Abstract
The ATR/CHK1-dependent intra-S checkpoint inhibits replicon initiation and replication fork progression in response to DNA damage caused by UV (UV) radiation. It has been proposed that this signaling cascade protects against UV-induced mutations by reducing the probability that damaged DNA will be replicated before it can be repaired. Normal human fibroblasts (NHF) were depleted of ATR or CHK1, or treated with the CHK1 kinase inhibitor TCS2312, and the UV-induced mutation frequency at the HPRT locus was measured. Despite clear evidence of S-phase checkpoint abrogation, neither ATR/CHK1 depletion nor CHK1 inhibition caused an increase in the UV-induced HPRT mutation frequency. These results question the premise that the UV-induced intra-S checkpoint plays a prominent role in protecting against UV-induced mutagenesis.
Introduction
The capacity to replicate and segregate the entire genome with high fidelity is a hallmark of normal division cycles. Furthermore, preventing replication of damaged DNA decreases the potential for mutations.Citation1 Cellular DNA constantly experiences damage, both as a consequence of normal cellular metabolism and respiration, and as a result of environmental insults. In order to cope with such genomic damage, cells have evolved a network of DNA damage responses (DDR),Citation2 consisting of DNA repair mechanisms, damage tolerance pathways, and cell cycle checkpoints.
Cell cycle checkpoints are systems in place to recognize damaged DNA, or aberrant DNA structures, and activate signaling cascades designed to halt or slow progression through the cell cycle, thus maximizing the time to repair such damage before DNA is replicated or segregated to progeny. Indeed, checkpoint failures have been shown to result in chromosomal abnormalities, developmental defects, and predisposition to cancer.Citation3,Citation4 Upon sensing damaged DNA, cell cycle checkpoints inhibit progression out of the G1 and G2 phases of the cell cycle, while the intra-S checkpoint slows progression through S-phase.Citation4
The intra-S checkpoint can be divided into two main signaling cascades that respond to different forms of DNA damage. The first recognizes double strand breaks in DNA, such as those induced by ionizing radiation (IR). The intra-S checkpoint response to IR requires the proteins ATM, MRE-11, RAD50 and NBS1 and involves activation of the checkpoint kinases CHK1 and CHK2, promoting proteolytic degradation of CDC25A. Loss of the CDC25A phosphatase prevents removal of the inhibitory phosphorylation at tyr15 of CDK2, thus maintaining this kinase inactive, and preventing CDC45 loading and initiation at new replication origins.Citation4 ATM also maintains the intra-S checkpoint response by signaling through the cohesion subunits Smc1 and Smc3.Citation4,Citation5 The second main signaling cascade recognizes bulky DNA adducts, such as those induced by UV (UV) radiation or benzo[a]pyrene diolepoxide. This checkpoint response to UV was found to require the activity of checkpoint proteins ataxia telangiectasia and Rad3-related (ATR) kinase, and its target CHK1, to act on downstream substrate(s) to inhibit the firing of new origins of replicationCitation6-Citation8 and to reduce the rate of DNA chain elongation.Citation9-Citation11 At sub-lethal fluences of UV, the ATR/CHK1 pathway has been shown not to depend on degradation of CDC25A, but rather it has been proposed to act on the DBF4-dependent kinase to prevent CDC45-dependent activation of the MCM helicase, thus inhibiting new replicon initiation.Citation12,Citation13
It is well established that loss of functional ATM increases IR-induced mutation frequencyCitation14 and predisposes mammals to lymphomas and other malignancies.Citation15 Although ATM is widely recognized as a tumor-suppressor gene, it is unclear whether this function depends primarily on the activation of the ATM-dependent intra-S checkpoint or on ATM’s roles in other DNA damage responses. In contrast, loss of Atr causes embryonic lethality in mice,Citation16,Citation17 while Seckel syndrome is a consequence of an ATR hypomorphic splice mutation. Seckel syndrome manifests as a developmental disease, with patients exhibiting dwarfism, microcephaly, and mental retardation, but cancer is not typically associated with the disorder.Citation3 Abrogation of the ATR/CHK1 pathway has clearly been shown to inhibit the intra-S checkpoint to UV,Citation6,Citation8 and to lead to an increase in chromosomal instability.Citation18 Despite these observations, it is less clear whether abrogation of the ATR/CHK1-dependent S-phase checkpoint influences mutagenesis. To address this question, we employed siRNA-mediated depletion of ATR or CHK1, or pharmacological inhibition of CHK1 kinase function, to inactivate the intra-S checkpoint and measured mutation frequency at the HPRT locus in UV-irradiated human fibroblasts. The results described here support the conclusion that in the presence or absence of the rapid activation of intra-S checkpoint responses, UV-irradiated normal human fibroblasts acquire the same burden of mutations.
Results
Depletion of ATR or CHK1 abrogated the intra-S phase checkpoint triggered by UV
The activation of the intra-S checkpoint in UV-irradiated NHF1 was assessed by examining changes in the steady-state distribution of sizes of nascent DNA pulse-labeled with 3H-thymidine. Responses were compared in populations pre-treated with a non-targeted control (NTC) siRNA and those with siRNA-mediated depletion of ATR or CHK1. illustrates the siRNA-mediated depletion of ATR, which in the irradiated cells was accompanied by a severe reduction in phosphorylation of its substrate CHK1, a known mediator of UV-induced S-phase checkpoint activation.Citation6-Citation8 Velocity sedimentation analyses showed that fibroblasts pre-treated with the NTC-siRNA () displayed a stereotypical reduction in the abundance of small molecular weight (MW) nascent DNA when exposed to a low fluence of UVC. The selective reduction in abundance of this size class of origin-proximal, newly synthesized DNA represents a functional indicator of the inhibition of initiation of replication origins that were scheduled to fire post-irradiation.Citation8,Citation19,Citation20 Depletion of ATR () interfered with this selective inhibition of the synthesis of small MW nascent DNA, thus demonstrating the abrogation of the intra-S checkpoint response of inhibition of replicon initiation. Similarly, inhibition of this S-phase checkpoint response was confirmed following siRNA-mediated depletion of CHK1 ()
Figure 1. Depletion of ATR abrogates the intra-S checkpoint response: (A) NHF1 cells were electroporated with NTC or ATR siRNA and UV-irradiated (sham or 2.5 J/m2) 48 h following electroporation. Cells were harvested and protein extracts examined by immunoblotting 1 h after irradiation. (B and C) Velocity sedimentation analysis of nascent DNA from NHF1 cells exposed to UVC (sham-closed squares or 2.5 J/m2-open diamonds) 48 h post-electroporation with (B) NTC or (C) ATR siRNA; the irradiated cells were incubated for 30 min and then pulse labeled with 3H-thymidine for 15 min.
