
Abstract
The E1a gene from adenovirus is known to be a potent inducer of chemo/radiosensitivity in a wide range of tumors. However, the molecular bases of its radiosensitizer properties are still poorly understood. In an attempt to study this effect, U87MG cells, derived from a radio-resistant tumor as glioblastoma, where infected with lentivirus carrying E1a gene developing an acute sensitivity to ionizing radiation. The induction of radiosensitivity correlated with a marked G2/M phase accumulation and a potent apoptotic response. Our findings demonstrate that c-Myc plays a pivotal role in E1a-associated radiosensitivity through the induction of a replicative stress situation, as our data support by genetic approaches, based in interference and overexpression in U87MG cells. In fact, we present evidence showing that Chk1 is a novel transcriptional target of E1a gene through the effect exerted by this adenoviral protein onto c-Myc. Moreover, c-Myc upregulation also explains the marked phosphorylation of H2AX associated to E1a expression in the absence of DNA damage. Indeed, all these observations were applicable to other experimental models, such as T98G, LN-405 and A172, rendering the same pattern in terms of radiosensitivity, cell cycle distribution, upregulation of Chk1, c-Myc, and phosphorylation pattern of H2AX. In summary, our data propose a novel mechanism to explain how E1a mediates radiosensitivity through the signaling axis E1a→c-Myc→ replicative stress situation. This novel mechanism of E1a-mediated radiosensitivity could be the key to open new possibilities in the current therapy of glioblastoma.
Introduction
E1a, one of the early genes of adenovirus, has been related to cancer with a dual role, allowing this gene to act as an oncogene or as a tumor suppressor.Citation1 As a transforming gene, E1a has been used as a potent tool to study cooperation between oncogenes such as Ras, Sis, or PML.Citation2-Citation4 This oncogenic side of E1a seems to be mediated through the ability to interfere with molecular mechanisms (signaling pathways and cellular proteins) implicated in tumor suppression and senescence.Citation5-Citation8 Its tumor suppressor activity has been related to several mechanisms, such as downregulation of oncogenic proteins like Her2/neu or EGFRCitation9,Citation10 and, more recently, deregulation of microRNAs.Citation11 Moreover, the proapoptotic activity of E1a and its effects in angiogenesisCitation12 make this adenoviral gene an ideal candidate for cancer therapy.Citation13E1a has another important therapeutic implication due to its ability to promote chemo- and radiosensitivity. Regarding its role as a chemosensitizer agent, it has been proved in response to different type of drugs, such as DNA damaging agents or histone deacetylases,Citation14,Citation15 as well as in different experimental models, such as breast cancer or myeloid leukemia cells.Citation13,Citation16 However, the ability of E1a as a radio sensitizer agent, described in the mid 90s,Citation17 seems to be less studied. In this regard, it has been proposed that inhibition of NFκB in an AKT-independent fashion is a critical event in this radiosensitizer effect.Citation18 Interestingly, other cellular targets of E1a, such as pRB, p400, or Fox-3a, have been studied in terms of induction of chemosensitivity,Citation19-Citation21 but no role has been proposed in terms of radio response. Therefore, all the previous indicates that the molecular basis of E1a-associated radiosensitivity still needs to be fully elucidated.
Glioblastoma multiforme (GBM) is the most common primary tumor of the central nervous system as well as the most aggressive. The mean survival of GBM patients is about 12 mo, with treatment based in the use of surgery, radiotherapy, and chemotherapy. Although the improved imaging techniques and the use of novel targeted therapies such as TK inhibitors, mAbs, etc., are helping classical therapy,Citation22 resistance of glioblastoma to ionizing radiation (IR) remains as a therapeutic challenge.Citation23 Therefore, the search for new therapeutic strategies to sensitize GBM to IR would be a novel approach to improve the effectiveness of current treatments.
In this scenario, we decided to study if E1a was able to promote radiosensitivity in an experimental model of GBM and the molecular basis of this potential therapeutic use. Our data demonstrates that E1a promotes a replicative stress situation through the deregulation of c-Myc signaling, as evaluation of Chk1 and Histone H2AX (H2AX) indicate. This novel mechanism could explain the radiosensitivity associated with E1a, offering new therapeutic windows in GBM.
Results
E1a induces radiosensitivity in GBM cell lines
To investigate whether E1a could induce radiosensitivity in GBM-derived cells lines, 2 different cell lines, U87MG and T98G, were infected with a lentivirus-carrying E1a gene. Selected pools were analyzed in terms of E1a expression and challenged against IR (). As expected, cells expressing E1a showed a considerable increase in radiosensitivity compared with control cells () according to previous reports.Citation17,Citation24
Figure 1. E1a induces radiosensitivity in GBM-derived cell lines. (A) U87MG and T98G were transduced with empty vector (U87 E.V., T98 E.V.) or E1a (U87 E1a, T98 E1a) Fifty micrograms of total cell lysates (TCL) were blotted against E1a. Membranes were reproved against tubulin as loading control. (B) Cells were exposed to indicated doses of IR. Viability of the cells was evaluated by using the violet crystal method 6 d later. Data represent average ± SD. (C) Cell cycle profile of U87 E.V. and U87 E1a cell lines was determined by flow cytometry after exposure to IR (10 Gy). Graphs represent the % of cells in G2/M phase at the indicated times. Image shows a representative experiment out of 3. (D) The same experiment as in (C) for T98 E.V. and T98 E1a cell lines. (E) Annexin V assay in U87 E.V. and U87 E1a cells were analyzed 48 h after 10 Gy exposure. Between parentheses, percentage of apoptotic population. Image shows a representative experiment out of 3. (F) Caspase 3/7 activity assay in U87 E.V and E1a cells 24 h after 10 Gy exposure. Histogram data are the average of 3 independent experiments performed in triplicate cultures.
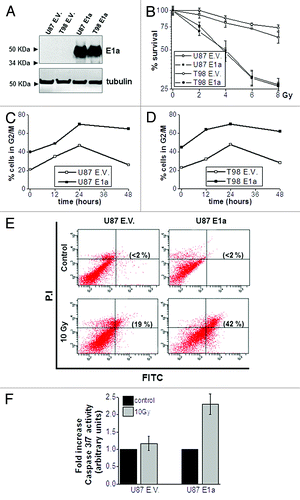
To further investigate the radiosensitizer effect of E1a, we analyzed cell cycle perturbations in response to IR as putative markers of radiosensitivity.Citation25 Interestingly, cells expressing E1a showed a marked increase in G2/M phase in basal condition, correlating with increased levels of cyclin B1 (S1), which was sustained at late time points after IR exposure (). This accumulation in G2/M phase correlates with a marked increases in the apoptotic response triggered by IR as judged by Annexin V staining () and Caspase 3/7 activity (). In summary, this set of experiments suggests that radiosensitivity associated to E1a correlates with deregulation in the cell cycle, rendering an increase in the apoptotic response.
