miR-29b inhibitors
To obtain cells stably expressing miR-29b inhibitory sequences, HEK293T cells were co-transfected with 10 μg of the lentiviral pGreenPuro expression vector or the miRZip-29b anti-miR-29b construct (System Biosciences), 10 μg of pCMV-VSVG, and 4 μg of Delta 8.9 plasmids as previously described.Citation80 Supernatants containing GFP-expressing lentivirus were collected 24 h after the transfection and then filtered through 0.45 μM filters; 1 mL of supernatants was used for overnight transduction of MM cells (0.5 × 106) in the presence of 8 μg/ml of polybrene (Sigma-Aldrich). Two days after transduction, selection with 1 μg/ml puromycin was performed for 3 d.
Gene expression profiling
Gene expression profiles were analyzed on the GeneChip® Human Gene 1.0 ST chip platform (Affymetrix Inc). The raw intensity expression values were processed by robust multi-array average (RMA) complete procedure (Irizarry Biostatistics 2003) with the re-mapped chip definition files (CDF) from BrainArray libraries version 15.0.0 available at: http://brainarray.mbni.med.umich.edu/Brainarray/Database/CustomCDF/15.0.0/entrezg.asp
miRNA profiling
miRNA expression profiling was performed using Total RNA and Affymetrix GeneChip miRNA 1.0 according to manufacturer’s protocol. Expression values were extracted with Affymetrix miRNA QC tool (RMA normalized and log2-transformed).
In vitro transfection of MM cells with synthetic miR-29b mimics
Synthetic pre-miRNAs were purchased from Ambion (Applied Biosystems). 1.0 × 106 cells were electroporated with 200 nM scrambled (miR-NC) or synthetic pre-miR-29b (miR-29b) oligonucleotides, using the Neon® Transfection System (Invitrogen) with the following electroporation conditions: 1050 V, 30 ms, 2 pulse.
Quantitative real-time amplification of miRNAs and mRNAs
Total RNA from MM cells was extracted with the TRIzol® Reagent (Invitrogen) according to manufacturer’s instructions. The single-tube TaqMan miRNA assays (Applied Biosystems, Assay id 000413) was used to detect and quantify mature miR-29b according to the manufacturer’s instructions, by the use Viia™ 7 Real-Time PCR detection system (Applied Biosystems). miR-29b expression was normalized on RNU44 (Applied Biosystems, Assay Id 001094). For mRNA dosage, oligo-dT-primed cDNA was obtained using the High-Capacity cDNA Reverse Transcription Kit (Applied Biosystems) and then used as template to quantify VEGF-A (AB Hs00900055_m1), IL-8 (AB Hs00174103_m1), SOCS-1 (AB Hs00705164_s1), IL-6 (AB Hs00985639_m1), and MMP2 (AB Hs01548727_m1) levels by TaqMan assay (Applied Biosystems); normalization was performed with GAPDH (AB Hs03929097_g1). Comparative real-time polymerase chain-reaction (RT-PCR) was performed in triplicate, including no-template controls. Relative expression was calculated using the comparative cross threshold (Ct) method.Citation81
Primer design for EpiTYPER assay
Primers were designed according to Sequenom Standard EpiPanel. A T7-promoter tag (CAGTAATACG ACTCACTATA GGGAGAAGGC T) was added to the reverse primer and a 10-mer tag sequence (AGGAAGAGAG) was added to the forward primer to balance the PCR primer length. Primer sequences are reported in Table S1.
Bisulfite treatment and PCR conditions
Bisulfite conversion of each DNA sample was performed by using EZ-96 DNA Methylation-Gold™ kit (Zymo Research), according to the manufacturer's protocol. Briefly, 1 µg of genomic DNA was added to 130 µl of CT Conversion Reagent in a final volume of 150 µl. The mix was incubated at 98 °C for 10 min and, successively, at 64 °C for 2.5 h. After adding 400 µl of M-Binding Buffer to the wells of the Silicon-ATM Binding plate, each sample was loaded into the wells and centrifuged at 3000 g for 5 min. After adding of 400 µl of M-Wash Buffer to the wells and a centrifugation at 3000 g for 5 min, 200 µl of M-Desulphonation Buffer were added to each well and incubated at room temperature for 20 min. Then, the solution was removed by centrifugation at 3000 g for 5 min, and the wells were washed twice with 400 µl of M-Wash Buffer. Deaminated DNA was eluted in 30 µl of M-Elution Buffer.
