Abstract
The pentose phosphate pathway (PPP) provides ribose and NADPH that support biosynthesis and antioxidant defense. Our recent findings suggest that the p53-related protein TAp73 enhances the PPP flux. TAp73 stimulates the expression of glucose-6-phophate dehydrogenase (G6PD), the rate-limiting enzymes of the PPP. Through this regulation, TAp73 promotes the accumulation of macromolecules and increases cellular capability to withstand oxidative stresses. TAp73 also regulates other metabolic enzymes, and the relative importance of these targets in TAp73-mediated cell growth is not well understood. Here we show that, like in other cell lines, TAp73 is required for supporting proliferation and maintaining the expression of G6PD in the human lung cancer H1299 cells. Restoration of G6PD expression almost fully rescues the defects in cell growth caused by TAp73 knockdown, suggesting that G6PD is the major proliferative target of TAp73 in these cells. G6PD expression is elevated in various tumors, correlating with the upregulation of TAp73. These results indicate that TAp73 may function as an oncogene, and that G6PD is likely a focal point of regulation in oncogenic growth.
Introduction
p73 is a member of the p53 family, and it shares extensive similarity in sequence and structural organization to p53 ().Citation1 However, while p53 is a well-established tumor suppressor,Citation2,Citation3 the role of p73 in tumorigenesis is still a matter of debate.Citation4-Citation7 The complex role of p73 is, in part, due to the expression of 2 major p73 isoform classes: the transactivation-proficient (TA) and the transactivation-deficient (ΔN) isoform, through the use of alternative promoters.Citation4-Citation7 In transfection assays TAp73 often behaves like p53 and regulates genes involved in apoptosis and cell cycle arrest, while ΔNp73 exerts a dominant-negative effect on TAp73 and p53, likely through the formation of non-functional hetero-complexes and/or the competition for DNA binding. Consistent with an essential role of p53 in tumor suppression, p53-deficient mice seem to develop normally but almost inevitably die of tumors within 6 mo of birth.Citation8,Citation9 In contrast, mice deficient in both p73 isoforms show profound defects in development, but no obvious increase in tumor incidence,Citation10 suggesting an essential function of these isoforms in development, but either dispensable or opposing roles of these isoforms in tumor suppression. Mice deficient specifically in the TAp73 isoform displayed an intermediate phenotype between p53- and total p73-deficient mice in terms of both developmental defects and tumor incidence,Citation11 suggesting a role for TAp73 in tumor suppression. This is, in part, attributable for a function for TAp73 in maintaining genome stability.Citation11,Citation12 Mice deficient in the ΔNp73 isoform, on the other hand, have a normal lifespan, albeit with signs of neurodegeneration, and cells derived from these mice show an increase in sensitivity to DNA damage agents and in p53-mediated apoptosis.Citation13 The developmental phenotypes of the p73-deficient mice are much more severe compared with the TAp73- or ΔNp73-specific knockout mice. These results suggest a model in which TAp73 suppresses tumor formation, while ΔNp73 promotes it. Moreover, these isoforms may play a partially redundant role in development. Nevertheless, while p53 is mutated in 50–70% of all human tumors, making it the most frequently mutated gene in tumors, TAp73 is almost never mutated in human tumors.Citation4,Citation6,Citation7 Rather, it is overexpressed, often along with ΔNp73, in a wide range of tumor types, and the frequency of TAp73 overexpression can be as high as 30%. These observations raise important questions as to whether TAp73 confers an advantage on tumor cells and, if so, what may be the molecular mechanism.
Figure 1. p73 isoforms and the pentose phosphate pathway. (A) Schematic representation of p73 isoforms and p53. Each p73 isoform class comprises various splicing variants (α, β, γ, etc.) that differ in their C-terminal regions. TA, transactivation domain; DBD, DNA-binding domain; OD, oligomerization domain; SAM, sterile α motif. (B) The pentose phosphate pathway and glycolysis. FBP, fructose 1,6-biphosphate; PEP, phosphoenolpyruvate; ROS, reactive oxygen species. Glycolytic pathway is indicated in blue, and oxidative pentose phosphate pathway is indicated in green.