Chk1 mediates cell cycle alterations and is a novel target of E1a
In light of the observed cell cycle alterations after E1a expression, we decided to explore the putative mechanism to explain the increase in G2/M phase. Considering that the late time points of G2/M arrest after IR seem to be ATM-independent,Citation26 we decided to focus on the ATR→ Chk1 signaling axis, known to promote G2/M arrest.Citation27,Citation28 First, we evaluated Chk1 expression level in the presence/absence of E1a in our experimental model of U87MG cells. Interestingly, we found that Chk1 protein and its phosphorylation levels at Ser 345 were upregulated in the presence of E1a (). Next, we investigated if E1a was affecting Chk1 at the transcriptional level. To this end, q-RT-PCR assays were performed, showing a marked increase in the mRNA level of Chk1 in E1a-expressing cells (). To discard a stabilization of RNA levels, we analyzed the promoter activity of Chk1 in the absence/presence of E1a in U87MG. As it is shown (), co-transfection of E1a with a luciferase reporter under the control of human Chk1 promoter clearly increases the transcriptional activity of this promoter, suggesting that Chk1 is a novel transcriptional target of the E1a gene. Indeed, similar results were obtained in T98G cell line (S2).
Figure 2. Chk1 is a novel target of E1a in GBM cells. (A) Fifty micrograms of total cell lysates (TCL) from U87MG cells infected with E1a or empty vector were blotted against Chk1. Tubulin was used as loading control. (B) Chk1 mRNA levels in U87 E.V. and U87 E1a were analyzed by qRT- PCR. Graph shows the relative levels of Chk1 mRNA referred to U87 E.V. Data represents mean ± SD. (C) Cell lines were cotransfected with 100 ng of pGL3-F0 and increasing amounts of pLESIP E1A up to 2 μg. Luciferase activity was normalized by Renilla. Data represents mean ± SD fold induction. (D) Percentage of cells in G2/M phase in U87 E1a cells transfected with siRNA scrambled or against Chk1. As a control, U87 empty vector cells were also evaluated. Data represents mean ± SD of 3 independent experiments.
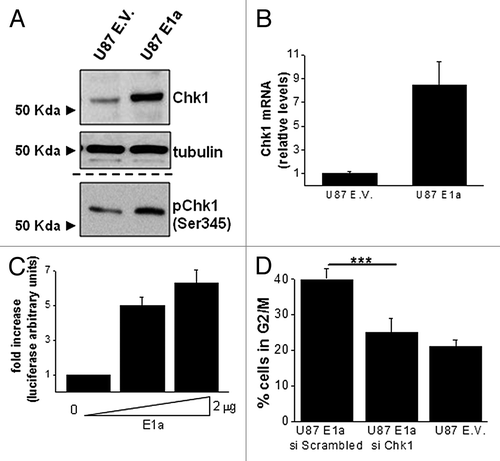
To study the role of Chk1 in cell cycle alterations associated with E1a expression we knocked down the expression of Chk1 by siRNA (S3), and the effects in cell cycle were evaluated. As shown in , in the absence of Chk1, the prominent G2/M accumulation observed in E1a-expressing cells was abrogated, rendering a similar pattern to control cells. Interestingly, overexpression of Chk1 promotes radiosensitivity in U87MG cells (S3), suggesting a putative implication of this protein in the E1a-associated radiosensitivity that needs to be fully addressed in future researches.
In summary, these results suggest a pivotal role of Chk1 in E1a-mediated cell cycle alterations and demonstrate that this kinase is a novel target of the E1a at the transcriptional level.
c-Myc meditates E1a effects on Chk1
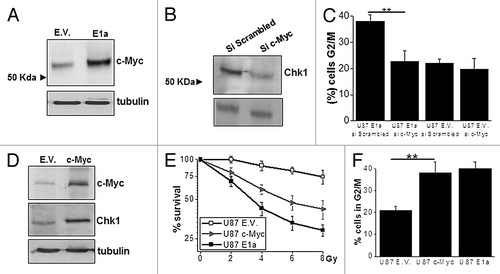
To analyze the molecular basis of the control exerted by E1a onto Chk1, we decided to further investigate the role of c-Myc, which has been recently proposed as a key molecule in the control of Chk1 levelsCitation29 and is also known to be implicated in E1a-associated biological effects.Citation30 Initially, expression levels of c-Myc were evaluated in E1a-expressing cells, showing the expected increase in c-Myc level () according to previous reports.Citation31 In order to study the contribution of c-Myc to Chk1 upregulation, we decided to perform an interference assay against c-Myc. Abrogation of c-Myc at the RNA and protein level in E1a-expressing cells (S4) induced a decrease in Chk1 expression (). Furthermore, interference of c-Myc restores cell cycle distribution in E1a-expressing cells with a marked decrease in G2/M population (). Finally, to fully elucidate the role of c-Myc in E1a-expressing cells, we decided to mimic E1a in U87MG cell by overexpression of c-Myc, rendering the same pattern as E1a in terms of Chk1 upregulation () radiosensitivity () and cell cycle distribution ().
Figure 3. c-Myc upregulates Chk1 in E1a-expressing cells. (A) Fifty micrograms of total cell lysate (TCL) of U87 E.V. and U87 E1a cells were blotted against c-Myc. Tubulin was used as loading control. (B) Expression of Chk1 was evaluated in U87 E1a transfected with siRNA Scrambled or against c-Myc by western blot as in . (C) Percentage of cells in G2/M phase in U87 E1a cells transfected with siRNA scrambled or against c-Myc. As a control, U87 empty vector cells were also evaluated. Data represents mean ± SD of 3 independent experiments. (D) Fifty μg of TCL from U87 E.V. and U87 overexpressing c-Myc (c-Myc) were blotted against c-Myc and Chk1. Tubulin was used as loading control. (E) Viability of U87MG cells overexpressing c-Myc (c-Myc), E1a (E1a) or empty vector (E.V.) was measured in response to IR as in . Data represents mean ± SD of 3 independent experiments. (F) Percentage of cells in G2/M phase in U87 E.V., U87 c-Myc, and U87 E1a cell lines. Data represents mean ± SD of 3 independent experiments.
E1a promotes replicative stress through c-Myc
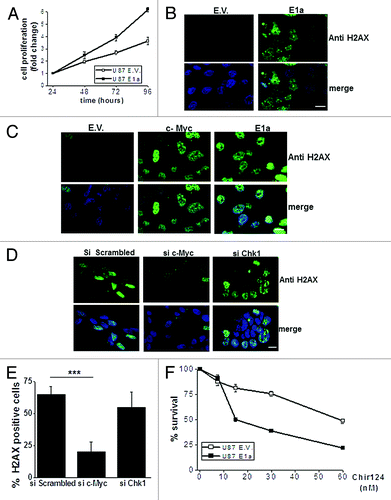
All the previous prompted us to investigate if E1a was promoting a situation consistent with replicative stress (RS) associated to c-Myc upregulation.Citation32,Citation33 First, we proved that E1a increases proliferation in our experimental model of U87MG, with an increase of almost the double at late time points (). Then, we further analyzed if the phosphorylation of H2AX at Ser139, a well-established marker of RS and DNA damage,Citation29,Citation34-Citation36 was affected by E1a expression. While control cells showed a null positivity for H2AX, E1a-expressing cells showed a marked phosphorylation (>65%) in basal conditions (). Indeed, a similar result was observed in our experimental model of T98G cells (data not shown). Although a previous report connect the deregulated H2AX phosphorylation in the presence of E1a with ATM,Citation37 preliminary data discard the implication of this protein in our experimental model (S5). Therefore, to elucidate the molecular mechanism of the altered activation pattern of H2AX in E1a-expressing cells, we decided to evaluate the role of c-Myc. Interestingly, c-Myc overexpression rendered a similar effect to E1a expression, with a marked upregulation of Η2AX phosphorylation (). Furthermore, interference of c-Myc in E1a-expressing cells lead to a marked reduction of H2AX phosphorylation, while Chk1 abrogation in these cells did not significantly modify H2AX phosphorylation (). Considering recent evidences showing how Chk1 inhibitors selectively kill cells with a marked RS,Citation32,Citation38 we decided to challenge E1a-expressing cells to Chir-124, a known inhibitor for Chk1.Citation39 Interestingly, E1a cells showed a higher toxicity in response to Chir-124 (). In fact similar results were obtained with UCN-O1 (data not shown), as it has been shown in experimental models with c-Myc upregulation.Citation32 In summary, this set of experiments support that E1a expression promotes a situation consistent with RS.