The PCR reactions were performed in a total volume of 5 µl using 1 µl of bisulfite-treated DNA, EpiTaq PCR Buffer 1×, 0.4 µM of each primer, 0.3 mM dNTP mixture, 2.5 mM of MgCl2, 0.005 U TaKaRa EpiTaq HS (TaKaRa). The thermal profile used for the reaction included a 4 min heat activation of the enzyme at 95 °C, followed by 45 cycles of denaturation at 94 °C for 20 s, annealing (for temperature see Table S1) for 30 s, extension at 72 °C for 1 min, then 1 cycle at 72 °C for 3 min. 0.5 μl of each PCR product were electrophoresed on 1.5% agarose gel to confirm successful PCR amplification and amplification specificity. The length of each amplicon of SOCS1 gene is reported in Table S1.
Dephosphorylation of unincorporated deoxynucleotide triphosphates and in vitro transcription and RNase A cleavage
Unincorporated dNTPs in the amplification products were dephosphorylated by adding 1.7 µl DNase-free water and 0.3 μl (0.5 U) shrimp alkaline phosphatase (SAP) (Sequenom, Inc). Each reaction was incubated at 37 °C for 40 min and SAP was then heat inactivated for 5 min at 85 °C. Subsequently, samples were incubated for 3 h at 37 °C with 5 µl of T-Cleavage reaction mix (Sequenom) containing 3.21 μl RNase-free water, 0.89 μl 5× T7 polymerase buffer, 0.22 μl T cleavage mix, 0.22 μl 100 mM DTT, 0.40 μl T7 RNA, and DNA polymerase and 0.06 μl RNase A, for concurrent in vitro transcription and base-specific cleavage. The samples of cleaved fragments were then diluted with 20 μl water. Conditioning of the cleavage reaction was performed by adding 6 mg of clean resin.
Mass spectrometry
Ten nl of the resultant cleavage reactions were spotted onto silicon matrix-preloaded chips (Spectro-CHIP, Sequenom) using a MassARRAY nanodispenser (Sequenom), and analyzed using the MassARRAY Compact System matrix-assisted laser desorption/ionization-time-of-flight mass spectrometer (MALDI-TOF) (Sequenom). The spectra’s methylation ratios were calculated using EpiTYPER software v1.0 (Sequenom). The method yields quantitative results for each of the sequence-defined analytic units, referred to as CpG units, which contain either 1 individual CpG site or an aggregate of downstream CpG sites. Triplicate independent analyses from sodium bisulfite-treated DNA sample were undertaken. The effectiveness of the entire experimental procedure was also assayed by analyzing CpGenome™ Universal Unmethylated DNA (Chemicon) and CpGenome™ Universal Methylated DNA (Chemicon) as control. Poor-quality and non-valuable data for the quantitative methylation of each CpG unit measured by MALDI-TOF-MS were excluded.
Statistical analyses were performed using SPSS 15.0 statistical software (SPSS Inc). The difference in the methylation rates of each CpG units as well as in global DNA methylation analysis between untreated and treated samples was analyzed by one-way analysis of variance (ANOVA) and Student t test, with a significance level defined as α = 0.05.
Global DNA methylation analysis
Global DNA methylation was evaluated using the Methylamp™ Global DNA Methylation Quantification Kit following instructions provided by the manufacturer (Epigentek). Briefly, 150 ng of genomic DNA extracted from cell samples were immobilized in the strip well specifically treated to have high affinity to DNA at 37 °C for 90 min. Then, wells were blocked by adding 150 µl of Block solution (GU4), incubated at 37 °C for 30 min and washed for 3 times in wash buffer 1× (GU1). The methylated DNA fraction was recognized by sequential incubation with Capture Antibody (GU5) for 60 min at room temperature (RT), and with Detector Antibody (GU6) for 60 min at RT. After 5 consecutive washes in GU1, wells were first incubated with 150 µl of Enhancer Solution (GU7) at RT for 30 min, then in Developer solution (GU8) at RT for 5 min away from light. A colorimetric reaction allowed methylated DNA quantification at 450 nM with microplate reader. A methylated DNA was used as positive control. The analysis was repeated twice.
The amount of methylated DNA is proportional to the optical density (OD) intensity, and the degree of DNA methylation was calculated according the following formula:
Methylation % = ([Sample OD – Negative Control OD]/X*)/(Positive Control OD – Negative Control OD) *10
Where negative control is buffer without DNA, positive control is methylated control DNA, and X* is the human genomic GC content. Two independent triplicate experiments were performed.