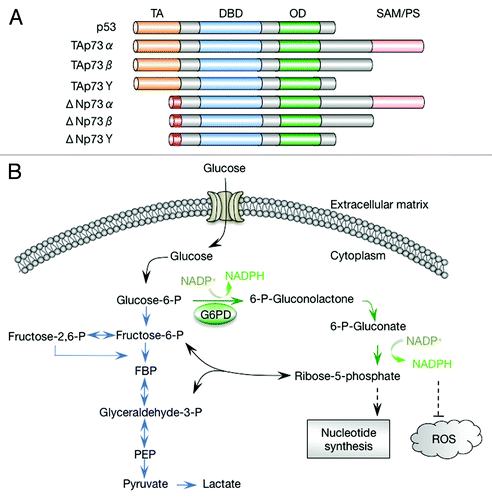
A long-recognized hallmark capacity of cancer cells is the reprogramming of the metabolic pathways. As early as in the 1920s, Otto Warburg observed that cancer cells consumed a much larger amount of glucose and derived more energy from glycolysis compared with their normal counterparts, even in the presence of appropriate levels of oxygen.Citation14,Citation15 This phenomenon (aerobic glycolysis or the Warburg effect) is part of major re-organization of various metabolic fluxes in tumor cells. The metabolic “rewiring” in tumor cells likely ensures sufficient bioenergetics, a favorite redox state that is stimulatory for proliferation while avoiding damages to cellular components, and an ample and balanced production of proteins, nucleic acids, lipids, and other macromolecules. The metabolic “rewiring” in cancer cells is brought about through via overlapping yet distinct mutations that result in hyperactive oncogenes or inactive tumor suppressors, and in limited cases, through mutations in metabolic enzymes themselves. Some aspects of this rewiring appear to be specific to certain tumor types, while others tend to be shared by many tumors.
A shared feature of tumor cell metabolism, in addition to the Warburg effect, appears to be a hyperactive flux through the pentose phosphate pathway (PPP) (). The PPP supplies cells with ribose, the obligate precursor for de novo synthesis of nucleotides and other critical metabolites, including ATP, NAD+, and NADP+.Citation16-Citation19 It is also a main pathway that reduces NADP+ to NADPH, which supplies electrons for reductive biosynthesis, including the synthesis of lipids, cholesterol, and deoxynucleotides. NADPH is also critical for antioxidant responses, generating reduced glutathione that protects against damages to proteins and providing the ultimate reducing power for the function of several other antioxidant systems. The PPP comprises 2 branches: an irreversible oxidative branch during which ribose and NADPH are generated, and a reversible non-oxidative branch during which ribose is inter-converted with the glycolytic intermediates. Through this inter-conversion and through the share of the same starting metabolite (glucose-6-phosphate or G6P), the PPP is closely linked to glycolysis. Consequently, the PPP can be modulated indirectly in tumor cells when glycolytic intermediates are accumulated.Citation20 Intriguingly, accumulation of 3-phosphoglycerate (3-PG), the substrate of the glycolytic enzyme phosphoglycerate mutase 1 (PGAM1), leads to inhibition of 6-phosphogluconate dehydrogenase in the oxidative branch of the PPP.Citation21 Genetic or pharmacological inhibition of PGAM1 results in increased 3-PG, as well as decreased glycolysis and PPP flux.
Perhaps more importantly, the PPP itself is directly regulated in response to the cellular demand for ROS detoxification and biosynthesis by multiple mechanisms, many of which appear to focus on the pacesetter of the oxidative PPP, glucose-6-phospate dehydrogenase (G6PD). G6PD is activated by NADP+, which is also its substrate, thereby enabling a homeostatic control of NADPH and ROS contents. The activity G6PD is increased by DNA damage signal through the ATM kinase-mediated phosphorylation of HSP27, an activator of G6PD.Citation22 This regulation may enhance the supplies of NADPH and ribose for repairing DNA damage and detoxifying ROS that are generated by DNA damaging agents. Interestingly, G6PD is inhibited by the tumor suppressor p53 through a mechanism involving direct protein–protein interactions instead of transcriptional regulation.Citation23 p53 binds to the active G6PD dimer and converts it to an inactive monomer. The majority of the cytoplasmic p53 is found to be associated with G6PD, suggesting that G6PD may be the primary target of p53 in this cellular compartment. While these post-translational modification regulatory permit rapid adjustment of the PPP flux, G6PD is also regulated at transcriptional levels, which provides a delayed but persistent change in the PPP flux. G6PD expression is strongly increased when cells are stimulated to proliferateCitation24 or are detached from matrix.Citation25 Nevertheless, the regulation of G6PD at the transcriptional levels is not well understood.