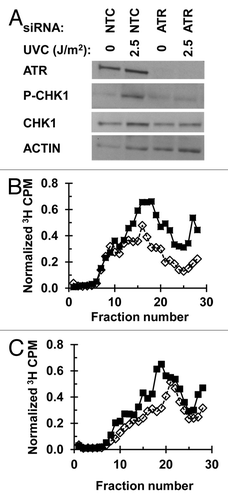
Figure 2. Depletion of CHK1 abrogates the intra-S checkpoint response: (A) NHF1 cells were electroporated with NTC or CHK1 siRNA. Cells were harvested 24 h later and protein extracts examined by immunoblotting. (B and C) Velocity sedimentation analysis of nascent DNA from NHF1 cells exposed to UVC (sham-closed squares or 2.5 J/m2-open diamonds), following electroporation with (B) NTC or (C) CHK1 siRNA. Cells were irradiated 24 h post-electroporation and 45 min later were pulse labeled with 3H-thymidine for 15 min.
Depletion of ATR or CHK1 did not increase the UV-induced mutation frequency at the HPRT locus
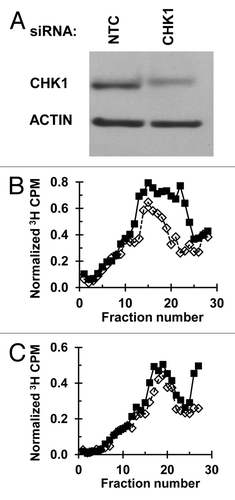
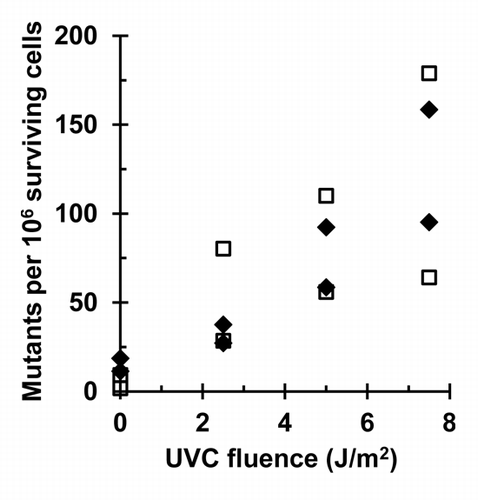
Mutation frequency at the HPRT locus was measured in NHF1 cells following depletion of ATR or CHK1 and irradiation with increasing fluences of UVC (). In cultured fibroblasts treated with the NTC-siRNA, UV induced the formation of 6-thioguanine resistant mutant colonies at frequencies above the background (measured in the sham-irradiated populations) (P < 0.05 for sham vs. 7.5 J/m2, one-sided Wilcoxon rank-sum test, or by linear regression of all fluences). As a pre-validation of the adopted methodology, under the laboratory conditions used in this study, NHF1 cultures were treated with siRNA targeting the translesion synthesis (TLS) DNA polymerase ηCitation21; an elevated recovery of HPRT mutant colonies was observed, as compared with the NTC siRNA-treated population, even at fluences ≤ 4 J/m2 (Fig. S1). In contrast, cells depleted of CHK1 or ATR did not display mutation frequencies higher than those treated with control siRNA ().
Figure 3. Depletion of ATR or CHK1 does not increase UV-induced mutation frequency at the HPRT locus: (A) Depletion of ATR in cells used for mutagenesis experiment. NHF1 cells were electroporated with NTC or ATR siRNA and irradiated (sham, 2.5, or 5 J/m2) 48 h following electroporation. Cells were harvested and protein extracts examined by immunoblotting 1 h after irradiation. (B) Depletion of CHK1 in cells used for mutagenesis experiment. NHF1 cells were electroporated with NTC or CHK1 siRNA. Cells were harvested 24 h later and protein extracts examined by immunoblotting. (C) UV-induced (0, 2.5, 5, or 7.5 J/m2) HPRT mutation frequencies in NHF1 following depletion of ATR (open triangle) or CHK1 (closed squares) or NTC (open diamonds) expressed as mutants per 106 surviving cells ± SEM. Values for sham or 7.5 J/m2 were slightly offset on figure to allow for clear illustration of error bars. In NTC-treated cells, UV induced a mutation frequency statistically higher than the sham irradiated cells (P < 0.05 for sham vs. 7.5 J/m2, one-sided Wilcoxon rank-sum test, or by linear regression of all fluences). Depletion of ATR or CHK1 did not cause a significant difference in UV-induced mutation frequency (P = 0.86 and 0.63, respectively, Wilcoxon rank-sum test).
Pharmacological inhibition of CHK1 with TCS2312 abrogated the UV-induced intra-S phase checkpoint
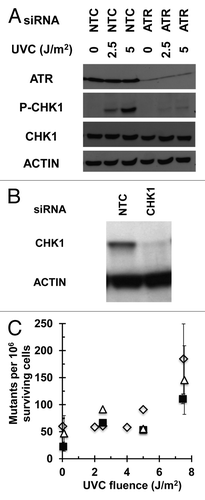
The results described above were confirmed by experiments in which an alternative approach was employed to inactivate the intra-S checkpoint. Instead of depleting the cells of the two major kinases involved in the activation of this checkpoint, a CHK1 inhibitor (TCS2312) was used to block the signaling pathway while retaining normal cellular protein levels. First, the effect of increasing concentrations of this inhibitor on overall DNA synthesis rates was measured. The lowest concentrations inhibiting DNA synthesis were tested for their effects on the intra-S checkpoint response of inhibition of replicon initiation (Fig. S2–4). As was observed following depletion of ATR or CHK1, treatment of fibroblasts with 1 µM TCS2312 reversed the UV-induced selective inhibition of small MW nascent DNA observed in the control population irradiated with 1 or 2.5 J/m2 UVC, indicating a blockage in the S-phase checkpoint signaling cascade (; Fig. S4).
Figure 4. CHK1 inhibitor TCS2312 abrogates the intra-S checkpoint response: Velocity sedimentation analysis of nascent DNA from NHF1 cells treated with the CHK1 inhibitor TCS2312. Cells were treated with (A) vehicle or (B) 1 µM TCS2312 30 min prior to irradiation with 0 (closed squares), 1 (open circles), or 2.5 (closed diamonds) J/m2 and inhibitor remained until cells were harvested. Cells were pulse-labeled with 3H-thymidine (15 min) 30 min post-irradiation.
CHK1 inhibition did not increase the UV-induced mutation frequency at the HPRT locus
Mutation frequencies at the HPRT locus were measured in NHF1 cells following treatment with 1 µM TCS2312 and irradiation with 0, 2.5, 5, or 7.5 J/m2 UVC (). NHF1 cells irradiated in the presence or absence of inhibitor formed 6-thioguanine resistant mutant colonies in a fluence-dependent manner (P < 0.005, linear regression analysis). Despite abrogation of the S-phase checkpoint through pharmacological inhibition of CHK1, HPRT mutation frequencies were no different than those observed in cells not receiving the inhibitor.