Chemotaxis assay
Chemotaxis was determined using 8 μm pore filters for the Transwell migration assay (BD Biosciences), according to the manufacturer’s instructions. Briefly, OPM2 cells (1.0 × 105/well) transfected with either scrambled (miR-NC) or synthetic pre-miR-29b (miR-29b) were washed and resuspended in RPMI 1640 medium supplemented with 1% FBS. Then cells were placed in the upper migration chambers, whereas the lower chamber contained RPMI 1640 medium supplemented with 10% FBS in the presence or absence of 10 ng/mL of IL-8 (R&D Systems). After 5 h of incubation at 37 °C, 5% CO2, cells migrated to the lower chambers were determined by a Trypan-Blue exclusion assay. HUVEC cells (7.0 × 103 /well) were resuspended in serum-free RPMI 1640 medium supplemented with 0.1% BSA in transwell chemotaxis (BD Biosciences) above 8 μm-pore filters and exposed to conditioned medium from multiple myeloma cells transfected with either scrambled (miR-NC) or synthetic pre-miR-29b (miR-29b). Filters were removed after 6 h, fixed in methanol, and stained with Diff-Quick (Medion Diagnostics GmbH). Two independent experiments were performed in triplicate; cells from 5 different fields were counted for each condition.
Adhesion assay
HS-5 stromal cells were grown to confluence in 12-well plates and fixed with glutaraldehyde 0.0125% (Agar Scientific Ltd). After fixation, cells were treated with 10 mM ethanolamine to block aldehydic groups and washed several times before plating MM cells. OPM2 cells (0.5 × 106/well) transfected with either scrambled (NC) or synthetic pre-miR-29b (miR-29b) were added to each well and incubated for 4 h. Non-adherent cells were washed, and adhesion was detected by hematoxylin/eosin staining. Each test group was assayed in triplicate; five fields were counted for each condition.
Western blotting
SDS-PAGE and western blotting (WB) were performed according to standard protocols.Citation82 Whole-cell lysates (40 μg per lane) were separated using 4–12% Novex Bis-Tris SDS-acrylamide gels (Invitrogen, Life Technologies), electro-transferred on nitrocellulose membranes (Bio-Rad), and immunoblotted with the rabbit anti-SOCS-1, anti-STAT3, anti-STAT3 phosphorylated at tyrosine 705, anti-IκBα, anti-IκBα phosphorylated at serine 32 (all from Cell Signaling), or anti-GAPDH (Santa Cruz Biotechnology) antibodies.
Luciferase assay for NFκB activity
MM cells (1.0 × 106) were electroporated as above indicated with 200 nM of synthetic miR-29b mimics or NC together with 10 μg of the PGL3-NF-κB-reporter vector (Promega) and 5 μg of pRL-TK Renilla vector (Promega) to normalize different transfection efficiencies. Firefly and Renilla luciferase activities were measured consecutively using the dual-luciferase assay kit and the GloMax® 96 Microplate Luminometer (Promega) 24 h after transfection.
Statistical analysis
Student t test, 2-tailed, was used to calculate all reported P values using GraphPad software (http://www.graphpad.com). Graphs were obtained using SigmaPlot version 11.0.
Additional material
Download Zip (610.8 KB)Acknowledgments
This work has been supported by the Italian Association for Cancer Research (AIRC), PI: PT. “Special Program Molecular Clinical Oncology - 5 per mille” n. 9980, 2010/15.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26585
Related Research Data
References
- Anderson KC. The 39th David A. Karnofsky Lecture: bench-to-bedside translation of targeted therapies in multiple myeloma. J Clin Oncol 2012; 30:445 - 52; http://dx.doi.org/10.1200/JCO.2011.37.8919; PMID: 22215754
- Vicente-Dueñas C, Romero-Camarero I, García-Criado FJ, Cobaleda C, Sánchez-García I. The cellular architecture of multiple myeloma. Cell Cycle 2012; 11:3715 - 7; http://dx.doi.org/10.4161/cc.22178; PMID: 22983005