We recently reported that TAp73 enhances the expression of G6PD.Citation26 This function is unique to TAp73, and is not shared by p53 or the other p53 family protein, p63. It is required for maintaining G6PD expression in unstressed cells, promoting glucose flux to the PPP for biosynthesis and antioxidant defense. We found that TAp73-deficient tumor cells grow slowly and form smaller tumors compared with TAp73-proficient cells. Supplement of both nucleosides and an ROS scavenger, but not either one alone, can restore the growth of TAp73-deficient cells, indicating that the proliferative effect of TAp73 relates to both an increase in nucleotides synthesis and a reduction in ROS. These results suggest that TAp73 enhances cell proliferation, at least in part, through the increase in glucose flux through the PPP. Nevertheless, in a few cell lines that were tested, overexpression of G6PD only partially rescued the growth defect of TAp73-deficient cells, suggesting that in these cells, G6PD is not the only proliferative target of TAp73. Interestingly, Rufini et al. revealed another proliferative targets of the PPP, the mitochondrial complex IV (cytochrome C oxidase) subunit 4 (Cox4i1).Citation27 In this study, we examined the role of TAp73-mediated G6PD expression in additional cell lines. TAp73 deficiency primarily leads to the formation of lung tumors,Citation11 and hence we used a human lung cancer cell line, H1299. We found that, as in other cells, TAp73 stimulates the expression of G6PD in H1299 cells. Interestingly, however, overexpression of G6PD in this cell line almost completely restores the growth of TAp73-knockdown cells, suggesting that G6PD is a major proliferative target of TAp73 in these cells.
Results
p73 regulates G6PD expression in H1299 cells
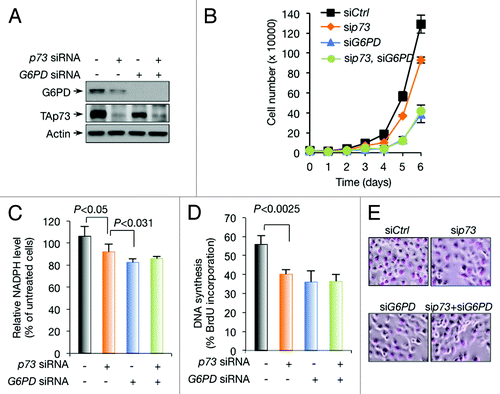
p73 silencing reduces G6PD expression and cell proliferation in a number of cell lines, including human osteosarcoma U2OS cells, mouse embryonic fibroblast cells (MEFs), and isogenic human colon cancer HCT116 cells that are wild-type for both p53 and its target gene p21 or null for either one of them.Citation26 Similarly, knockdown of p73 reduced G6PD expression in H1299 cells and impeded cell proliferation (). PPP is a major source of cellular NADPH and ribose.Citation28 In H1299 cells, knockdown of p73 reduced NADPH levels () and DNA synthesis (). As expected, G6PD knockdown also resulted in a decline in NADPH levels, DNA synthesis, and cell proliferation, to extents similar to or greater than p73 knockdown (). Of note, in G6PD knockdown H1299 cells, silencing TAp73 no longer affected these parameters (). These results suggest that, in H1299 cells, p73 controls the expression of G6PD and promotes cell proliferation.
Figure 2. Regulation of G6PD expression, NADPH levels, and DNA synthesis by p73 in H1299 cells (A and B) H1299 cells were transfected with control (−), p73, and G6PD siRNA as indicated. Protein expression was analyzed (A) and cell proliferation is shown (B). (C) NADPH levels in H1299 cells treated with control, p73, and G6PD siRNA as indicated. (D and E) H1299 cells were transfected with control, p73, and G6PD siRNA as indicated. Cells were assayed for BrdU incorporation (D). Representative images of cells stained with BrdU (E).
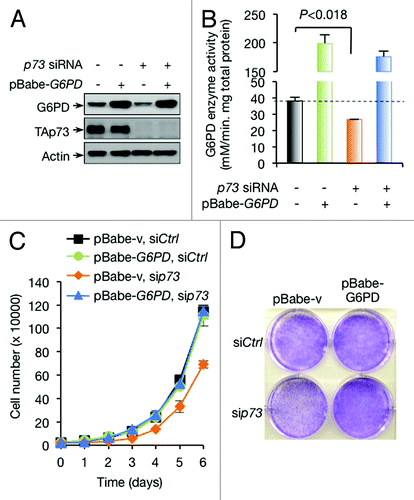
G6PD overexpression completely restores the growth of p73-depleted H1299 cells
To further assess the importance of TAp73-mediated G6PD expression in H1299 cells, we forced the expression of G6PD (). As expected, this restored the activity of G6PD (). Interestingly, unlike in the other cell lines, where G6PD overexpression only partially restored the growth of p73-depleted cells,Citation26 p73-depleted H1299 cells expressing exogenous G6PD grew nearly as well as control cells (). This data suggests that induction of G6PD expression is both necessary and sufficient for TAp73-mediated cell proliferation in H1299 cells.