Figure 5. CHK1 inhibitor TCS2312 does not increase UV-induced mutation frequency at the HPRT locus: UV-induced (0, 2.5, 5, or 7.5 J/m2) HPRT mutation frequencies in NHF1 following treatment with vehicle (closed diamonds) or 1 µM TCS2312 (open squares) expressed as mutants per 106 surviving cells (n = 2). In both vehicle and TCS2312 treated cells, UV induced a fluence-dependent increase in mutation frequency (P < 0.005, linear regression analysis). The UV-induced mutation frequency was not different between cells treated with vehicle or inhibitor (P = 0.97, linear regression analysis).
Colony forming efficiency and UV-induced cytotoxicity following ATR/CHK1 depletion or CHK1 inhibition
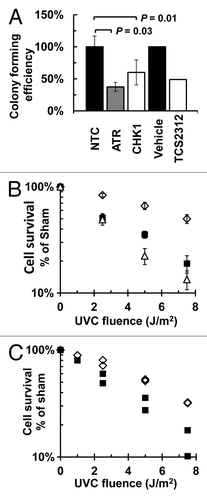
In the course of performing the mutagenesis studies described above, colony forming assays were done in each experiment to assess the effect of siRNA-mediated depletion of CHK1 or ATR, or inhibition of CHK1, on overall cell survival and on UV-induced cytotoxicity. Depletion of either ATR (P = 0.03, one-sided Wilcoxon rank-sum test) or CHK1 (P = 0.01, one-sided Wilcoxon rank-sum test) decreased colony forming efficiency in the absence of UV by 63% or 40%, respectively, as compared with the colony forming efficiency of NTC siRNA-treated cells; similarly, inhibition of CHK1 by TCS2312 reduced colony forming efficiency by 51% compared with the vehicle treated cells (). Loss of ATR or CHK1 (), or treatment with TCS2312 (), also sensitized NHF1 to UV-induced cytotoxicity (P < 0.0001 for both siRNA depletions, P < 0.001 for inhibitor, by linear regression analysis).
Figure 6. Influence of ATR or CHK1 depletion, or CHK1 inhibition, on colony forming efficiency or UV-induced cytotoxicity: (A) Colony forming efficiency of cells depleted of CHK1 (n = 4) or ATR (n = 6), or treated with 1 µM TCS2312 (n = 2), as compared with either the NTC (n = 10) or vehicle (n = 2) treated cells. Depletion of ATR (p = 0.03, one-sided Wilcoxon rank-sum test) or CHK1 (P = 0.01, one-sided Wilcoxon rank-sum test) decreased colony forming efficiency from that of NTC treated cells. (B) UV-induced cytotoxicity of NHF1 after siRNA-mediated depletion of NTC (open diamonds), ATR (open triangles) or CHK1 (closed squares) as measured by colony forming efficiency. Depletion of ATR or CHK1 increased UV-induced cytotoxicity (p < 0.0001 for both depletions, linear regression analysis). (C) UV-induced cytotoxicity of NHF1 after treatment with vehicle (closed diamonds), or 1 µM TCS2312 (open squares) as measured by colony forming efficiency. All values n = 2. The UV-induced cytotoxicity in cells treated with inhibitor was greater than that of vehicle treated cells (P < 0.001, linear regression analysis).
Discussion
Nucleotide excision repair, DNA polymerase η-dependent translesion synthesis (TLS), and cell cycle checkpoints are known to minimize the consequences of UV-induced DNA damage. Xeroderma pigmentosum (XP) is a genetic disorder characterized by dramatic UV sensitivity and a > 1000-fold predisposition to skin cancers in areas exposed to solar radiation.Citation22 XP is primarily caused by mutations in nucleotide excision repair genes, highlighting the importance of this pathway in protecting against UV-induced mutagenesis and carcinogenesis.Citation23 Similarly, XP patients of the variant complementation group (XPV) lack functional DNA polymerase η, which is required for efficient synthesis across UV-induced photoproducts.Citation24 XPV patients also display increased sensitivity to sunlight, typically developing skin cancers between the ages of 20 and 30.Citation25 To date it has not been established that aberrant cell cycle checkpoints contribute directly to UV-induced carcinogenesis. Although not proven to be cancer drivers, there is evidence that the G1 and G2 checkpoints are defective in some melanoma cell lines,Citation26-Citation28 but there is no evidence that the intra-S checkpoint is abrogated in cancers. The prevailing paradigm has been that checkpoints decrease the probability that damaged DNA will be replicated, allowing time for nucleotide excision repair to remove template lesions. The intra-S checkpoint, in particular, has been hypothesized to minimize mutagenesis by inhibiting the replication of damaged DNA,Citation5 or by improving the efficiency of TLS across DNA photoproducts by facilitating PCNA ubiquitinationCitation5,Citation29,Citation30 and recruitment of DNA polymerase η.Citation31,Citation32
Absent or malfunctioning components of the IR-induced DDR that regulate cell cycle checkpoint pathways lead to a number of human disorders, including ataxia telangiectasia (AT), AT-like disorder, Nijmegen breakage syndrome, and Li Fraumeni syndrome. Mutations in ATM, NBS1, MRE11, and CHK2 have been linked to increased cancer susceptibility, and ATM-deficient cells have been shown to be hypermutable in response to IR.Citation14 The proteins coded by these genes are involved in multiple facets of the IR-induced DDR, including double strand break repair and multiple cell cycle checkpointsCitation4; hence, it is not clear if loss of the intra-S checkpoint contributes to the increase in susceptibility to malignancy. Mutations identified as causing Seckel syndrome, either in ATR, ATRIP, or PCNT, result in impaired UV-induced DDR,Citation33-Citation35 but elevated cancer risk is not typically associated with this disease. It is unclear whether a malfunctioning ATR/CHK1-dependent intra-S checkpoint results in increased mutation frequencies in cells exposed to agents that induce bulky DNA lesions (e.g., UV or benzo[a]pyrene diolepoxide). Results shown here confirmed that upon depletion of ATR or CHK1 the intra-S checkpoint was not activated by UV. Similarly, pharmacologic inhibition of the CHK1 kinase abrogated the UV-induced inhibition of replicon initiation. However, the UV-induced mutation frequency at the HPRT locus was not increased relative to matched controls. These results suggest that loss of ATR/CHK1 signaling, at least during the first S phase following UV exposure when cells must cope with the highest density of DNA photoproducts, does not result in higher mutation frequencies, even at a locus (HPRT) where point mutations, as well as small or large deletions, result in the same phenotype (6-thioguanine resistance).