Figure 4. E1a promotes c-Myc driven replicative stress (A) Proliferation of U87 E1a and E.V. was measured by MTT assay. Values at 24 h after plating were referred as 1 for each cell line. Data represent the average ± SD of O.D. at 570 nm. (B) Immunocytochemistry against phospo H2AX (Ser 139) was performed in U87 E.V. and U87 E1a cells. Nuclei were counterstained with DAPI. Scale bar represents 25 μm. Image shows a representative field out of 5 of at least 3 experiments. (C) Immunocytochemistry against phospo H2AX (Ser 139) in U87 E.V. cells and U87 overexpressing c-Myc (c-Myc) and E1a (E1a). Nucleus was counterstained with DAPI. Scale bar represents 25 μm, Image shows a representative field out of 5 of at least 2 experiments. (D) U87 E1a cells were transfected with scrambled siRNA, siRNA against c-Myc, or Chk1, and 48 h post-transfection immunocitochemistry against phospo H2AX (Ser 139) was assessed. Scale bar represents 25 μm. Image shows a representative field out of 10 obtained from 2 independent experiments. (E) Quantification of the above mentioned experiments (10 fields for each condition). (F) Viability assay (MTT based assay) in U87 E.V. and E1a cells in response to Chir-124 at indicated doses for 48 h. Viability was referred to untreated cells as 100%. Data represent the average ± SD.
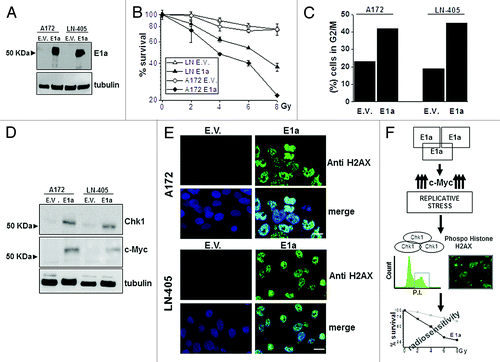
Finally, to fully support our observations mainly obtained in U87MG cells, we decided to challenge other experimental models, such as A172 and LN405. These new cell lines were infected with lentivirus carrying E1a (), and radioresponse was evaluated, showing the expected pattern of radiosensitivity (). In addition, cell cycle, H2AX phosphorylation, Chk1, and c-Myc expressions () were almost identical to the one observed in U87MG cells, indicating the universal character of our observations in GBM-derived cell lines
Figure 5. Effects of E1a in different GBM derived cell lines. (A) A172 and LN-405 GBM cell lines were infected with lentivirus carrying E1a (E1a) and empty vector (E.V). E1a expression was assessed as in . Tubulin was used as loading control. (B) Viability in response to IR in A172, LN-405 empty vector (A172 E.V., LN-405 E.V.) or E1a expressing cells (A172 E1a, LN-405 E1a) was evaluated as in . Data represent the average ± SD. (C) Percentage of cells in G2/M phase was assessed as in . Image shows a representative experiment out of 2. (D) 50 μg of total cell lysate of A172 E.V. A172 E1a, LN-405 E.V., and LN-405 E1a cells, were blotted against Chk1 and c-Myc. Tubulin was used as a loading control. (E) Immunocytochemistry against phospo H2AX (Ser 139) was performed in A172 E.V. A172 E1a, LN-405 E.V., and LN-405 E1a cells as in . Scale bar represents 25 μm. Image shows a representative field out of 5 of at least 2 experiments. (F) Proposed model for the effects elicited by E1a in GBM cells.
Discussion
Several conclusions can be obtained from the present report. The first conclusion is the universal character of E1a as a potent inducer of radiosensitivity in GBM. Interestingly, our data support the use of E1a as a radiosensitazer agent even in tumors with alterations in key protein/signaling pathways in the response to IR as p53 or PI3K/AKT, as the use of cell lines with mutant p53 (e.g., T98G) or pTEN alteration (e.g., U87MG) indicates. This observation suggests that GBM could be a novel member of the growing list of tumors that could benefit from E1a-based therapy (for a review, see refs. Citation40 and Citation41). In this regard, oncolytic adenoviruses are considered as a novel therapeutic approach in GBM disease, with the ability to promote radiosensitivity (for a review, see ref. Citation42) in which E1a could be a key element, as has been proposed, for chemotherapy.Citation43
Regarding the molecular basis of E1a-associated radiosensitivity, our initial observation showed a marked correlation with a prominent increase in G2/M phase, which is a well-established radiosensitive phase of the cell cycle.Citation44 Indeed, cells arrested in G2/M phase for a long time period usually died, probably because this arrest is the preamble of mitotic catastrophe that could render apoptosis.Citation45 In this sense, our data propose a novel mechanism to explain the characteristic distribution of the cell cycle mediated by E1a through the upregulation of Chk1. This kinase is one of the best-characterized substrates for ATRCitation46 implicated in the control of G2/M checkpoint.Citation47 Our data shows how the upregulation of Chk1 correlates with an increase in the activation of this kinase (Ser345) and also with an increase in cyclin B1 levels, consistent with mitotic arrest. However, other possibilities, such as reduction in the length of G1 phase, as has been proposed in quiescent cells due to the effects of E1a onto c-Myc and pRBCitation48 through specific mechanisms,Citation49 should be considered to fully understand the effect of E1a onto cell cycle progression. In addition, we demonstrate that E1a mediates upregulation of Chk1 at the transcriptional level trough c-Myc (see below), as q-RT- PCR and luciferase assays supports. Therefore, Chk1 could be included in the list of genes controlled by E1a at the transcriptional level. It is noteworthy that a previous report suggests a connection between adenovirus infection and Chk1, in which deregulated proliferation associated to E1a triggers the ATR-Chk1 signaling pathway.Citation50 Whether our observation, based on overexpression of E1a alone, could apply to the whole process of adenovirus infection that requires other adenoviral proteins (e.g., E1b), needs to be further investigated. Nonetheless, in spite of the mechanisms that controls Chk1 phosphorylation, it has been described other mechanisms to regulate both activation and activityCitation51 that need to be further explored to fully evaluate the effects exerted by E1a onto Chk1.