Figure 3. Overexpression of G6PD rescues growth defects of p73-depleted cells. (A and B) Protein expression (A) and G6PD activity (B) in H1299 cells stably overexpressing G6PD or vector control in the presence or absence of p73 siRNA are shown. (C and D) H1299 cells stably overexpressing G6PD or vector control were treated with p73 or control siRNA as indicated. Cell proliferation (C) and representative images of cells stained with crystal violet at day 6 (D) are shown
p73 fails to affect ROS homeostasis in H1299 cells
Our recent data using a number of cancer cell lines indicates that ROS detoxification and biosynthesis of nucleosides are essential for TAp73-mediated cell growth. Since depletion of either p73 or G6PD in H1299 cells resulted in a strong reduction in DNA synthesis (), both p73 and G6PD may play an important role in nucleotide synthesis in these cells. We measured the effect of p73 and G6PD on ROS content using the cell-permeably radical dye 2’,7’-dichlorofluorescein diacetate (DCFDA). While G6PD-depleted cells displayed an increasing content of ROS as expected, p73-depleted cells failed to accumulate ROS (). This result suggests that in p73-depleted cells, the expression of G6PD might not have been reduced to a level that affected cellular ROS. Forced expression of G6PD in either control cells or p73-depleted cells had minimal effect on ROS levels (), consistent with the notion that the levels of endogenous G6PD in these cells might be sufficient for ROS detoxification.
Figure 4. p73 depletion fails to affect ROS in H1299 cells (A) ROS levels in H1299 cells transfected with indicated siRNA. (B) H1299 stably overexpressing G6PD or vector control were transfected with p73 siRNA or control siRNA as indicated. ROS levels were analyzed. (C) H1299 stably overexpressing G6PD or vector control were transfected with p73 siRNA or control siRNA as indicated. Cells were treated with or without 100 µM or 250 µM H2O2 for 24 h, and cell viability was analyzed by trypan blue staining.
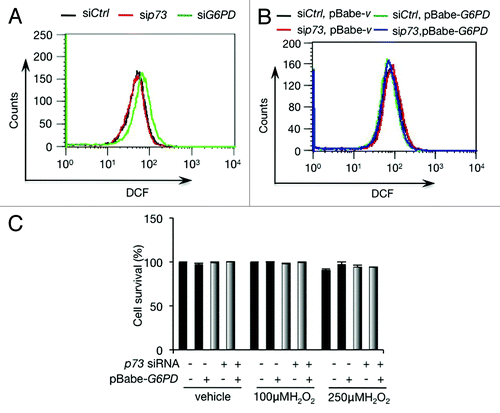
To extend these analyses, we examined the effect of p73 on cellular capacity to withstand oxidative stresses. H1299 cells were highly resistant to oxidative stress, even when they were treated with 250 µM H2O2 for 24 h (). Silencing p73 or enforcing the expression of G6PD had minimal impact on cell survival in the presence of H2O2 (). Taken together, these data suggest that H1299 cells possess a robust antioxidant capacity, even when G6PD levels are reduced, and that p73 has a minimal role in maintaining the redox state of these cells, although it is important for DNA synthesis.
G6PD is overexpressed in many human cancers
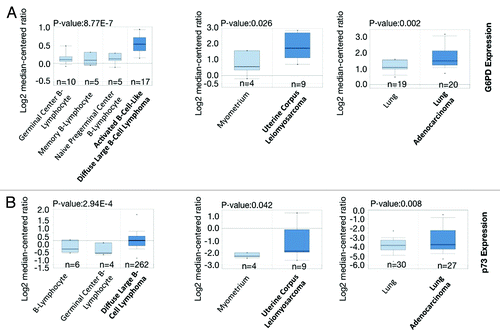
Given the importance of G6PD in cell proliferation, we analyzed public microarray data (http://www.oncomine.org) to determine whether it is highly overexpressed in human cancers. As shown in , G6PD expression is significantly higher in many cancers including B-cell lymphoma, uterine cancer and lung tumor samples. Consistent with previous findings, p73 is also highly expressed in these cancers (). These data suggest that G6PD expression is abnormally increased in many human cancers, correlating with p73 expression and likely promoting cancer growth.
Figure 5. G6PD and p73 are frequently overexpressed in many human cancers (A and B) Box plot comparing TP73 and G6PD transcript levels in diffuse large B-cell lymphoma,Citation36,Citation37 uterine corpus leiomyosarcoma,Citation38 lung adenocarcinoma,Citation39,Citation40 and their counterparts. The graphs were derived from published data available through the ONCOMINE database. The differential gene expression data are centered on the median of expression levels and plotted on a log2 scale. The P value was calculated using a 2-sample t test. Whiskers indicate minimum and maximum data values that are not outliers. The number of samples (n) in each class is indicated.