To date, little information is available regarding the role of the ATR/CHK1 pathway as a tumor suppressor. However, some insight can be derived from research describing germline or somatic mutations that are presumed to affect this pathway, or by examining data from animal models targeting ATR/CHK1. A germline mutation in the FAT domain of ATR in an autosomal-dominant oropharyngeal cancer syndrome,Citation36 or a mutation in the checkpoint mediator CLSPN in the germline of a familial breast cancer case,Citation37 did not impair intra-S checkpoint activation as measured by CHK1 phosphorylation. Somatic mutations in ATR and CHK1 have also been reported in endometrial, colorectal, and stomach cancers, particularly in mismatch repair deficient cells with microsatellite instability (MSI).Citation38-Citation40 It is unclear whether or not these mutations compromise the activation of the intra-S checkpoint, and whether they are cancer drivers or passenger mutations. One MSI+ colon cancer cell line, heterozygous for an ATR truncation mutation, was shown to be impaired in the UV-induced phosphorylation of CHK1 and the late-S/G2 arrest induced by topotecan.Citation41 In this same study, chronic myelogenous leukemia cells transiently transfected with an ATR truncation mutant allele displayed a similar phenotype while retaining expression of endogenous ATR, suggesting that the truncation mutation acts in a dominant negative manner. We cannot rule out the possibility that a dominant negative ATR or CHK1 could influence UV-induced mutation frequency. However, such a mechanism would most likely not depend on the inhibition of CHK1 kinase activity, as we did not observe a change in UV-induced mutation frequency upon pharmacological inhibition of CHK1 kinase.
Animal models for ATR or CHK1 heterozygosity have also shown enhanced tumorigenesisCitation17,Citation42-Citation45 or tumor progression.Citation42 The reported effects, however, often require synergistic combination with a cancer-driver mutation. For example, loss of one allele of Chk1 or p53 in mouse mammary tissue did not result in tumor formation, but when combined, the loss of one allele at each locus resulted in 60% tumor formation.Citation46 Similarly, Atr+/−, Mlh−/− mice developed cancer at much higher rates (71%) than their Atr+/− (0%) or Mlh−/− (17%) littermates.Citation44 It is known that Atr and Chk1 knockout mice are not viableCitation16,Citation17,Citation45 and that loss of either protein enhances cytotoxicity; therefore, the tumorigenic effects described above might depend on a threshold of expression of these proteins. The implications to our findings would be that below this threshold, cell survival is compromised to the point that enhanced mutagenesis may not be observed in our in vitro system. It has yet to be determined if moderately depleted levels of ATR or CHK1, such as those observed in heterozygous knockout animals, would allow cell survival at the cost of genomic instability and increased mutagenesis.
There is evidence that intra-S checkpoint signaling affects TLS,Citation29,Citation31,Citation32,Citation47 and it is conceivable that intra-S checkpoint inhibition is also inhibiting efficient TLS. Because mutagenesis requires replication of the damaged DNA, error-prone TLS is considered a prominent driver of UV-induced mutagenesis. Thus, even if intra-S checkpoint inhibition was allowing newly fired replication forks to encounter UV-induced DNA damage sites, TLS would be required to replicate across the photolesions, and to potentially induce mutations. If ATR or CHK1 were important for efficient bypass synthesis (in addition to their known checkpoint functions), it stands to reason that the depletion of these enzymes might inhibit TLS and reduce mutation frequency. It has been suggested that ATR, CHK1, and other components of the replication fork protection complex, not only participate in the activation of the intra-S checkpoint, but also possess separate functions for preservation of intrinsic chromosomal stability,Citation18 particularly at common fragile sites.Citation48,Citation49 Common fragile sites tend to be replicated late in S phase; the formation of secondary structures within the fragile sites is thought to slow progression of replication forks.Citation49,Citation50 It is of note that the active HPRT gene is replicated early in S-phase.Citation51 Perhaps the primary function of the intra-S checkpoint proteins is to preserve genomic stability at difficult to replicate regions and/or regions replicated late in S-phase, and that inhibition of replicon initiation in response to UV is not of primary importance in minimizing UV-induced mutations in regions of the genome that are replicated more easily or early in S-phase.
The results reported here show that severe depletion of ATR/CHK1 did not increase UV-induced mutagenesis, perhaps as a consequence of preferential cytotoxicity in cells most depleted of CHK1 or ATR, or decreased TLS efficiency. There has been increasing interest in developing ATR or CHK1 kinase inhibitors as therapeutics designed to sensitize cancer cells to the cytotoxic effects of clinical drugs.Citation52 Our results agree with many others showing that CHK1 inhibition sensitizes cells to DNA damaging agents, including cisplatin in non-small cell lung carcinoma cellsCitation53 and camptothecin in colon cancer cells.Citation54 An attractive idea has been that some cancer cells are selectively sensitive to ATR/CHK1 inhibition in combination with DNA damaging agents, perhaps because of loss of the p53-dependent G1 checkpoint.Citation55 Although our results indicate that loss of ATR/CHK1 or inhibition of CHK1 does not increase UV-induced mutagenesis, it remains to be determined if this would hold true for other DNA-damaging chemotherapeutics. If so, our results would suggest that the combination of ATR/CHK1 inhibitors and chemotherapy would have the additional advantage of not enhancing the probability of drug resistant clones or secondary, chemotherapy-induced cancers emerging from an increase in mutagenesis.
Materials and methods
Cell culture and UV treatment
The normal human fibroblast cell line NHF1-hTERT was derived from neonatal foreskin fibroblastsCitation56 and immortalized by ectopic expression of the catalytic subunit of human telomerase.Citation6 Cells were cultured in minimum essential medium (GIBCO) supplemented with 10% fetal calf serum and 2 mM L-glutamine, and were maintained in humidified 95% air and 5% CO2 at 37 °C. For UV irradiations, cells were rinsed in phosphate buffered saline, all extra liquid removed, and the culture plates irradiated without the lids. Irradiation was performed with a fluorescent germicidal lamp emitting primarily 254 nm UVC (Sylvania G8T5, 90% emission at 254 nm). Control plates were treated under identical conditions, but were not exposed to UV (sham controls). Lamp irradiance was measured using a UVX Digital Radiometer (Ultra-Violet Products Inc) using the UVX-25 sensor (250–290 nm range, calibrated at 254 nm).
Protein depletion
siRNA was introduced by electroporation (100 pmol siRNA per 106 cells), using the normal human dermal fibroblast kit VDP-1001 (Lonza) and program U-23 on a Nucleofector 2b electroporation device (Lonza). ON-TARGET plus duplex siRNAs were purchased from Dharmacon: NTC (D-001210–2), CHK1 (J-003255), and ATR SMART pool (L-003202). Experiments were conducted 24 h post electroporation with CHK1 or 48 h post-electroporation with ATR.