In addition, our data support how the transcription factor c-Myc, a known target of E1aCitation31 with a critical role in some of the biological effects of this adenoviral protein,Citation30 is a key player in the molecular basis of E1a-associated radiosensitivity. Interestingly, although no previous study has related c-Myc with the chemo- and radio-sensitizer effects of E1a, it has been reported that a mutant form of E1a unable to bind p400 (e.g., Δ26–35), and theoretically without c-Myc stabilization, is also defective in the induction of chemosensitivity to DNA damaging agents as Doxorrubicin.Citation21 Furthermore, it’s demonstrated that c-Myc controls Chk1 expression, at the transcriptional level.Citation29,Citation52 Therefore, it seems to be plausible that c-Myc through RS (see below) would be implicated in the radiosensitivity associated to E1a. In this regard, it has been recently reported that c-Myc, through the transcriptional upregulation of Chk1/2, could be implicated in the radioresistant phenotype of cancer stem cell derived from nasopharyngeal carcinomas,Citation52 indicating that the effects of the c-Myc→Chk1 signaling axis could be dependent on the cell type (e.g., stem cell) or the genetic background (e.g., presence of E1a).
One of the most challenging questions that opens this study is the implication of the RS in the biological effects associated to E1a expression, including radiosensitivity. This observation is inferred from the ability of E1a to promote DNA synthesis, rendering a deregulated proliferation, and the marked increase in the H2AX phosphorylation observed in our experimental model of GBM. In this situation, the c-Myc stabilization mediated by E1a is a critical event, as our interference data demonstrate discarding a direct role for Chk1 in agreement with recent reports.Citation53 Therefore, a c-Myc-dependent RS in E1a-expressing cells could be a real possibility, especially considering that c-Myc could mediate an aberrant DNA synthesis and activation of DNA damage response in a p300-impaired context.Citation33 In addition, our data provides a novel explanation for the observed pattern of H2AX phosphorylation associated to E1a expression trough RS mediated by c-Myc rather than through ATM-dependent mechanism.Citation37 Finally, regarding the role of Chk1 in E1a-expressing cells, our data support the idea that this could be a key element to maintain viability in these cells, as has been shown in c-Myc-driven tumorgenesis or in melanoma,Citation32,Citation38 and even could be implicated in the transforming properties of E1a as has been shown in other experimental systems.Citation54
In summary, this report presents a novel mechanism to explain E1a-mediated radiosensitivity through a RS situation mediated by c-Myc upregulation, explaining the observed deregulation of Chk1 and the H2AX phosphorylation associated to E1a expression (). Whether our proposed mechanism could apply to other types of tumors, the putative implications in the therapy of GBM, as well as the role of the different components of this signaling axis in the radiosensitizer effect of E1a, needs to be further investigated.
Materials and Methods
Cell lines and plasmids
GBM-derived cell lines U87MG and T98G were purchased from ATCC (http://www.lgcstandards-atcc.org/en/Products/Cells_and_Microorganisms/Cell_Lines/Human.aspx). LN-405 and A172 were kindly provided by Dr Manuel Guzmán (Universidad Complutense de Madrid). Cells were maintained in 5% CO2 and 37 °C. Cells were grown in Dulbecco modified Eagle medium supplemented with 10% fetal bovine serum, 2 mM glutamine plus antibiotics. All cell culture reagents were provided by Lonza (http://www.lonza.com/products-services/bio-research/cell-culture-products).
pcDNA4-Chk1-flag was obtained from Dr A Sancar through Addgene (Plasmid #22894Citation55) (http://www.addgene.org/search/?q=22894#q=22894&p=true) and pcDNA3-Flag-Myc was generous gift of Dr Víctor Sanchez Arevalo (CNIO).
E1a isoform 13s was obtained by PCR from cDNA of 293T cells and cloned in PCDNA3.1. Primers used were: forward 5′-GTGGATCCAT GAGACATATT ATCTGCC-3 and reverse 5-GGGAATTCTT ATGGCCTGGG GCGTTTAC-3′. PCR conditions were 94 °C 30 s during the first cycle and then, 30 cycles (94 °C 30 s, 52 °C 1 min and 72 °C 3 min) with a final extension of 72 °C during 10 min. After sequencing and expression assay, E1a 13s was subcloned, using the BamH1/EcoR1 sites, onto pLESIP vector kindly provided by Dr S Gutkind (NIDCR, NIH).
The sequence of the genomic Chk1 promoter (−1845–284) was cloned upstream luciferase gene specifically in the restriction sites MluI and BglII of the final vector pGL3-luciferase basic (Promega Biotech Ibérica, http://www.promega.es/products/reporter-assays-and-transfection/reporter-vectors-and-cell-lines/pgl3-luciferase-reporter-vectors/). The genomic DNA was extracted from the HCT116 line, and the fragment upstream Chk1 gene was amplified by using the oligonucleotides FW 5′GGACGCGTAA GCCATTCTCC TGCCTCGC 3′and Rv 5′GGAGATCTCC GGCGAACGAC TGGGGAAG 3′, PCR conditions were 94 °C 30 s during the first cycle, and then 35 cycles (94 °C 30 s, 66 °C 1 min and 68 °C 3 min) with a final extension of 72 °C during 10 min. Then ligated into the intermediate vector pGEMT-easy (Promega, http://www.promega.es/products/pcr/pcr-cloning/pgem_t-easy-vector-systems/), sequenced, digested, and finally cloned into pGL3-vector. The construction was called pGL3-F0.
Chemicals and antibodies
Antibodies against H2AX phosphorylated at Ser 139, Chk1, and phospho-Chk1 (Ser 345) were purchased from Cell Signaling Technologies (http://www.cellsignal.com). Antibodies against E1a, c-Myc, cyclin B, and tubulin were purchased from Santa Cruz Technology (http://www.scbt.com). Antibody against active ATM (ser1981) was from Rockland Immunochemicals Inc. (http://www.rockland-inc.com/Product.aspx?id=41061). Poly-L-lysine and DAPI were obtained from Sigma-Aldrich (http://www.sigmaaldrich.com/sigma-aldrich/home.html). UCN-01(Calbiochem, http://www.merckmillipore.com/spain/life-science-research/ucn-01/EMD_BIO-539644/p_R66b.s1LTrMAAAEWx2EfVhTm?WFSimpleSearch_NameOrID=UCN-01&BackButtonText=search+results KU55933 (Tocris,http://www.tocris.com/dispprod.php?ItemId=226718#.UZ4AI6KSJMQ) and Chir-124 (Selleckchem, http://www.selleckchem.com/products/chir-124.html) were dissolved in DMSO and stored at −20 °C.
Transfections and infections
Lentiviral production and infection was performed as previously described.Citation56 Briefly, HEK293T cells were transfected overnight by using calcium phosphate with 9 μg of pLESIP-E1a or empty vector, 6 μg of PSPAX2, and 3 μg of plasmid encoding the viral envelope protein, VSVG. Supernatants were collected 48 h after transfection and added to the GBM cells for 16 h in the presence of polybrene at 8 μg/ml. Forty-eight hours post-infection cells expressing E1a were selected with 0.5 μg/ml of puromycin (Sigma-Aldrich, http://www.sigmaaldrich.com/catalog/product/sigma/p8833?lang=en®ion=US) for 72 h.