Discussion
Metabolism in cancer cells is commonly dysregulated. The results presented here and in our recent report reveal a role for TAp73, a gene that is frequently upregulated in tumors, in stimulating the expression of G6PD, the pacesetter of the PPP. These results suggest that TAp73 plays a previously unrecognized function in cell proliferation, and that G6PD is a focal point of regulation for tumor growth.
TAp73 is required for the expression of G6PD in unstressed cells and the maintenance of appropriate basal levels of PPP flux.Citation26 The effect of TAp73 on G6PD is highly specific and is not shared by either p53 or p63, despite the structural similarity among these proteins. This selectivity appears to be determined by the sequence of the TAp73 response element within the G6PD genes, instead of the context of the chromatin structure, as shown by a reporter gene assay.Citation26 It would be interesting to determine the precise sequence within this response element that underlines this specificity. We also found that G6PD expression is substantially enhanced in cells treated with genotoxic agents.Citation26 Under these conditions, the demand for both NADPH and ribose likely increases to counter the oxidative stress and DNA damage inflicted by these agents. This upregulation is almost completely dependent on TAp73. And in the absence of TAp73, the expression levels of G6PD may actually decline in cells experiencing DNA damage.Citation26 These observations indicate that the TAp73-G6PD pathway is an integral part of the homeostatic control of cellular ROS and biosynthesis.
Along with enhanced incidences of tumorigenesis and neuronal defects, TAp73 deficiency exacerbates aging-related phenotypes in mice and accelerates the onset of senescence in cultured cells.Citation27 An important target of TAp73 is likely the complex IV subunit Cox4i1; the reduced expression of Cox4il in TAp73-deficient animals likely contributes to the dysfunction of the electron transport chain and enhanced production of ROS. Like Cox4i1, downregulation of G6PD also leads to senescence.Citation26 Therefore, TAp73 may modulate cellular ROS homeostasis and aging through regulating both Cox4il and G6PD. The relative contributions of these 2 targets may vary dependent on cellular context, and the results presented here indicate that, at least in H1299 lung cancer cells, G6PD appears to be the main proliferative target of TAp73. We found that the addition of both ROS scavengers and nucleosides, but not each one alone, almost completely restores the growth of TAp73-deficient cells, suggesting that the function of TAp73 in stimulating ribose synthesis via G6PD is important for cell proliferation. Of note, TAp73-deficient mice display a highly organ-specific tumor formation, primarily in the lung.Citation11 It would be interesting to further investigate the role of the PPP in lung cancer. Aside from G6PD and Cox4i1, TAp73 may regulate other metabolic enzymes. A recent study using inducible TAp73 cell lines has revealed that multiple metabolic pathways are also enhanced upon TAp73 induction. One of the TAp73 targets identified in this study is 6-phosphogluconolactonase (PGLS), the second enzyme of the PPP pathway, suggesting an intriguing possibility that the activity of this enzyme may also influence the rate of the PPP flux.Citation29 For the other pathways that are enhanced in TAp73-overexpressing cells, it remains to be determined whether these pathways are directly controlled by TAp73, or indirectly due to the known effect of TAp73 on the PPP and mitochondrial respiration.
We previously found that p53 suppresses G6PD activity through a distinct mechanism involving protein–protein interactions.Citation23 This function is lost in tumor-associated p53 mutants. The prevalence of p53 mutations and TAp73 overexpression in many human cancer cells supports the notion that G6PD is tightly regulated in normal cells and the increasing flux through G6PD is essential for tumor growth. This notion is supported by the observation that G6PD is highly expressed in many human cancers (), and that blockage of PPP attenuates cell proliferation and tumor growth.Citation21,Citation26 The other major NADPH-generating enzymes include malic enzymes, which convert the tricarboxylic acid cycle (TCA) intermediate malate to the common TCA cycle substrate pyruvate, with the concomitant reduction of NADP+.Citation30,Citation31 Of note, p53 suppresses all 3 malic enzymes,Citation32 which are localized in either the cytoplasm or the mitochondria. These observations suggest that NADPH production may be a rate-limiting step and a focal point of regulation in cell proliferation. The NADPH-generating enzymes, including G6PD and malic enzymes, may be valuable targets for tumor therapy.
Materials and Methods
Cell culture and gene knockdown with siRNA
U2OS cells were maintained in McCoy 5A Medium, IMR90 cells in MEMα medium, and H1299 cells in RPMI1640 medium (Life Technologies). All mediums were supplemented with 10% fetal bovine serum (FBS). The following siRNAs were purchased from Invitrogen (Carlsbad) with sequences indicated: human G6PD, 5′-AAACCCACUC UCUUCAUCAGCUCGU-3′; human p73, 5′-GAGCUCGGGA GGGACUUCAA CGAAG-3′.