CHK1 inhibitor TCS2312
The CHK1 inhibitor TCS2312 (Tocris) was dissolved in sterile water (1 mM stock solution) and added to cell culture medium at the indicated concentrations. For velocity sedimentation experiments, inhibitor was added 30 min prior to irradiation and kept in the medium during the subsequent 30 min incubation and the 15 min 3H-thymidine pulse-labeling period. For mutagenesis and cytotoxicity experiments, inhibitor was added 30 min prior to irradiation and removed six hours after the UV exposure, at which time the cells were rinsed with buffered saline and fed with fresh culture medium.
Western blotting and antibodies
One hour after irradiation, cells were harvested by trypsinization, pelleted and flash frozen in liquid nitrogen. Frozen pellets were resuspended in lysis buffer (8 M urea, 5 M NaH2PO4, 1 M Tris-HCl pH 8) on ice for 30 min and then clarified by centrifugation (16 000 × g and 4 °C). The Bio-Rad Protein Assay kit was used to determine protein concentration. Equal volumes of loading buffer (125 mM Tris pH 6.8, 20% glycerol, 10% β-mercaptoethanol, 4% sodium dodecyl sulfate, 0.05% bromo-phenol blue) were combined with protein lysates and samples were boiled for 5 min. Equal amounts of protein were loaded onto BioRad Criterion-TGX 4–15% gradient gels (approximately 150–200 V, 2–4 h). Size-separated proteins were transferred overnight (100 mA) onto a nitrocellulose membrane. After blocking in 5% powdered milk in TBST (0.5% Tween-20, 10 mM Tris-HCl pH 7.4, 10 mM NaCl,), primary antibodies were applied. The following antibodies were used: rabbit anti-P-CHK1 S345 (Cell Signaling), mouse anti-CHK1, goat anti-ATR, and goat anti-Actin (Santa Cruz). Following application of secondary antibody conjugated with horseradish peroxidase (Amersham), bands were visualized on film by enhanced chemiluminescence (ECL). An Alpha Innotech Fluor-Chem HD2 was used to measure the densitometry of film bands.
Velocity sedimentation
As previously describedCitation19,Citation57 the steady-state size distribution of nascent DNA was determined by centrifugation in an alkaline sucrose density gradient 45 min after UV irradiation. Cells were uniformly pre-labeled during logarithmic growth by incubation with 14C-thymidine (5–10 nCi/mL) for at least 36 h. Medium containing 14C-thymidine was removed overnight and cells treated the following day. Thirty minutes after UV-irradiation, cells were incubated with 25 µCi/mL 3H-thymidine for 15 min. Cells were rinsed in PBS then scraped on ice into 0.1 M NaCl, 0.01 M EDTA (pH 8) and then passed through a 22G needle 5×. An equal volume of cell suspension was added to 0.5 mL of lysis buffer (1 M NaOH, 0.02 M EDTA) already on top of an alkaline sucrose gradient (5–20% sucrose in 0.4 M NaOH, 2 M NaCl, 0.01 M EDTA). These gradients were left under fluorescent lighting for 45 min and then centrifuged in a Beckman SW32Ti rotor for 5 h at 25 000 rpm and 20 °C. Gradients were fractioned through a hole punctured in the bottom of the centrifuge tube and acid-precipitable material filtered onto glass microfiber filters (Whatman GF/C 24 mm). The glass microfiber filters were then analyzed by liquid scintillation counting. All experiments included cells pre-labeled with 14C-thymidine but not pulse-labeled with 3H-thymidine. These samples were used to measure the 14C CPM values detected in the 3H channel during liquid scintillation counting. Normalized 3H CPM were the 3H CPM in each fraction, corrected for the 14C spillover, and divided by the total 14C in the gradient (proportional to the number of added cells).
HAT selection
Pre-selection for functional HPRT in cells used for mutagenesis experiments was performed by expanding cultures in medium supplemented with 1 × HAT (100× lyophilized HAT contains 10 mM sodium hypoxanthine, 40 M aminopterin, and 1.6 mM thymidine) for 10–14 d.Citation58 A new aliquot of frozen and stored HAT selected cells was used in each HPRT mutagenesis experiment. Cell cultures were expanded in normal medium without HAT for 10–14 d prior to any treatment.
Cytotoxicity experiments
Concurrent with the mutagenesis experiments, cytotoxicity was determined by colony forming assay at the time of treatment. Cells depleted of CHK1 (and their matched NTC controls) were plated at a density of 1000 cells per 10-cm dish and irradiated 24 h after electroporation. Cells depleted of ATR (and their matched NTC controls) were placed in culture for 24 h post-electroporation and re-seeded at a density of 500 cells per 10-cm dish, and irradiated 48 h after electroporation. Cells to be treated with 1 µM TCS2312 or vehicle (sterile water) were seeded at a density of 500 cells per 10-cm dish and the inhibitor or vehicle added 30 min prior to irradiation (24 h post-electroporation). The inhibitor remained on the cultures for 6 h post-irradiation. The selected cell densities resulted in approximately 100 colonies per dish for the vehicle or NTC-treated, sham-irradiated plates, and 3–6 plates per condition were counted. Cells were fed weekly and colonies fixed in 3:1 methanol:acetic acid and stained with 0.05% crystal violet approximately two weeks later. Colonies with more than 50 cells were counted.
Mutagenesis
Mutagenesis experiments were started by plating electroporated cells at 1 × 106 per dish or 5 × 105 for cells to be treated with vehicle/inhibitor (2 plates per condition). Cells were irradiated 24 h (CHK1) or 48 h post-electroporation (ATR). In the inhibitor experiments, 1 µM TCS2312 or an equal volume of sterile water was added 30 min before the cells were irradiated (24 h after plating). Cells were maintained in logarithmic growth for 4–6 population doublings before HPRT mutants were selected by plating 4 × 104 cells per 10-cm dish (55 dishes per treatment) in medium containing 40 µM 6-thioguanine. At the time of selection, colony forming efficiency was determined by plating 400 cells per dish in medium lacking 6-thioguanine. Approximately two weeks post-seeding, colonies in all plates were fixed in 3:1 methanol:acetic acid and stained with 0.05% crystal violet, and those with more than 50 cells were counted. Mutation frequencies were calculated from the number of plates without any mutant colonies, using the Poisson distribution as follows: (-ln [number of plates without 6-thioguanine resistant colonies/total number of plates])/([number of cells plated for selection] × [colony-forming efficiency at time of selection]).