For stable expression of flag-tagged Chk1 or c-Myc, U87MG cells were transfected with 2 μg of the plasmids by using lipofectamine 2000 (Invitrogen, http://products.invitrogen.com/ivgn/product/11668019?ICID=search-product). After 72 h, cells were selected with G418 at 600 μg/ml, for at least 10 d, prior any assay.
Western blotting
Cells were collected in lysis buffer (100 mM HEPES, pH 7.5, 50 mM NaCl, 0,1% Triton X-100, 5 mM EDTA, 0,125M EGTA). Protease and phosphatase inhibitors (0.2 μg/ml Leupeptin, 2 μg/ml, Aprotinin, 1 mM PMSF, and 0.1 mM Na3VO4) were added prior to lysis. Protein quantification was performed by using the BCA Protein Assay Kit (Pierce, http://www.piercenet.com/browse.cfm?fldID=02020101) following the manufacturer’s instructions. Indicated amounts of protein were loaded onto 10% SDS-PAGE, transferred to PVDF filters and blotted against different proteins using specific antibodies.
Antibody detection was achieved by enhanced chemiluminescence (Amersham, GE Health Care) in a LAS-3000 system (FujiFilm, http://www.fujifilm.com/products/). Results show a representative blot out of 3 with nearly identical results. Tubulin was used as a loading control
Immunocytochemistry
Cells were grown onto glass coverslips treated with poly-L-lysine. If required, cells were then transfected as described. Samples were fixed, permeabilized as previously described.Citation57 Then samples were incubated with antibody against H2AX phosphorylated at Ser 139 (1/500) overnight and, after extensive wash, incubated 60 min with Alexafluor 488 conjugated anti-rabbit antibody (Invitrogen Molecular Probes, http://products.invitrogen.com/ivgn/product/A21441). Samples were mounted with Fluorsave (DAKO, http://www.dako.com/es/ar38/p107570/prod_products.htm). Positive immuno-fluorescence was detected using a Zeiss LSM-710 (http://www.zeiss.es/C12574750032E818/EmbedTitelIntern/LSM710NLO_Specification/$File/60-1-0004_e_TechDat.pdf) confocal microscope. Images were acquired and processed using Zen 2009 Light Edition program. Image shows a representative field out of 5. The scale bar represents 25 μm.
RNA isolation, reverse transcription, and real-time quantitative PCR
Total RNA was obtained, and reverse transcription was performed as previously described.Citation57Changes in the mRNA expression of Chk1 and c-Myc were examined by Quantitative Real-Time PCR using an ABIPrism 7500 FAST Sequence Detection System (Applied Biosystems, http://www.invitrogen.com/site/us/en/home/Products-and-Services/Applications/PCR/real-time-pcr/real-time-pcr-instruments/7500-fast-real-time-pcr-system.html). cDNA was amplified using SYBR1 Green PCR Master Mix (Applied Biosystems, https://products.invitrogen.com/ivgn/product/4385612?ICID=cvc-sybr-realtime-c2b1) in the presence of specific oligonucleotides. The PCR conditions and quantification was performed as previously described 57Primers for all target sequences were designed using the Primer Express software specially provided with the 7500 Sequence Detection System (Applied Biosystems, http://www.invitrogen.com/). PCR primers were purchase from Bonsai Technologies (http://www.bonsaiadvanced.com/).
For Chk1: forward 5′-GGCCCCTGAA AGAATAAACC C-3′, reverse 5′- CGAAGGATGG CCAACTCAAT C-3′; for c-Myc: forward 5′-GACCTGGAGC GGCTGACA-3′ - reverse 5′- TACCATCAAA AGCTGAGATG AAACA-3′; and for GAPDH: forward 5′-TCGTGGAAGG ACTCATGACC A-3′, reverse 5′-CAGTCTTCTG GGTGGCAGTG A-3′. Data are the average of at least 3 independent experiments performed in triplicates.
Luciferase assay
U87MG and T98G cells were seeded in 24-well plates (5 × 103 cells/well), and 24 h later cells were transfected (lipofectamine 2000) with 100 ng of the indicated reporter plasmid (F0-pGL3). plus 10 ng of a plasmid coding for Renilla in the presence of the indicated amounts of pLESIP E1a 13s.The total amount of DNA per well was adjusted by adding pLESIP empty vector. Twenty-four hours after transfection, cells were harvested in lysis buffer, and Renilla luciferase activities were determined using the dual luciferase system from Promega (http://www.promega.es/products/reporter-assays-and-transfection/reporter-assays/dual_luciferase-reporter-assay-system/) in a Monolight 3096 (BD PharMingen, http://www.bd.com/). Firefly luciferase activity was normalized based on the renilla luciferase activity. Data are the average of at least 3 independent experiments performed in duplicate cultures.
Interference assays
siRNA for Chk1 and c-Myc were purchased from Dharmacon (http://www.thermoscientificbio.com/search/?term=siRNA%20) (ON-TARGET plus SMART pool Human Chk1 Cat. L-003255–00, ON-TARGET plus SMART pool Human c-Myc Cat. L-003282–00 and ON-TARGET plus CONTROL pool Cat. D-001810-10-05) and used following the manufacturer’s recommendations. For siRNA assays, cells were transfected by using lipofectamine 2000 (Invitrogene). q-RT-PCR and western blotting experiments were performed 48 h and 72 h post-transfection, respectively.
Irradiation and dose response assays
Cells were irradiated by the technical staff of the Radiotherapy Unit at the University General Hospital of Albacete in a Clinac Low Energy 600C linear electron accelerator from Varian (http://www.varian.com/) at a dose rate of 600 cGy/min in a radiation field of 40 × 40 cm. For dose response assays, 6 × 103 cells/well were seeded 24 h prior to irradiation. Culture medium was replaced 16–24 h after IR and refreshed every 3 d until the end of the experiment. Viability was evaluated by the crystal violet method 6 d after IR exposure. Briefly, cells were washed with 1× PBS and incubated with 0.5% glutaraldehide for 10 min at room temperature in mild rocking. Crystal violet reactant (10 mg/ml in distilled water) was then added to the cells and incubated for 30 min. Colorant was recovered with 10% acetic acid and the optical density was evaluated at 595 nm. Values were referred to untreated controls, set at 100%. Data are the average of at least 3 independent experiments performed in triplicate cultures.
Flow cytometry
For cell cycle analysis, cells were trypsinized, washed with PBS, fixed with cold 70% ethanol in PBS at −20 °C and extensively washed in cold 1× PBS. The cells were then incubated with 10 µg/ml propidium iodide (PI) and 20 µg/ml RNase for 20 min on darkness. Cells were plated in 6-well plates and, if required, transfected as described and processed at least 48 h later. In the case of IR treatments, cells were seeded 24 h before the exposure to IR, and samples were collected at the indicated times.
For apoptosis evaluation, the Annexin V-FITC Apoptosis Detection Kit (Immunostep, http://www.immunostep.com/es/1092-annexin-v.html) was used as previously described.Citation57 Briefly, cells were collected, centrifuged for 5 min at 1200 rpm, washed twice in 1× PBS and resuspended at 106 cells/ml in 1× Annexin binding buffer (Immunostep, http://www.immunostep.com/es/2308-annexin-v-binding-buffer-10x.html). Cells were then stained (5 µl Annexin V-FITC and 5 µl PI per 100 µl of cell suspension) for 20 min on darkness.