Semi-quantitative PCR with reverse transcription PCR
Total RNA was isolated from cells by TRIzol reagent (Invitrogen), and 2 µg RNA of each sample was reversed to cDNA by the First-Strand cDNA Synthesis System (Marligen Biosciences). cDNA (0.2 µg) of each sample was used as a template to perform PCR. The primer pairs for human genes were: G6PD, 5′- ATGGCAGAGC AGGTGGCCCT-3′ and 5′-TCATGCAGGA CTCGTGAATG-3′; β-actin, 5′-GACCTGACTGACTACCTCAT GAAGAT-3′ and 5′-GTCACACTTC ATGATGGAGTTGAAGG-3′; TAp73, 5′-ATGGCCCAGTCCACCGCCAC C-3′ and 5′-TCGAAGGTGG AGCTGGG-3′.
G6PD enzyme activity, NADPH, and ROS levels
G6PD enzyme activity was determined as described previously.Citation33 NADPH levels were determined using the NADPH Quantification Kit (BioVision). ROS levels were analyzed as described previously.Citation34 Briefly, cells were incubated at 37 °C for 30 min in phosphate buffered saline (PBS) containing 10 µM 2’,7’-dichlorodihydrofluorescein diacetate (H2-DCFDA). Cells were then washed twice with PBS, treated with trypsin, and re-suspended in PBS. Fluorescence signal was immediately measured using a FACScan Flow Cytometer (Becton Dickinson).
BrdU incorporation assay
H1299 cells were treated with p73 siRNA, G6PD siRNA, or control siRNA. Then cells were pulse-labeled with BrdU for 1 h and analyzed by anti-BrdU immunostaining as described previously.Citation35 In brief, BrdU-labeled cells were washed with PBS and fixed with 2% paraformaldehyde and 70% ethanol at 4 °C. Afterwards, cells were permeabilized with 0.2% Triton X-100 and followed with TBS wash. After treatment of 4N HCl, 0.2% Triton X-100, staining with anti-BrdU antibody and FITC-labeled secondary antibody, the images were acquired with an Olympus DP71X microscope (Olympus). The FITC signal was converted to a red color for better visualization.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgment
This work is supported in part by the grants from NIH (CA088868), the US Department of Defense (W81XWH-13-1-0100), and Pennsylvania Commonwealth Universal Research Enhancement Program (CURE).
Related Research Data
References
- Kaghad M, Bonnet H, Yang A, Creancier L, Biscan JC, Valent A, Minty A, Chalon P, Lelias JM, Dumont X, et al. Monoallelically expressed gene related to p53 at 1p36, a region frequently deleted in neuroblastoma and other human cancers. Cell 1997; 90:809 - 19; http://dx.doi.org/10.1016/S0092-8674(00)80540-1; PMID: 9288759
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:307 - 10; http://dx.doi.org/10.1038/35042675; PMID: 11099028
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 31; http://dx.doi.org/10.1016/j.cell.2009.04.037; PMID: 19410540
- Melino G, De Laurenzi V, Vousden KH. p73: Friend or foe in tumorigenesis. Nat Rev Cancer 2002; 2:605 - 15; http://dx.doi.org/10.1038/nrc861; PMID: 12154353
- Yang A, Kaghad M, Caput D, McKeon F. On the shoulders of giants: p63, p73 and the rise of p53. Trends Genet 2002; 18:90 - 5; http://dx.doi.org/10.1016/S0168-9525(02)02595-7; PMID: 11818141
- Moll UM, Slade N. p63 and p73: roles in development and tumor formation. Mol Cancer Res 2004; 2:371 - 86; PMID: 15280445
- Deyoung MP, Ellisen LW. p63 and p73 in human cancer: defining the network. Oncogene 2007; 26:5169 - 83; http://dx.doi.org/10.1038/sj.onc.1210337; PMID: 17334395
- Donehower LA, Harvey M, Slagle BL, McArthur MJ, Montgomery CA Jr., Butel JS, Bradley A. Mice deficient for p53 are developmentally normal but susceptible to spontaneous tumours. Nature 1992; 356:215 - 21; http://dx.doi.org/10.1038/356215a0; PMID: 1552940
- Jacks T, Remington L, Williams BO, Schmitt EM, Halachmi S, Bronson RT, Weinberg RA. Tumor spectrum analysis in p53-mutant mice. Curr Biol 1994; 4:1 - 7; http://dx.doi.org/10.1016/S0960-9822(00)00002-6; PMID: 7922305