Statistical methods
Statistical comparisons were performed in order to determine whether UV-induced mutation frequencies, UV-induced cytotoxicity, or colony forming efficiency following siRNA-mediated depletion of ATR or CHK1, or pharmacological inhibition of CHK1, varied significantly from the NTC or vehicle controls. The linear regression model was used to carry out data analysis for estimating various parameters of interest with appropriate 95% confidence intervals, and hypothesis testing. Specifically, the linear model was used to model the mutation frequency and cell survival percentage as response variables, using UVC fluence level and siRNA-mediated protein depletion or inhibitor treatment as covariates. The interaction effect between covariates was also considered to make the model more flexible. Wald statistics were used to determine the statistical significance of the comparisons. The Wilcoxon rank sum test was used to do group-wise comparisons at different UVC fluences and with different protein depletions to test for statistical differences in mutation frequency or colony forming efficiency. Exact test statistics were used to determine the statistical significance of the comparisons. All statistical analyses were performed using SAS 9.3 (SAS Institute Inc).
| Abbreviations: | ||
| ATM | = | ataxia-telangiectasia mutated |
| ATR | = | ataxia-telangiectasia mutated and Rad3-related |
| CHK1 | = | checkpoint kinase 1 |
| DDR | = | DNA damage response |
| HPRT | = | hypoxanthine phosphoribosyltransferase |
| IR | = | ionizing radiation |
| NHF | = | normal human fibroblasts |
| NTC | = | non-targeted control |
| siRNA | = | small interfering RNA |
| TLS | = | trans-lesion synthesis |
| UV | = | ultraviolet |
| XP | = | Xeroderma pigmentosum |
| XPV | = | xeroderma pigmentosum variant |
Additional material
Download Zip (590.3 KB)Acknowledgments
We would like to acknowledge valuable input regarding the use of CHK1 inhibitors from Dr. Timothy Heffernan, and Jenna Brophy for technical help.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
National Institutes of Health (RO1 ES015856 to MCS, T32 ES007126 to the UNC Curriculum in Toxicology-support for CS, P30 CA16086 to the Lineberger Comprehensive Cancer Center, P30 ES10126 to the Center for Environmental Health and Susceptibility)
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26590
References
- Konze-Thomas B, Hazard RM, Maher VM, McCormick JJ. Extent of excision repair before DNA synthesis determines the mutagenic but not the lethal effect of UV radiation. Mutat Res 1982; 94:421 - 34; http://dx.doi.org/10.1016/0027-5107(82)90305-0; PMID: 7110182
- Giglia-Mari G, Zotter A, Vermeulen W. DNA damage response. Cold Spring Harb Perspect Biol 2011; 3:a000745; http://dx.doi.org/10.1101/cshperspect.a000745; PMID: 20980439
- Kerzendorfer C, O’Driscoll M. Human DNA damage response and repair deficiency syndromes: linking genomic instability and cell cycle checkpoint proficiency. DNA Repair (Amst) 2009; 8:1139 - 52; http://dx.doi.org/10.1016/j.dnarep.2009.04.018; PMID: 19473885
- Kastan MB, Bartek J. Cell-cycle checkpoints and cancer. Nature 2004; 432:316 - 23; http://dx.doi.org/10.1038/nature03097; PMID: 15549093
- Kaufmann WK. The human intra-S checkpoint response to UVC-induced DNA damage. Carcinogenesis 2010; 31:751 - 65; PMID: 19793801
- Heffernan TP, Simpson DA, Frank AR, Heinloth AN, Paules RS, Cordeiro-Stone M, Kaufmann WK. An ATR- and Chk1-dependent S checkpoint inhibits replicon initiation following UVC-induced DNA damage. Mol Cell Biol 2002; 22:8552 - 61; http://dx.doi.org/10.1128/MCB.22.24.8552-8561.2002; PMID: 12446774
- Chastain PD 2nd, Heffernan TP, Nevis KR, Lin L, Kaufmann WK, Kaufman DG, Cordeiro-Stone M. Checkpoint regulation of replication dynamics in UV-irradiated human cells. Cell Cycle 2006; 5:2160 - 7; http://dx.doi.org/10.4161/cc.5.18.3236; PMID: 16969085
- Miao H, Seiler JA, Burhans WC. Regulation of cellular and SV40 virus origins of replication by Chk1-dependent intrinsic and UVC radiation-induced checkpoints. J Biol Chem 2003; 278:4295 - 304; http://dx.doi.org/10.1074/jbc.M204264200; PMID: 12424256
- Unsal-Kaçmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol 2007; 27:3131 - 42; http://dx.doi.org/10.1128/MCB.02190-06; PMID: 17296725
- Seiler JA, Conti C, Syed A, Aladjem MI, Pommier Y. The intra-S-phase checkpoint affects both DNA replication initiation and elongation: single-cell and -DNA fiber analyses. Mol Cell Biol 2007; 27:5806 - 18; http://dx.doi.org/10.1128/MCB.02278-06; PMID: 17515603
- Petermann E, Maya-Mendoza A, Zachos G, Gillespie DA, Jackson DA, Caldecott KW. Chk1 requirement for high global rates of replication fork progression during normal vertebrate S phase. Mol Cell Biol 2006; 26:3319 - 26; http://dx.doi.org/10.1128/MCB.26.8.3319-3326.2006; PMID: 16581803
- Heffernan TP, Unsal-Kaçmaz K, Heinloth AN, Simpson DA, Paules RS, Sancar A, Cordeiro-Stone M, Kaufmann WK. Cdc7-Dbf4 and the human S checkpoint response to UVC. J Biol Chem 2007; 282:9458 - 68; http://dx.doi.org/10.1074/jbc.M611292200; PMID: 17276990
- Lee AY, Chiba T, Truong LN, Cheng AN, Do J, Cho MJ, Chen L, Wu X. Dbf4 is direct downstream target of ataxia telangiectasia mutated (ATM) and ataxia telangiectasia and Rad3-related (ATR) protein to regulate intra-S-phase checkpoint. J Biol Chem 2012; 287:2531 - 43; http://dx.doi.org/10.1074/jbc.M111.291104; PMID: 22123827