- Calimeri T, Battista E, Conforti F, Neri P, Di Martino MT, Rossi M, Foresta U, Piro E, Ferrara F, Amorosi A, et al. A unique three-dimensional SCID-polymeric scaffold (SCID-synth-hu) model for in vivo expansion of human primary multiple myeloma cells. Leukemia 2011; 25:707 - 11; http://dx.doi.org/10.1038/leu.2010.300; PMID: 21233838
- Tassone P, Neri P, Burger R, Di Martino MT, Leone E, Amodio N, Caraglia M, Tagliaferri P. Mouse models as a translational platform for the development of new therapeutic agents in multiple myeloma. Curr Cancer Drug Targets 2012; 12:814 - 22; http://dx.doi.org/10.2174/156800912802429292; PMID: 22671927
- Schüler J, Ewerth D, Waldschmidt J, Wäsch R, Engelhardt M. Preclinical models of multiple myeloma: a critical appraisal. Expert Opin Biol Ther 2013; 13:Suppl 1 S111 - 23; http://dx.doi.org/10.1517/14712598.2013.799131; PMID: 23742200
- Tassone P, Neri P, Carrasco DR, Burger R, Goldmacher VS, Fram R, Munshi V, Shammas MA, Catley L, Jacob GS, et al. A clinically relevant SCID-hu in vivo model of human multiple myeloma. Blood 2005; 106:713 - 6; http://dx.doi.org/10.1182/blood-2005-01-0373; PMID: 15817674
- Tassone P, Tagliaferri P, Rossi M, Calimeri T, Bulotta A, Abbruzzese A, Caraglia M, Neri P. Challenging the current approaches to multiple myeloma-related bone disease: from bisphosphonates to target therapy. Curr Cancer Drug Targets 2009; 9:854 - 70; http://dx.doi.org/10.2174/156800909789760393; PMID: 20025573
- DeWeerdt S. Animal models: Towards a myeloma mouse. Nature 2011; 480:S38 - 9; http://dx.doi.org/10.1038/480S38a; PMID: 22169799
- Neri P, Tagliaferri P, Di Martino MT, Calimeri T, Amodio N, Bulotta A, Ventura M, Eramo PO, Viscomi C, Arbitrio M, et al. In vivo anti-myeloma activity and modulation of gene expression profile induced by valproic acid, a histone deacetylase inhibitor. Br J Haematol 2008; 143:520 - 31; PMID: 18986388
- Misso G, Zappavigna S, Castellano M, De Rosa G, Di Martino MT, Tagliaferri P, Tassone P, Caraglia M. Emerging pathways as individualized therapeutic target of multiple myeloma. Expert Opin Biol Ther 2013; 13:Suppl 1 S95 - 109; http://dx.doi.org/10.1517/14712598.2013.807338; PMID: 23738692
- Rossi M, Di Martino MT, Morelli E, Leotta M, Rizzo A, Grimaldi A, Misso G, Tassone P, Caraglia M. Molecular targets for the treatment of multiple myeloma. Curr Cancer Drug Targets 2012; 12:757 - 67; http://dx.doi.org/10.2174/156800912802429300; PMID: 22671925
- Munshi NC, Anderson KC. New strategies in the treatment of multiple myeloma. Clin Cancer Res 2013; 19:3337 - 44; http://dx.doi.org/10.1158/1078-0432.CCR-12-1881; PMID: 23515406
- Morgan GJ, Walker BA, Davies FE. The genetic architecture of multiple myeloma. Nat Rev Cancer 2012; 12:335 - 48; http://dx.doi.org/10.1038/nrc3257; PMID: 22495321
- Levacque Z, Rosales JL, Lee KY. Level of cdk5 expression predicts the survival of relapsed multiple myeloma patients. Cell Cycle 2012; 11:4093 - 5; http://dx.doi.org/10.4161/cc.21886; PMID: 22987154
- Podar K, Anderson KC. A therapeutic role for targeting c-Myc/Hif-1-dependent signaling pathways. Cell Cycle 2010; 9:1722 - 8; http://dx.doi.org/10.4161/cc.9.9.11358; PMID: 20404562
- Tassone P, Tagliaferri P, Rossi M, Gaspari M, Terracciano R, Venuta S. Genetics and molecular profiling of multiple myeloma: novel tools for clinical management?. Eur J Cancer 2006; 42:1530 - 8; http://dx.doi.org/10.1016/j.ejca.2006.04.005; PMID: 16820292
- Heuck CJ, Mehta J, Bhagat T, Gundabolu K, Yu Y, Khan S, Chrysofakis G, Schinke C, Tariman J, Vickrey E, et al. Myeloma is characterized by stage-specific alterations in DNA methylation that occur early during myelomagenesis. J Immunol 2013; 190:2966 - 75; http://dx.doi.org/10.4049/jimmunol.1202493; PMID: 23408834
- Cheng X, Blumenthal RM. Coordinated chromatin control: structural and functional linkage of DNA and histone methylation. Biochemistry 2010; 49:2999 - 3008; http://dx.doi.org/10.1021/bi100213t; PMID: 20210320