- Yang A, Walker N, Bronson R, Kaghad M, Oosterwegel M, Bonnin J, Vagner C, Bonnet H, Dikkes P, Sharpe A, et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000; 404:99 - 103; http://dx.doi.org/10.1038/35003607; PMID: 10716451
- Tomasini R, Tsuchihara K, Wilhelm M, Fujitani M, Rufini A, Cheung CC, Khan F, Itie-Youten A, Wakeham A, Tsao MS, et al. TAp73 knockout shows genomic instability with infertility and tumor suppressor functions. Genes Dev 2008; 22:2677 - 91; http://dx.doi.org/10.1101/gad.1695308; PMID: 18805989
- Talos F, Nemajerova A, Flores ER, Petrenko O, Moll UM. p73 suppresses polyploidy and aneuploidy in the absence of functional p53. Mol Cell 2007; 27:647 - 59; http://dx.doi.org/10.1016/j.molcel.2007.06.036; PMID: 17707235
- Wilhelm MT, Rufini A, Wetzel MK, Tsuchihara K, Inoue S, Tomasini R, Itie-Youten A, Wakeham A, Arsenian-Henriksson M, Melino G, et al. Isoform-specific p73 knockout mice reveal a novel role for delta Np73 in the DNA damage response pathway. Genes Dev 2010; 24:549 - 60; http://dx.doi.org/10.1101/gad.1873910; PMID: 20194434
- Warburg O, Posener K, Negelein E. Ueber den Stoffwechsel der Tumoren. Biochem Z 1924; 152:319 - 44
- Warburg O. On the origin of cancer cells. Science 1956; 123:309 - 14; http://dx.doi.org/10.1126/science.123.3191.309; PMID: 13298683
- Luzzatto. L, Mehta. A, eds. Glucose-6-phoshate dehydrogenase deficiency. London: McGraw-Hill, 1995
- Pandolfi PP, Sonati F, Rivi R, Mason P, Grosveld F, Luzzatto L. Targeted disruption of the housekeeping gene encoding glucose 6-phosphate dehydrogenase (G6PD): G6PD is dispensable for pentose synthesis but essential for defense against oxidative stress. EMBO J 1995; 14:5209 - 15; PMID: 7489710
- Ursini MV, Parrella A, Rosa G, Salzano S, Martini G. Enhanced expression of glucose-6-phosphate dehydrogenase in human cells sustaining oxidative stress. Biochem J 1997; 323:801 - 6; PMID: 9169615
- Ferri P, Biagiotti E, Ambrogini P, Santi S, Del Grande P, Ninfali P. NADPH-consuming enzymes correlate with glucose-6-phosphate dehydrogenase in Purkinje cells: an immunohistochemical and enzyme histochemical study of the rat cerebellar cortex. Neurosci Res 2005; 51:185 - 97; http://dx.doi.org/10.1016/j.neures.2004.11.002; PMID: 15681036
- Anastasiou D, Poulogiannis G, Asara JM, Boxer MB, Jiang JK, Shen M, Bellinger G, Sasaki AT, Locasale JW, Auld DS, et al. Inhibition of pyruvate kinase M2 by reactive oxygen species contributes to cellular antioxidant responses. Science 2011; 334:1278 - 83; http://dx.doi.org/10.1126/science.1211485; PMID: 22052977
- Hitosugi T, Zhou L, Elf S, Fan J, Kang HB, Seo JH, Shan C, Dai Q, Zhang L, Xie J, et al. Phosphoglycerate mutase 1 coordinates glycolysis and biosynthesis to promote tumor growth. Cancer Cell 2012; 22:585 - 600; http://dx.doi.org/10.1016/j.ccr.2012.09.020; PMID: 23153533
- Cosentino C, Grieco D, Costanzo V. ATM activates the pentose phosphate pathway promoting antioxidant defence and DNA repair. EMBO J 2011; 30:546 - 55; http://dx.doi.org/10.1038/emboj.2010.330; PMID: 21157431
- Jiang P, Du W, Wang X, Mancuso A, Gao X, Wu M, Yang X. p53 regulates biosynthesis through direct inactivation of glucose-6-phosphate dehydrogenase. Nat Cell Biol 2011; 13:310 - 6; http://dx.doi.org/10.1038/ncb2172; PMID: 21336310
- Tian WN, Braunstein LD, Pang J, Stuhlmeier KM, Xi QC, Tian X, Stanton RC. Importance of glucose-6-phosphate dehydrogenase activity for cell growth. J Biol Chem 1998; 273:10609 - 17; http://dx.doi.org/10.1074/jbc.273.17.10609; PMID: 9553122
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, Brugge JS. Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 2009; 461:109 - 13; http://dx.doi.org/10.1038/nature08268; PMID: 19693011