Samples were analyzed on a BD FACS Canto II flow cytometer (Becton Dickinson, http://www.bd.com/es/). Data were analyzed by using FACSDiva (Becton Dickinson) and FlowJo (Tree Star Inc., http://www.flowjo.com/) software packages.
Caspase 3/7 activity
Caspase 3 and 7 activity was evaluated by using caspase 3/7 Glo from Promega (http://www.promega.es/products/cell-health-assays/apoptosis-assays/luminescent-caspase-assays/caspase_glo-3_7-assay-systems/). Briefly, 10 000 cells were seeded 24 h prior to IR (10 Gy). After 24–30 h cells were processed in lysis buffer from Promega and incubated for 60 to 90 min. Then, luminescence of each sample was measured using a Monolight 3096 (BD PharMingen, http://www.bdbiosciences.com/). Activity of untreated cells for each cell line was refered to as 1.
Cell proliferation measurements
Sub-confluent monolayer cultures were trypsinized, and cells were plated in 24-well plates at a density of 104 cells/well. Cell proliferation was analyzed at 1, 2, 3 and 4 d after plating by an MTT-based assay (http://www.sigmaaldrich.com/catalog/product/sigma/m5655?lang=es®ion=ES) Briefly, MTT at 0.5 mg/ml was added to the medium in each well, and plates were returned to the incubator for 1 h. The medium MTT was then removed, 500 μl of DMSO were added to each well, and the plate was kept in agitation for 5 min in the dark to dissolve the MTT–formazan crystals. The absorbance of the samples was then recorded at 570 nm. Wells containing medium plus MTT but no cells were used as blanks. For UCN-01 experiments, cells were treated 24 h after plating at indicated dose during indicated times and processed as described above. Data are the average of 3 independent experiments performed in triplicate cultures.
Statistical analysis
Data are presented as mean ± standard deviation (S.D). Statistical significance was evaluated by Student t test using the GraphPad Prism 5.00 software (http://www.graphpad.com/scientific-software/prism/). The statistical significance of differences was indicated in figures by asterisks as follows: *P < 0.05, ** P < 0.01, and *** P < 0.001.
| Abbreviations: | ||
| E1a | = | adenovirus early region 1A |
| Chk1 | = | serin/treonin kinase Chk1, also named CHEK1 |
| H2AX | = | Histone 2AX |
| GBM | = | glioblastoma multiforme |
| c-Myc | = | cellular Myc |
| ATR | = | Ataxia telangiectasia and Rad3-related protein |
| ATM | = | Ataxia telangiectasia mutated protein |
Additional material
Download Zip (1.7 MB)Acknowledgments
This work was supported by grants from Fundación Leticia Castillejo Castillo, JCCM (PPII10-0141-040) and Ministerio de Economia y Competitividad (SAF2012-30862) to RSP. Instituto de Salud Carlos III FIS PS09/1988 and CCG10-UAM/BIO-5871 to ISP. RSP Research Institute, and the work performed in his laboratory receive support from the European Community through the regional development funding program (FEDER).
We appreciate the comment and suggestions of Drs V Sánchez-Arévalo and Santiago Ramón y Cajal
This work is dedicated to the memory of Dr Carlos Parada-Cobo.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Supplemental Materials
Supplemental materials may be found here: www.landesbioscience.com/journals/cc/article/26754
Related Research Data
References
- Frisch SM, Mymryk JS. Adenovirus-5 E1A: paradox and paradigm. Nat Rev Mol Cell Biol 2002; 3:441 - 52; http://dx.doi.org/10.1038/nrm827; PMID: 12042766
- Zerler B, Moran B, Maruyama K, Moomaw J, Grodzicker T, Ruley HE. Adenovirus E1A coding sequences that enable ras and pmt oncogenes to transform cultured primary cells. Mol Cell Biol 1986; 6:887 - 99; PMID: 3022137
- Ragozzino MM, Kuo A, DeGregori J, Kohl N, Ruley HE. Mechanisms of oncogene cooperation: activation and inactivation of a growth antagonist. Environ Health Perspect 1991; 93:97 - 103; http://dx.doi.org/10.1289/ehp.919397; PMID: 1837777
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:2015 - 27; PMID: 10950866
- Callejas-Valera JL, Guinea-Viniegra J, Ramírez-Castillejo C, Recio JA, Galan-Moya E, Martinez N, Rojas JM, Ramón y Cajal S, Sánchez-Prieto R. E1a gene expression blocks the ERK1/2 signaling pathway by promoting nuclear localization and MKP up-regulation: implication in v-H-Ras-induced senescence. J Biol Chem 2008; 283:13450 - 8; http://dx.doi.org/10.1074/jbc.M709230200; PMID: 18316372
- Lowe SW, Ruley HE. Stabilization of the p53 tumor suppressor is induced by adenovirus 5 E1A and accompanies apoptosis. Genes Dev 1993; 7:535 - 45; http://dx.doi.org/10.1101/gad.7.4.535; PMID: 8384579
- Fuchs M, Gerber J, Drapkin R, Sif S, Ikura T, Ogryzko V, Lane WS, Nakatani Y, Livingston DM. The p400 complex is an essential E1A transformation target. Cell 2001; 106:297 - 307; http://dx.doi.org/10.1016/S0092-8674(01)00450-0; PMID: 11509179
- Chan HM, Narita M, Lowe SW, Livingston DM. The p400 E1A-associated protein is a novel component of the p53 --> p21 senescence pathway. Genes Dev 2005; 19:196 - 201; http://dx.doi.org/10.1101/gad.1280205; PMID: 15655109
- Flinterman M, Gäken J, Farzaneh F, Tavassoli M. E1A-mediated suppression of EGFR expression and induction of apoptosis in head and neck squamous carcinoma cell lines. Oncogene 2003; 22:1965 - 77; http://dx.doi.org/10.1038/sj.onc.1206190; PMID: 12673202
- Yu D, Suen TC, Yan DH, Chang LS, Hung MC. Transcriptional repression of the neu protooncogene by the adenovirus 5 E1A gene products. Proc Natl Acad Sci U S A 1990; 87:4499 - 503; http://dx.doi.org/10.1073/pnas.87.12.4499; PMID: 1972274
- Su JL, Chen PB, Chen YH, Chen SC, Chang YW, Jan YH, Cheng X, Hsiao M, Hung MC. Downregulation of microRNA miR-520h by E1A contributes to anticancer activity. Cancer Res 2010; 70:5096 - 108; http://dx.doi.org/10.1158/0008-5472.CAN-09-4148; PMID: 20501832