- Delcuve GP, Rastegar M, Davie JR. Epigenetic control. J Cell Physiol 2009; 219:243 - 50; http://dx.doi.org/10.1002/jcp.21678; PMID: 19127539
- Bird A, Taggart M, Frommer M, Miller OJ, Macleod D. A fraction of the mouse genome that is derived from islands of nonmethylated, CpG-rich DNA. Cell 1985; 40:91 - 9; http://dx.doi.org/10.1016/0092-8674(85)90312-5; PMID: 2981636
- Cedar H, Bergman Y. Programming of DNA methylation patterns. Annu Rev Biochem 2012; 81:97 - 117; http://dx.doi.org/10.1146/annurev-biochem-052610-091920; PMID: 22404632
- Han H, Cortez CC, Yang X, Nichols PW, Jones PA, Liang G. DNA methylation directly silences genes with non-CpG island promoters and establishes a nucleosome occupied promoter. Hum Mol Genet 2011; 20:4299 - 310; http://dx.doi.org/10.1093/hmg/ddr356; PMID: 21835883
- Deaton AM, Bird A. CpG islands and the regulation of transcription. Genes Dev 2011; 25:1010 - 22; http://dx.doi.org/10.1101/gad.2037511; PMID: 21576262
- Miranda TB, Jones PA. DNA methylation: the nuts and bolts of repression. J Cell Physiol 2007; 213:384 - 90; http://dx.doi.org/10.1002/jcp.21224; PMID: 17708532
- Kaiser MF, Johnson DC, Wu P, Walker BA, Brioli A, Mirabella F, Wardell CP, Melchor L, Davies FE, Morgan GJ. Global methylation analysis identifies prognostically important epigenetically inactivated tumor suppressor genes in multiple myeloma. Blood 2013; 122:219 - 26; http://dx.doi.org/10.1182/blood-2013-03-487884; PMID: 23699600
- Zaidi SK, Van Wijnen AJ, Lian JB, Stein JL, Stein GS. Targeting deregulated epigenetic control in cancer. J Cell Physiol 2013; 228:2103 - 8; http://dx.doi.org/10.1002/jcp.24387; PMID: 23589100
- Verma M, Srivastava S. Epigenetics in cancer: implications for early detection and prevention. Lancet Oncol 2002; 3:755 - 63; http://dx.doi.org/10.1016/S1470-2045(02)00932-4; PMID: 12473517
- Laird PW. The power and the promise of DNA methylation markers. Nat Rev Cancer 2003; 3:253 - 66; http://dx.doi.org/10.1038/nrc1045; PMID: 12671664
- Migliore C, Giordano S. MiRNAs as new master players. Cell Cycle 2009; 8:2185 - 6; http://dx.doi.org/10.4161/cc.8.14.9113; PMID: 19617713
- Tagliaferri P, Rossi M, Di Martino MT, Amodio N, Leone E, Gulla A, Neri A, Tassone P. Promises and challenges of MicroRNA-based treatment of multiple myeloma. Curr Cancer Drug Targets 2012; 12:838 - 46; http://dx.doi.org/10.2174/156800912802429355; PMID: 22671926
- Amodio N, Leotta M, Bellizzi D, Di Martino MT, D’Aquila P, Lionetti M, Fabiani F, Leone E, Gullà AM, Passarino G, et al. DNA-demethylating and anti-tumor activity of synthetic miR-29b mimics in multiple myeloma. Oncotarget 2012; 3:1246 - 58; PMID: 23100393
- Pitari MR, Amodio N, Di Martino MT, Conforti F, Leone E, Botta C, Paolino FM, Del Giudice T, Iuliano E, et al. miR-29b negatively regulates human osteoclastic cell differentiation and function: Implications for the treatment of multiple myeloma-related bone disease. J Cell Physiol 2013; 228:1506 - 15; http://dx.doi.org/10.3109/10428194.2013.800199; PMID: 23254643
- Rossi M, Botta C, Correale P, Tassone P, Tagliaferri P. Immunologic microenvironment and personalized treatment in multiple myeloma. Expert Opin Biol Ther 2013; 13:Suppl 1 S83 - 93; http://dx.doi.org/10.1517/14712598.2013.799130; PMID: 23692463
- Deng S, Calin GA, Croce CM, Coukos G, Zhang L. Mechanisms of microRNA deregulation in human cancer. Cell Cycle 2008; 7:2643 - 6; http://dx.doi.org/10.4161/cc.7.17.6597; PMID: 18719391
- Hagen JW, Lai EC. microRNA control of cell-cell signaling during development and disease. Cell Cycle 2008; 7:2327 - 32; PMID: 18677099
- Weber B, Stresemann C, Brueckner B, Lyko F. Methylation of human microRNA genes in normal and neoplastic cells. Cell Cycle 2007; 6:1001 - 5; http://dx.doi.org/10.4161/cc.6.9.4209; PMID: 17457051