- Xue L, Yu D, Furusawa Y, Cao J, Okayasu R, Fan S. ATM-dependent hyper-radiosensitivity in mammalian cells irradiated by heavy ions. Int J Radiat Oncol Biol Phys 2009; 75:235 - 43; http://dx.doi.org/10.1016/j.ijrobp.2009.04.088; PMID: 19695441
- Reiman A, Srinivasan V, Barone G, Last JI, Wootton LL, Davies EG, Verhagen MM, Willemsen MA, Weemaes CM, Byrd PJ, et al. Lymphoid tumours and breast cancer in ataxia telangiectasia; substantial protective effect of residual ATM kinase activity against childhood tumours. Br J Cancer 2011; 105:586 - 91; http://dx.doi.org/10.1038/bjc.2011.266; PMID: 21792198
- de Klein A, Muijtjens M, van Os R, Verhoeven Y, Smit B, Carr AM, Lehmann AR, Hoeijmakers JH. Targeted disruption of the cell-cycle checkpoint gene ATR leads to early embryonic lethality in mice. Curr Biol 2000; 10:479 - 82; http://dx.doi.org/10.1016/S0960-9822(00)00447-4; PMID: 10801416
- Brown EJ, Baltimore D. ATR disruption leads to chromosomal fragmentation and early embryonic lethality. Genes Dev 2000; 14:397 - 402; PMID: 10691732
- Smith-Roe SL, Patel SS, Zhou Y, Simpson DA, Rao S, Ibrahim JG, Cordeiro-Stone M, Kaufmann WK. Separation of intra-S checkpoint protein contributions to DNA replication fork protection and genomic stability in normal human fibroblasts. Cell Cycle 2013; 12:332 - 45; http://dx.doi.org/10.4161/cc.23177; PMID: 23255133
- Kaufmann WK, Cleaver JE, Painter RB. Ultraviolet radiation inhibits replicon initiation in S phase human cells. Biochim Biophys Acta 1980; 608:191 - 5; http://dx.doi.org/10.1016/0005-2787(80)90147-1; PMID: 7388031
- Kaufmann WK, Cleaver JE. Mechanisms of inhibition of DNA replication by ultraviolet light in normal human and xeroderma pigmentosum fibroblasts. J Mol Biol 1981; 149:171 - 87; http://dx.doi.org/10.1016/0022-2836(81)90297-7; PMID: 7310880
- Choi JH, Pfeifer GP. The role of DNA polymerase eta in UV mutational spectra. DNA Repair (Amst) 2005; 4:211 - 20; http://dx.doi.org/10.1016/j.dnarep.2004.09.006; PMID: 15590329
- Cleaver JE. Cancer in xeroderma pigmentosum and related disorders of DNA repair. Nat Rev Cancer 2005; 5:564 - 73; http://dx.doi.org/10.1038/nrc1652; PMID: 16069818
- Lehmann AR, McGibbon D, Stefanini M. Xeroderma pigmentosum. Orphanet J Rare Dis 2011; 6:70; http://dx.doi.org/10.1186/1750-1172-6-70; PMID: 22044607
- Masutani C, Kusumoto R, Yamada A, Dohmae N, Yokoi M, Yuasa M, Araki M, Iwai S, Takio K, Hanaoka F. The XPV (xeroderma pigmentosum variant) gene encodes human DNA polymerase eta. Nature 1999; 399:700 - 4; http://dx.doi.org/10.1038/21447; PMID: 10385124
- Kannouche P, Stary A. Xeroderma pigmentosum variant and error-prone DNA polymerases. Biochimie 2003; 85:1123 - 32; http://dx.doi.org/10.1016/j.biochi.2003.10.009; PMID: 14726018
- Kaufmann WK, Nevis KR, Qu P, Ibrahim JG, Zhou T, Zhou Y, Simpson DA, Helms-Deaton J, Cordeiro-Stone M, Moore DT, et al. Defective cell cycle checkpoint functions in melanoma are associated with altered patterns of gene expression. J Invest Dermatol 2008; 128:175 - 87; http://dx.doi.org/10.1038/sj.jid.5700935; PMID: 17597816
- Carson C, Omolo B, Chu H, Zhou Y, Sambade MJ, Peters EC, Tompkins P, Simpson DA, Thomas NE, Fan C, et al. A prognostic signature of defective p53-dependent G1 checkpoint function in melanoma cell lines. Pigment Cell Melanoma Res 2012; 25:514 - 26; http://dx.doi.org/10.1111/j.1755-148X.2012.01010.x; PMID: 22540896
- Omolo B, Carson C, Chu H, Zhou Y, Simpson DA, Hesse JE, Paules RS, Nyhan KC, Ibrahim JG, Kaufmann WK. A prognostic signature of G(2) checkpoint function in melanoma cell lines. Cell Cycle 2013; 12:1071 - 82; http://dx.doi.org/10.4161/cc.24067; PMID: 23454897
- Bi X, Slater DM, Ohmori H, Vaziri C. DNA polymerase kappa is specifically required for recovery from the benzo[a]pyrene-dihydrodiol epoxide (BPDE)-induced S-phase checkpoint. J Biol Chem 2005; 280:22343 - 55; http://dx.doi.org/10.1074/jbc.M501562200; PMID: 15817457
- Yang XH, Zou L. Dual functions of DNA replication forks in checkpoint signaling and PCNA ubiquitination. Cell Cycle 2009; 8:191 - 4; http://dx.doi.org/10.4161/cc.8.2.7357; PMID: 19158510
- Speroni J, Federico MB, Mansilla SF, Soria G, Gottifredi V. Kinase-independent function of checkpoint kinase 1 (Chk1) in the replication of damaged DNA. Proc Natl Acad Sci U S A 2012; 109:7344 - 9; http://dx.doi.org/10.1073/pnas.1116345109; PMID: 22529391
- Göhler T, Sabbioneda S, Green CM, Lehmann AR. ATR-mediated phosphorylation of DNA polymerase η is needed for efficient recovery from UV damage. J Cell Biol 2011; 192:219 - 27; http://dx.doi.org/10.1083/jcb.201008076; PMID: 21242293
- Ogi T, Walker S, Stiff T, Hobson E, Limsirichaikul S, Carpenter G, Prescott K, Suri M, Byrd PJ, Matsuse M, et al. Identification of the first ATRIP-deficient patient and novel mutations in ATR define a clinical spectrum for ATR-ATRIP Seckel Syndrome. PLoS Genet 2012; 8:e1002945; http://dx.doi.org/10.1371/journal.pgen.1002945; PMID: 23144622
- Griffith E, Walker S, Martin CA, Vagnarelli P, Stiff T, Vernay B, Al Sanna N, Saggar A, Hamel B, Earnshaw WC, et al. Mutations in pericentrin cause Seckel syndrome with defective ATR-dependent DNA damage signaling. Nat Genet 2008; 40:232 - 6; http://dx.doi.org/10.1038/ng.2007.80; PMID: 18157127
- Alderton GK, Joenje H, Varon R, Børglum AD, Jeggo PA, O’Driscoll M. Seckel syndrome exhibits cellular features demonstrating defects in the ATR-signalling pathway. Hum Mol Genet 2004; 13:3127 - 38; http://dx.doi.org/10.1093/hmg/ddh335; PMID: 15496423