- Zhou Z, Zhou RR, Guan H, Bucana CD, Kleinerman ES. E1A gene therapy inhibits angiogenesis in a Ewing’s sarcoma animal model. Mol Cancer Ther 2003; 2:1313 - 9; PMID: 14707272
- Liao Y, Yu D, Hung MC. Novel approaches for chemosensitization of breast cancer cells: the E1A story. Adv Exp Med Biol 2007; 608:144 - 69; http://dx.doi.org/10.1007/978-0-387-74039-3_11; PMID: 17993239
- Sánchez-Prieto R, Quintanilla M, Cano A, Leonart ML, Martin P, Anaya A, Ramón y Cajal S. Carcinoma cell lines become sensitive to DNA-damaging agents by the expression of the adenovirus E1A gene. Oncogene 1996; 13:1083 - 92; PMID: 8806698
- Yamaguchi H, Chen CT, Chou CK, Pal A, Bornmann W, Hortobagyi GN, Hung MC. Adenovirus 5 E1A enhances histone deacetylase inhibitors-induced apoptosis through Egr-1-mediated Bim upregulation. Oncogene 2010; 29:5619 - 29; http://dx.doi.org/10.1038/onc.2010.295; PMID: 20676141
- Stiewe T, Parssanedjad K, Esche H, Opalka B, Pützer BM. E1A overcomes the apoptosis block in BCR-ABL+ leukemia cells and renders cells susceptible to induction of apoptosis by chemotherapeutic agents. Cancer Res 2000; 60:3957 - 64; PMID: 10919674
- Marchetti E, Romero J, Sanchez R, Vargas J, Dominguez C, Lacal J, Cajal S. Oncogenes and cellular-sensitivity to radiotherapy - a study on murine keratinocytes transformed by v-h-ras, v-myc, v-Neu, adenovirus e1a and mutant p53. Int J Oncol 1994; 5:611 - 8; PMID: 21559621
- Shao R, Tsai EM, Wei K, von Lindern R, Chen YH, Makino K, Hung MC. E1A inhibition of radiation-induced NF-kappaB activity through suppression of IKK activity and IkappaB degradation, independent of Akt activation. Cancer Res 2001; 61:7413 - 6; PMID: 11606372
- Su JL, Cheng X, Yamaguchi H, Chang YW, Hou CF, Lee DF, Ko HW, Hua KT, Wang YN, Hsiao M, et al. FOXO3a-Dependent Mechanism of E1A-Induced Chemosensitization. Cancer Res 2011; 71:6878 - 87; http://dx.doi.org/10.1158/0008-5472.CAN-11-0295; PMID: 21911455
- Samuelson AV, Lowe SW. Selective induction of p53 and chemosensitivity in RB-deficient cells by E1A mutants unable to bind the RB-related proteins. Proc Natl Acad Sci U S A 1997; 94:12094 - 9; http://dx.doi.org/10.1073/pnas.94.22.12094; PMID: 9342368
- Samuelson AV, Narita M, Chan HM, Jin J, de Stanchina E, McCurrach ME, Narita M, Fuchs M, Livingston DM, Lowe SW. p400 is required for E1A to promote apoptosis. J Biol Chem 2005; 280:21915 - 23; http://dx.doi.org/10.1074/jbc.M414564200; PMID: 15741165
- Van Meir EG, Hadjipanayis CG, Norden AD, Shu HK, Wen PY, Olson JJ. Exciting new advances in neuro-oncology: the avenue to a cure for malignant glioma. CA Cancer J Clin 2010; 60:166 - 93; http://dx.doi.org/10.3322/caac.20069; PMID: 20445000
- Noda SE, El-Jawahri A, Patel D, Lautenschlaeger T, Siedow M, Chakravarti A. Molecular advances of brain tumors in radiation oncology. Semin Radiat Oncol 2009; 19:171 - 8; http://dx.doi.org/10.1016/j.semradonc.2009.02.005; PMID: 19464632
- Shen LF, Chen J, Zeng S, Zhou RR, Zhu H, Zhong MZ, Yao RJ, Shen H. The superparamagnetic nanoparticles carrying the E1A gene enhance the radiosensitivity of human cervical carcinoma in nude mice. Mol Cancer Ther 2010; 9:2123 - 30; http://dx.doi.org/10.1158/1535-7163.MCT-09-1150; PMID: 20587666
- Pawlik TM, Keyomarsi K. Role of cell cycle in mediating sensitivity to radiotherapy. Int J Radiat Oncol Biol Phys 2004; 59:928 - 42; http://dx.doi.org/10.1016/j.ijrobp.2004.03.005; PMID: 15234026
- Xu B, Kim ST, Lim DS, Kastan MB. Two molecularly distinct G(2)/M checkpoints are induced by ionizing irradiation. Mol Cell Biol 2002; 22:1049 - 59; http://dx.doi.org/10.1128/MCB.22.4.1049-1059.2002; PMID: 11809797
- Yarden RI, Pardo-Reoyo S, Sgagias M, Cowan KH, Brody LC. BRCA1 regulates the G2/M checkpoint by activating Chk1 kinase upon DNA damage. Nat Genet 2002; 30:285 - 9; http://dx.doi.org/10.1038/ng837; PMID: 11836499
- Kaneko YS, Watanabe N, Morisaki H, Akita H, Fujimoto A, Tominaga K, Terasawa M, Tachibana A, Ikeda K, Nakanishi M. Cell-cycle-dependent and ATM-independent expression of human Chk1 kinase. Oncogene 1999; 18:3673 - 81; http://dx.doi.org/10.1038/sj.onc.1202706; PMID: 10391675
- Höglund A, Nilsson LM, Muralidharan SV, Hasvold LA, Merta P, Rudelius M, Nikolova V, Keller U, Nilsson JA. Therapeutic implications for the induced levels of Chk1 in Myc-expressing cancer cells. Clin Cancer Res 2011; 17:7067 - 79; http://dx.doi.org/10.1158/1078-0432.CCR-11-1198; PMID: 21933891
- Chakraborty AA, Tansey WP. Adenoviral E1A function through Myc. Cancer Res 2009; 69:6 - 9; http://dx.doi.org/10.1158/0008-5472.CAN-08-3026; PMID: 19117980
- Tworkowski KA, Chakraborty AA, Samuelson AV, Seger YR, Narita M, Hannon GJ, Lowe SW, Tansey WP. Adenovirus E1A targets p400 to induce the cellular oncoprotein Myc. Proc Natl Acad Sci U S A 2008; 105:6103 - 8; http://dx.doi.org/10.1073/pnas.0802095105; PMID: 18413597
- Murga M, Campaner S, Lopez-Contreras AJ, Toledo LI, Soria R, Montaña MF, D’Artista L, Schleker T, Guerra C, Garcia E, et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat Struct Mol Biol 2011; 18:1331 - 5; http://dx.doi.org/10.1038/nsmb.2189; PMID: 22120667
- Sankar N, Kadeppagari RK, Thimmapaya B. c-Myc-induced aberrant DNA synthesis and activation of DNA damage response in p300 knockdown cells. J Biol Chem 2009; 284:15193 - 205; http://dx.doi.org/10.1074/jbc.M900776200; PMID: 19332536
- Murga M, Bunting S, Montaña MF, Soria R, Mulero F, Cañamero M, Lee Y, McKinnon PJ, Nussenzweig A, Fernandez-Capetillo O. A mouse model of ATR-Seckel shows embryonic replicative stress and accelerated aging. Nat Genet 2009; 41:891 - 8; http://dx.doi.org/10.1038/ng.420; PMID: 19620979