- Di Martino MT, Leone E, Amodio N, Foresta U, Lionetti M, Pitari MR, Cantafio ME, Gullà A, Conforti F, Morelli E, et al. Synthetic miR-34a mimics as a novel therapeutic agent for multiple myeloma: in vitro and in vivo evidence. Clin Cancer Res 2012; 18:6260 - 70; http://dx.doi.org/10.1158/1078-0432.CCR-12-1708; PMID: 23035210
- Leone E, Morelli E, Di Martino MT, Amodio N, Foresta U, Gullà A, Rossi M, Neri A, Giordano A, Munshi NC, et al. Targeting miR-21 inhibits in vitro and in vivo multiple myeloma cell growth. Clin Cancer Res 2013; 19:2096 - 106; http://dx.doi.org/10.1158/1078-0432.CCR-12-3325; PMID: 23446999
- Amodio N, Di Martino MT, Foresta U, Leone E, Lionetti M, Leotta M, Gullà AM, Pitari MR, Conforti F, Rossi M, et al. miR-29b sensitizes multiple myeloma cells to bortezomib-induced apoptosis through the activation of a feedback loop with the transcription factor Sp1. Cell Death Dis 2012; 3:e436; http://dx.doi.org/10.1038/cddis.2012.175; PMID: 23190608
- Di Martino MT, Gullà A, Cantafio ME, Lionetti M, Leone E, Amodio N, Guzzi PH, Foresta U, Conforti F, Cannataro M, et al. In vitro and in vivo anti-tumor activity of miR-221/222 inhibitors in multiple myeloma. Oncotarget 2013; 4:242 - 55; PMID: 23479461
- Amodio N, Di Martino MT, Neri A, Tagliaferri P, Tassone P. Non-coding RNA: a novel opportunity for the personalized treatment of multiple myeloma. Expert Opin Biol Ther 2013; 13:Suppl 1 S125 - 37; http://dx.doi.org/10.1517/14712598.2013.796356; PMID: 23692413
- Fabbri M, Calin GA. Epigenetics and miRNAs in human cancer. Adv Genet 2010; 70:87 - 99; http://dx.doi.org/10.1016/B978-0-12-380866-0.60004-6; PMID: 20920746
- Wang Y, Zhang X, Li H, Yu J, Ren X. The role of miRNA-29 family in cancer. Eur J Cell Biol 2013; 92:123 - 8; http://dx.doi.org/10.1016/j.ejcb.2012.11.004; PMID: 23357522
- Fabbri M, Garzon R, Cimmino A, Liu Z, Zanesi N, Callegari E, Liu S, Alder H, Costinean S, Fernandez-Cymering C, et al. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc Natl Acad Sci U S A 2007; 104:15805 - 10; http://dx.doi.org/10.1073/pnas.0707628104; PMID: 17890317
- Garzon R, Liu S, Fabbri M, Liu Z, Heaphy CE, Callegari E, Schwind S, Pang J, Yu J, Muthusamy N, et al. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009; 113:6411 - 8; http://dx.doi.org/10.1182/blood-2008-07-170589; PMID: 19211935
- Croker BA, Kiu H, Nicholson SE. SOCS regulation of the JAK/STAT signalling pathway. Semin Cell Dev Biol 2008; 19:414 - 22; http://dx.doi.org/10.1016/j.semcdb.2008.07.010; PMID: 18708154
- Calò V, Migliavacca M, Bazan V, Macaluso M, Buscemi M, Gebbia N, Russo A. STAT proteins: from normal control of cellular events to tumorigenesis. J Cell Physiol 2003; 197:157 - 68; http://dx.doi.org/10.1002/jcp.10364; PMID: 14502555
- Neuwirt H, Puhr M, Santer FR, Susani M, Doppler W, Marcias G, Rauch V, Brugger M, Hobisch A, Kenner L, et al. Suppressor of cytokine signaling (SOCS)-1 is expressed in human prostate cancer and exerts growth-inhibitory function through down-regulation of cyclins and cyclin-dependent kinases. Am J Pathol 2009; 174:1921 - 30; http://dx.doi.org/10.2353/ajpath.2009.080751; PMID: 19342366
- Strebovsky J, Walker P, Lang R, Dalpke AH. Suppressor of cytokine signaling 1 (SOCS1) limits NFkappaB signaling by decreasing p65 stability within the cell nucleus. FASEB J 2011; 25:863 - 74; http://dx.doi.org/10.1096/fj.10-170597; PMID: 21084693
- Galm O, Yoshikawa H, Esteller M, Osieka R, Herman JG. SOCS-1, a negative regulator of cytokine signaling, is frequently silenced by methylation in multiple myeloma. Blood 2003; 101:2784 - 8; http://dx.doi.org/10.1182/blood-2002-06-1735; PMID: 12456503
- Brakensiek K, Länger F, Schlegelberger B, Kreipe H, Lehmann U. Hypermethylation of the suppressor of cytokine signalling-1 (SOCS-1) in myelodysplastic syndrome. Br J Haematol 2005; 130:209 - 17; http://dx.doi.org/10.1111/j.1365-2141.2005.05590.x; PMID: 16029449