- Tanaka A, Weinel S, Nagy N, O’Driscoll M, Lai-Cheong JE, Kulp-Shorten CL, Knable A, Carpenter G, Fisher SA, Hiragun M, et al. Germline mutation in ATR in autosomal- dominant oropharyngeal cancer syndrome. Am J Hum Genet 2012; 90:511 - 7; http://dx.doi.org/10.1016/j.ajhg.2012.01.007; PMID: 22341969
- Zhang J, Song YH, Brannigan BW, Wahrer DC, Schiripo TA, Harris PL, Haserlat SM, Ulkus LE, Shannon KM, Garber JE, et al. Prevalence and functional analysis of sequence variants in the ATR checkpoint mediator Claspin. Mol Cancer Res 2009; 7:1510 - 6; http://dx.doi.org/10.1158/1541-7786.MCR-09-0033; PMID: 19737971
- Lewis KA, Bakkum-Gamez J, Loewen R, French AJ, Thibodeau SN, Cliby WA. Mutations in the ataxia telangiectasia and rad3-related-checkpoint kinase 1 DNA damage response axis in colon cancers. Genes Chromosomes Cancer 2007; 46:1061 - 8; http://dx.doi.org/10.1002/gcc.20486; PMID: 17879369
- Vassileva V, Millar A, Briollais L, Chapman W, Bapat B. Genes involved in DNA repair are mutational targets in endometrial cancers with microsatellite instability. Cancer Res 2002; 62:4095 - 9; PMID: 12124347
- Menoyo A, Alazzouzi H, Espín E, Armengol M, Yamamoto H, Schwartz S Jr.. Somatic mutations in the DNA damage-response genes ATR and CHK1 in sporadic stomach tumors with microsatellite instability. Cancer Res 2001; 61:7727 - 30; PMID: 11691784
- Lewis KA, Mullany S, Thomas B, Chien J, Loewen R, Shridhar V, Cliby WA. Heterozygous ATR mutations in mismatch repair-deficient cancer cells have functional significance. Cancer Res 2005; 65:7091 - 5; http://dx.doi.org/10.1158/0008-5472.CAN-05-1019; PMID: 16103057
- Tho LM, Libertini S, Rampling R, Sansom O, Gillespie DA. Chk1 is essential for chemical carcinogen-induced mouse skin tumorigenesis. Oncogene 2012; 31:1366 - 75; http://dx.doi.org/10.1038/onc.2011.326; PMID: 21804609
- Gilad O, Nabet BY, Ragland RL, Schoppy DW, Smith KD, Durham AC, Brown EJ. Combining ATR suppression with oncogenic Ras synergistically increases genomic instability, causing synthetic lethality or tumorigenesis in a dosage-dependent manner. Cancer Res 2010; 70:9693 - 702; http://dx.doi.org/10.1158/0008-5472.CAN-10-2286; PMID: 21098704
- Fang Y, Tsao CC, Goodman BK, Furumai R, Tirado CA, Abraham RT, Wang XF. ATR functions as a gene dosage-dependent tumor suppressor on a mismatch repair-deficient background. EMBO J 2004; 23:3164 - 74; http://dx.doi.org/10.1038/sj.emboj.7600315; PMID: 15282542
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Fishler T, Li YY, Wang RH, Kim HS, Sengupta K, Vassilopoulos A, Lahusen T, Xu X, Lee MH, Liu Q, et al. Genetic instability and mammary tumor formation in mice carrying mammary-specific disruption of Chk1 and p53. Oncogene 2010; 29:4007 - 17; http://dx.doi.org/10.1038/onc.2010.163; PMID: 20473325
- Yang XH, Shiotani B, Classon M, Zou L. Chk1 and Claspin potentiate PCNA ubiquitination. Genes Dev 2008; 22:1147 - 52; http://dx.doi.org/10.1101/gad.1632808; PMID: 18451105
- Casper AM, Nghiem P, Arlt MF, Glover TW. ATR regulates fragile site stability. Cell 2002; 111:779 - 89; http://dx.doi.org/10.1016/S0092-8674(02)01113-3; PMID: 12526805
- Durkin SG, Arlt MF, Howlett NG, Glover TW. Depletion of CHK1, but not CHK2, induces chromosomal instability and breaks at common fragile sites. Oncogene 2006; 25:4381 - 8; http://dx.doi.org/10.1038/sj.onc.1209466; PMID: 16732333
- Durkin SG, Glover TW. Chromosome fragile sites. Annu Rev Genet 2007; 41:169 - 92; http://dx.doi.org/10.1146/annurev.genet.41.042007.165900; PMID: 17608616
- Schmidt M, Migeon BR. Asynchronous replication of homologous loci on human active and inactive X chromosomes. Proc Natl Acad Sci U S A 1990; 87:3685 - 9; http://dx.doi.org/10.1073/pnas.87.10.3685; PMID: 2339112
- Chen T, Stephens PA, Middleton FK, Curtin NJ. Targeting the S and G2 checkpoint to treat cancer. Drug Discov Today 2012; 17:194 - 202; http://dx.doi.org/10.1016/j.drudis.2011.12.009; PMID: 22192883
- Mack PC, Gandara DR, Lau AH, Lara PN Jr., Edelman MJ, Gumerlock PH. Cell cycle-dependent potentiation of cisplatin by UCN-01 in non-small-cell lung carcinoma. Cancer Chemother Pharmacol 2003; 51:337 - 48; PMID: 12721762
- Furuta T, Hayward RL, Meng LH, Takemura H, Aune GJ, Bonner WM, Aladjem MI, Kohn KW, Pommier Y. p21CDKN1A allows the repair of replication-mediated DNA double-strand breaks induced by topoisomerase I and is inactivated by the checkpoint kinase inhibitor 7-hydroxystaurosporine. Oncogene 2006; 25:2839 - 49; http://dx.doi.org/10.1038/sj.onc.1209313; PMID: 16407843
- Toledo LI, Murga M, Fernandez-Capetillo O. Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol Oncol 2011; 5:368 - 73; http://dx.doi.org/10.1016/j.molonc.2011.07.002; PMID: 21820372
- Boyer JC, Kaufmann WK, Cordeiro-Stone M. Role of postreplication repair in transformation of human fibroblasts to anchorage independence. Cancer Res 1991; 51:2960 - 4; PMID: 1903328
- Day TA, Sproul C, Cordeiro-Stone M, Vaziri C. Analyzing DNA replication dynamics of genotoxin-treated cells using velocity sedimentation. Methods Mol Biol 2011; 782:159 - 70; http://dx.doi.org/10.1007/978-1-61779-273-1_11; PMID: 21870290
- Bassett E, King NM, Bryant MF, Hector S, Pendyala L, Chaney SG, Cordeiro-Stone M. The role of DNA polymerase eta in translesion synthesis past platinum-DNA adducts in human fibroblasts. Cancer Res 2004; 64:6469 - 75; http://dx.doi.org/10.1158/0008-5472.CAN-04-1328; PMID: 15374956