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886 - 95; http://dx.doi.org/10.1016/S0960-9822(00)00610-2; PMID: 10959836
- Gutiérrez-González A, Belda-Iniesta C, Bargiela-Iparraguirre J, Dominguez G, García Alfonso P, Perona R, Sanchez-Perez I. Targeting Chk2 improves gastric cancer chemotherapy by impairing DNA damage repair. Apoptosis 2013; 18:347 - 60; PMID: 23271172
- Hong S, Paulson QX, Johnson DG. E2F1 and E2F3 activate ATM through distinct mechanisms to promote E1A-induced apoptosis. Cell Cycle 2008; 7:391 - 400; http://dx.doi.org/10.4161/cc.7.3.5286; PMID: 18235226
- Brooks K, Oakes V, Edwards B, Ranall M, Leo P, Pavey S, Pinder A, Beamish H, Mukhopadhyay P, Lambie D, et al. A potent Chk1 inhibitor is selectively cytotoxic in melanomas with high levels of replicative stress. Oncogene 2013; 32:788 - 96; http://dx.doi.org/10.1038/onc.2012.72; PMID: 22391562
- Tse AN, Rendahl KG, Sheikh T, Cheema H, Aardalen K, Embry M, Ma S, Moler EJ, Ni ZJ, Lopes de Menezes DE, et al. CHIR-124, a novel potent inhibitor of Chk1, potentiates the cytotoxicity of topoisomerase I poisons in vitro and in vivo. Clin Cancer Res 2007; 13:591 - 602; http://dx.doi.org/10.1158/1078-0432.CCR-06-1424; PMID: 17255282
- Hung MC, Hortobagyi GN, Ueno NT. Development of clinical trial of E1A gene therapy targeting HER-2/neu-overexpressing breast and ovarian cancer. Adv Exp Med Biol 2000; 465:171 - 80; http://dx.doi.org/10.1007/0-306-46817-4_16; PMID: 10810625
- Hung MC, Yan DH, Zhang S. E1A-Mediated Gene Therapy. Methods Mol Med 2001; 39:775 - 82; PMID: 21340839
- Touchefeu Y, Vassaux G, Harrington KJ. Oncolytic viruses in radiation oncology. Radiother Oncol 2011; 99:262 - 70; http://dx.doi.org/10.1016/j.radonc.2011.05.078; PMID: 21704402
- Heise C, Sampson-Johannes A, Williams A, McCormick F, Von Hoff DD, Kirn DH. ONYX-015, an E1B gene-attenuated adenovirus, causes tumor-specific cytolysis and antitumoral efficacy that can be augmented by standard chemotherapeutic agents. Nat Med 1997; 3:639 - 45; http://dx.doi.org/10.1038/nm0697-639; PMID: 9176490
- Busse PM, Bose SK, Jones RW, Tolmach LJ. The action of caffeine on X-irradiated HeLa cells. III. Enhancement of X-ray-induced killing during G2 arrest. Radiat Res 1978; 76:292 - 307; http://dx.doi.org/10.2307/3574780; PMID: 156382
- Ganem NJ, Pellman D. Linking abnormal mitosis to the acquisition of DNA damage. J Cell Biol 2012; 199:871 - 81; http://dx.doi.org/10.1083/jcb.201210040; PMID: 23229895
- Zhao H, Piwnica-Worms H. ATR-mediated checkpoint pathways regulate phosphorylation and activation of human Chk1. Mol Cell Biol 2001; 21:4129 - 39; http://dx.doi.org/10.1128/MCB.21.13.4129-4139.2001; PMID: 11390642
- Liu Q, Guntuku S, Cui XS, Matsuoka S, Cortez D, Tamai K, Luo G, Carattini-Rivera S, DeMayo F, Bradley A, et al. Chk1 is an essential kinase that is regulated by Atr and required for the G(2)/M DNA damage checkpoint. Genes Dev 2000; 14:1448 - 59; PMID: 10859164
- Baluchamy S, Sankar N, Navaraj A, Moran E, Thimmapaya B. Relationship between E1A binding to cellular proteins, c-myc activation and S-phase induction. Oncogene 2007; 26:781 - 7; http://dx.doi.org/10.1038/sj.onc.1209825; PMID: 16862175
- Kadeppagari RK, Sankar N, Thimmapaya B. Adenovirus transforming protein E1A induces c-Myc in quiescent cells by a novel mechanism. J Virol 2009; 83:4810 - 22; http://dx.doi.org/10.1128/JVI.02145-08; PMID: 19279113
- Connell CM, Shibata A, Tookman LA, Archibald KM, Flak MB, Pirlo KJ, Lockley M, Wheatley SP, McNeish IA. Genomic DNA damage and ATR-Chk1 signaling determine oncolytic adenoviral efficacy in human ovarian cancer cells. J Clin Invest 2011; 121:1283 - 97; http://dx.doi.org/10.1172/JCI43976; PMID: 21383502
- Zhangand Y, Hunter T. Roles of Chk1 in cell biology and cancer therapy. Int J Cancer 2013; Ahead of Print
- Wang WJ, Wu SP, Liu JB, Shi YS, Huang X, Zhang QB, Yao KT. MYC regulation of CHK1 and CHK2 promotes radioresistance in a stem cell-like population of nasopharyngeal carcinoma cells. Cancer Res 2013; 73:1219 - 31; http://dx.doi.org/10.1158/0008-5472.CAN-12-1408; PMID: 23269272
- Gagou ME, Zuazua-Villar P, Meuth M. Enhanced H2AX phosphorylation, DNA replication fork arrest, and cell death in the absence of Chk1. Mol Biol Cell 2010; 21:739 - 52; http://dx.doi.org/10.1091/mbc.E09-07-0618; PMID: 20053681
- López-Contreras AJ, Gutierrez-Martinez P, Specks J, Rodrigo-Perez S, Fernandez-Capetillo O. An extra allele of Chk1 limits oncogene-induced replicative stress and promotes transformation. J Exp Med 2012; 209:455 - 61; http://dx.doi.org/10.1084/jem.20112147; PMID: 22370720
- Unsal-Kaçmaz K, Chastain PD, Qu PP, Minoo P, Cordeiro-Stone M, Sancar A, Kaufmann WK. The human Tim/Tipin complex coordinates an Intra-S checkpoint response to UV that slows replication fork displacement. Mol Cell Biol 2007; 27:3131 - 42; http://dx.doi.org/10.1128/MCB.02190-06; PMID: 17296725
- Galan-Moya EM, de la Cruz-Morcillo MA, Llanos Valero M, Callejas-Valera JL, Melgar-Rojas P, Hernadez Losa J, Salcedo M, Fernández-Aramburo A, Ramon y Cajal S, Sánchez-Prieto R. Balance between MKK6 and MKK3 mediates p38 MAPK associated resistance to cisplatin in NSCLC. PLoS One 2011; 6:e28406; http://dx.doi.org/10.1371/journal.pone.0028406; PMID: 22164285
- de la Cruz-Morcillo MA, Valero ML, Callejas-Valera JL, Arias-González L, Melgar-Rojas P, Galán-Moya EM, García-Gil E, García-Cano J, Sánchez-Prieto R. P38MAPK is a major determinant of the balance between apoptosis and autophagy triggered by 5-fluorouracil: implication in resistance. Oncogene 2012; 31:1073 - 85; http://dx.doi.org/10.1038/onc.2011.321; PMID: 21841826