- Saudemont A, Hamrouni A, Marchetti P, Liu J, Jouy N, Hetuin D, Colucci F, Quesnel B. Dormant tumor cells develop cross-resistance to apoptosis induced by CTLs or imatinib mesylate via methylation of suppressor of cytokine signaling 1. Cancer Res 2007; 67:4491 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-1627; PMID: 17483365
- Fukushima N, Sato N, Sahin F, Su GH, Hruban RH, Goggins M. Aberrant methylation of suppressor of cytokine signalling-1 (SOCS-1) gene in pancreatic ductal neoplasms. Br J Cancer 2003; 89:338 - 43; http://dx.doi.org/10.1038/sj.bjc.6601039; PMID: 12865927
- Oshimo Y, Kuraoka K, Nakayama H, Kitadai Y, Yoshida K, Chayama K, Yasui W. Epigenetic inactivation of SOCS-1 by CpG island hypermethylation in human gastric carcinoma. Int J Cancer 2004; 112:1003 - 9; http://dx.doi.org/10.1002/ijc.20521; PMID: 15386345
- Bagnyukova TV, Tryndyak VP, Muskhelishvili L, Ross SA, Beland FA, Pogribny IP. Epigenetic downregulation of the suppressor of cytokine signaling 1 (Socs1) gene is associated with the STAT3 activation and development of hepatocellular carcinoma induced by methyl-deficiency in rats. Cell Cycle 2008; 7:3202 - 10; http://dx.doi.org/10.4161/cc.7.20.6816; PMID: 18843197
- Bergsagel PL, Kuehl WM. Molecular pathogenesis and a consequent classification of multiple myeloma. J Clin Oncol 2005; 23:6333 - 8; http://dx.doi.org/10.1200/JCO.2005.05.021; PMID: 16155016
- Huang S. Regulation of metastases by signal transducer and activator of transcription 3 signaling pathway: clinical implications. Clin Cancer Res 2007; 13:1362 - 6; http://dx.doi.org/10.1158/1078-0432.CCR-06-2313; PMID: 17332277
- Gerlo S, Haegeman G, Vanden Berghe W. Transcriptional regulation of autocrine IL-6 expression in multiple myeloma cells. Cell Signal 2008; 20:1489 - 96; http://dx.doi.org/10.1016/j.cellsig.2008.04.004; PMID: 18502099
- Lee H, Herrmann A, Deng JH, Kujawski M, Niu G, Li Z, Forman S, Jove R, Pardoll DM, Yu H. Persistently activated Stat3 maintains constitutive NF-kappaB activity in tumors. Cancer Cell 2009; 15:283 - 93; http://dx.doi.org/10.1016/j.ccr.2009.02.015; PMID: 19345327
- de la Iglesia N, Konopka G, Lim KL, Nutt CL, Bromberg JF, Frank DA, Mischel PS, Louis DN, Bonni A. Deregulation of a STAT3-interleukin 8 signaling pathway promotes human glioblastoma cell proliferation and invasiveness. J Neurosci 2008; 28:5870 - 8; http://dx.doi.org/10.1523/JNEUROSCI.5385-07.2008; PMID: 18524891
- Kuai WX, Wang Q, Yang XZ, Zhao Y, Yu R, Tang XJ. Interleukin-8 associates with adhesion, migration, invasion and chemosensitivity of human gastric cancer cells. World J Gastroenterol 2012; 18:979 - 85; http://dx.doi.org/10.3748/wjg.v18.i9.979; PMID: 22408359
- Li A, Dubey S, Varney ML, Dave BJ, Singh RK. IL-8 directly enhanced endothelial cell survival, proliferation, and matrix metalloproteinases production and regulated angiogenesis. J Immunol 2003; 170:3369 - 76; PMID: 12626597
- Walker BA, Wardell CP, Chiecchio L, Smith EM, Boyd KD, Neri A, Davies FE, Ross FM, Morgan GJ. Aberrant global methylation patterns affect the molecular pathogenesis and prognosis of multiple myeloma. Blood 2011; 117:553 - 62; http://dx.doi.org/10.1182/blood-2010-04-279539; PMID: 20944071
- Griffiths EA, Gore SD. Epigenetic therapies in MDS and AML. Adv Exp Med Biol 2013; 754:253 - 83; http://dx.doi.org/10.1007/978-1-4419-9967-2_13; PMID: 22956506
- Ma X, Wang YW, Zhang MQ, Gazdar AF. DNA methylation data analysis and its application to cancer research. Epigenomics 2013; 5:301 - 16; http://dx.doi.org/10.2217/epi.13.26; PMID: 23750645
- Fratta E, Sigalotti L, Colizzi F, Covre A, Nicolay HJ, Danielli R, Fonsatti E, Altomonte M, Calabrò L, Coral S, et al. Epigenetically regulated clonal heritability of CTA expression profiles in human melanoma. J Cell Physiol 2010; 223:352 - 8; PMID: 20127705
- Shi XB, Tepper CG, deVere White RW. Cancerous miRNAs and their regulation. Cell Cycle 2008; 7:1529 - 38; http://dx.doi.org/10.4161/cc.7.11.5977; PMID: 18469525