- Du W, Jiang P, Mancuso A, Stonestrom A, Brewer MD, Minn AJ, Mak TW, Wu M, Yang X. TAp73 enhances the pentose phosphate pathway and supports cell proliferation. Nat Cell Biol 2013; 15:991 - 1000; http://dx.doi.org/10.1038/ncb2789; PMID: 23811687
- Rufini A, Niklison-Chirou MV, Inoue S, Tomasini R, Harris IS, Marino A, Federici M, Dinsdale D, Knight RA, Melino G, et al. TAp73 depletion accelerates aging through metabolic dysregulation. Genes Dev 2012; 26:2009 - 14; http://dx.doi.org/10.1101/gad.197640.112; PMID: 22987635
- Temple MD, Perrone GG, Dawes IW. Complex cellular responses to reactive oxygen species. Trends Cell Biol 2005; 15:319 - 26; http://dx.doi.org/10.1016/j.tcb.2005.04.003; PMID: 15953550
- D’Alessandro A, Marrocco C, Rinalducci S, Peschiaroli A, Timperio AM, Bongiorno-Borbone L, Finazzi Agrò A, Melino G, Zolla L. Analysis of TAp73-dependent signaling via omics technologies. J Proteome Res 2013; 12:4207 - 20; http://dx.doi.org/10.1021/pr4005508; PMID: 23919926
- Hsu RY. Pigeon liver malic enzyme. Mol Cell Biochem 1982; 43:3 - 26; http://dx.doi.org/10.1007/BF00229535; PMID: 7078548
- Chang GG, Tong L. Structure and function of malic enzymes, a new class of oxidative decarboxylases. Biochemistry 2003; 42:12721 - 33; http://dx.doi.org/10.1021/bi035251+; PMID: 14596586
- Jiang P, Du W, Mancuso A, Wellen KE, Yang X. Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 2013; 493:689 - 93; http://dx.doi.org/10.1038/nature11776; PMID: 23334421
- Leopold JA, Dam A, Maron BA, Scribner AW, Liao R, Handy DE, Stanton RC, Pitt B, Loscalzo J. Aldosterone impairs vascular reactivity by decreasing glucose-6-phosphate dehydrogenase activity. Nat Med 2007; 13:189 - 97; http://dx.doi.org/10.1038/nm1545; PMID: 17273168
- Cossarizza A, Ferraresi R, Troiano L, Roat E, Gibellini L, Bertoncelli L, Nasi M, Pinti M. Simultaneous analysis of reactive oxygen species and reduced glutathione content in living cells by polychromatic flow cytometry. Nat Protoc 2009; 4:1790 - 7; http://dx.doi.org/10.1038/nprot.2009.189; PMID: 20010930
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 501; http://dx.doi.org/10.1126/science.282.5393.1497; PMID: 9822382
- Compagno M, Lim WK, Grunn A, Nandula SV, Brahmachary M, Shen Q, Bertoni F, Ponzoni M, Scandurra M, Califano A, et al. Mutations of multiple genes cause deregulation of NF-kappaB in diffuse large B-cell lymphoma. Nature 2009; 459:717 - 21; http://dx.doi.org/10.1038/nature07968; PMID: 19412164
- Rosenwald A, Wright G, Chan WC, Connors JM, Campo E, Fisher RI, Gascoyne RD, Muller-Hermelink HK, Smeland EB, Giltnane JM, et al, Lymphoma/Leukemia Molecular Profiling Project. The use of molecular profiling to predict survival after chemotherapy for diffuse large-B-cell lymphoma. N Engl J Med 2002; 346:1937 - 47; http://dx.doi.org/10.1056/NEJMoa012914; PMID: 12075054
- Quade BJ, Wang TY, Sornberger K, Dal Cin P, Mutter GL, Morton CC. Molecular pathogenesis of uterine smooth muscle tumors from transcriptional profiling. Genes Chromosomes Cancer 2004; 40:97 - 108; http://dx.doi.org/10.1002/gcc.20018; PMID: 15101043
- Su LJ, Chang CW, Wu YC, Chen KC, Lin CJ, Liang SC, Lin CH, Whang-Peng J, Hsu SL, Chen CH, et al. Selection of DDX5 as a novel internal control for Q-RT-PCR from microarray data using a block bootstrap re-sampling scheme. BMC Genomics 2007; 8:140; http://dx.doi.org/10.1186/1471-2164-8-140; PMID: 17540040
- Stearman RS, Dwyer-Nield L, Zerbe L, Blaine SA, Chan Z, Bunn PA Jr., Johnson GL, Hirsch FR, Merrick DT, Franklin WA, et al. Analysis of orthologous gene expression between human pulmonary adenocarcinoma and a carcinogen-induced murine model. Am J Pathol 2005; 167:1763 - 75; http://dx.doi.org/10.1016/S0002-9440(10)61257-6; PMID: 16314486