- Rossi M, Amodio N, Di Martino MT, Caracciolo D, Tagliaferri P, Tassone P. From target therapy to miRNA therapeutics of human multiple myeloma: theoretical and technological issues in the evolving scenario. Curr Drug Targets 2013; 14:1144 - 9; http://dx.doi.org/10.2174/13894501113149990186; PMID: 23834146
- Reddy J, Shivapurkar N, Takahashi T, Parikh G, Stastny V, Echebiri C, Crumrine K, Zöchbauer-Müller S, Drach J, Zheng Y, et al. Differential methylation of genes that regulate cytokine signaling in lymphoid and hematopoietic tumors. Oncogene 2005; 24:732 - 6; http://dx.doi.org/10.1038/sj.onc.1208032; PMID: 15580314
- Richardson PG, Sonneveld P, Schuster MW, Irwin D, Stadtmauer EA, Facon T, Harousseau JL, Ben-Yehuda D, Lonial S, Goldschmidt H, et al, Assessment of Proteasome Inhibition for Extending Remissions (APEX) Investigators. Bortezomib or high-dose dexamethasone for relapsed multiple myeloma. N Engl J Med 2005; 352:2487 - 98; http://dx.doi.org/10.1056/NEJMoa043445; PMID: 15958804
- Molineaux SM. Molecular pathways: targeting proteasomal protein degradation in cancer. Clin Cancer Res 2012; 18:15 - 20; http://dx.doi.org/10.1158/1078-0432.CCR-11-0853; PMID: 22019514
- Ballabio E, Armesto M, Breeze CE, Manterola L, Arestin M, Tramonti D, Hatton CS, Lawrie CH. Bortezomib action in multiple myeloma: microRNA-mediated synergy (and miR-27a/CDK5 driven sensitivity)?. Blood Cancer J 2012; 2:e83; http://dx.doi.org/10.1038/bcj.2012.31; PMID: 22922378
- Liu S, Liu Z, Xie Z, Pang J, Yu J, Lehmann E, Huynh L, Vukosavljevic T, Takeki M, Klisovic RB, et al. Bortezomib induces DNA hypomethylation and silenced gene transcription by interfering with Sp1/NF-kappaB-dependent DNA methyltransferase activity in acute myeloid leukemia. Blood 2008; 111:2364 - 73; http://dx.doi.org/10.1182/blood-2007-08-110171; PMID: 18083845
- Chou J, Lin JH, Brenot A, Kim JW, Provot S, Werb Z. GATA3 suppresses metastasis and modulates the tumour microenvironment by regulating microRNA-29b expression. Nat Cell Biol 2013; 15:201 - 13; http://dx.doi.org/10.1038/ncb2672; PMID: 23354167
- Nelson EA, Sharma SV, Settleman J, Frank DA. A chemical biology approach to developing STAT inhibitors: molecular strategies for accelerating clinical translation. Oncotarget 2011; 2:518 - 24; PMID: 21680956
- Shi L, Wang S, Zangari M, Xu H, Cao TM, Xu C, Wu Y, Xiao F, Liu Y, Yang Y, et al. Over-expression of CKS1B activates both MEK/ERK and JAK/STAT3 signaling pathways and promotes myeloma cell drug-resistance. Oncotarget 2010; 1:22 - 33; PMID: 20930946
- Yan S, Li Z, Thiele CJ. Inhibition of STAT3 with orally active JAK inhibitor, AZD1480, decreases tumor growth in Neuroblastoma and Pediatric Sarcomas In vitro and In vivo. Oncotarget 2013; 4:433 - 45; PMID: 23531921
- Kline M, Donovan K, Wellik L, Lust C, Jin W, Moon-Tasson L, Xiong Y, Witzig TE, Kumar S, Rajkumar SV, et al. Cytokine and chemokine profiles in multiple myeloma; significance of stromal interaction and correlation of IL-8 production with disease progression. Leuk Res 2007; 31:591 - 8; http://dx.doi.org/10.1016/j.leukres.2006.06.012; PMID: 16879867
- Amodio N, Scrima M, Palaia L, Salman AN, Quintiero A, Franco R, Botti G, Pirozzi P, Rocco G, De Rosa N, et al. Oncogenic role of the E3 ubiquitin ligase NEDD4-1, a PTEN negative regulator, in non-small-cell lung carcinomas. Am J Pathol 2010; 177:2622 - 34; http://dx.doi.org/10.2353/ajpath.2010.091075; PMID: 20889565
- Livak KJ, Schmittgen TD. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods 2001; 25:402 - 8; http://dx.doi.org/10.1006/meth.2001.1262; PMID: 11846609
- Blotta S, Jakubikova J, Calimeri T, Roccaro AM, Amodio N, Azab AK, Foresta U, Mitsiades CS, Rossi M, Todoerti K, et al. Canonical and noncanonical Hedgehog pathway in the pathogenesis of multiple myeloma. Blood 2012; 120:5002 - 13; http://dx.doi.org/10.1182/blood-2011-07-368142; PMID: 22821765