
Abstract
Adiponectin, the most abundant protein secreted by adipose tissue, exhibits insulin-sensitizing, anti-inflammatory, antiatherogenic, and antiproliferative properties. In addition, it appears to play an important role also in the development and progression of several obesity-related malignancies, including breast cancer.
Here, we demonstrated that adiponectin induces a dichotomic effect on breast cancer growth. Indeed, it stimulates growth in ERα+ MCF-7 cells while inhibiting proliferation of ERα− MDA-MB-231 cells. Notably, only in MCF-7 cells adiponectin exposure exerts a rapid activation of MAPK phosphorylation, which is markedly reduced when knockdown of the ERα gene occurred. In addition, adiponectin induces rapid IGF-IR phosphorylation in MCF-7 cells, and the use of ERα siRNA prevents this effect. Moreover, MAPK activation induced by adiponectin was reversed by IGF-IR siRNA. Coimmunoprecipitation studies show the existence of a multiprotein complex involving AdipoR1, APPL1, ERα, IGF-IR, and c-Src that is responsible for MAPK signaling activation in ERα+ positive breast cancer cells. It is well known that in addition to the rapid effects through non-genomic mechanisms, ERα also mediates nuclear genomic actions. In this concern, we demonstrated that adiponectin is able to transactivate ERα in MCF-7 cells. We showed the classical features of ERα transactivation: nuclear localization, downregulation of mRNA and protein levels, and upregulation of estrogen-dependent genes. Thus, our study clarifies the molecular mechanism through which adiponectin modulates breast cancer cell growth, providing evidences on the cell-type dependency of adiponectin action in relationship to ERα status.
Introduction
Obesity is associated with an increase in white adipose tissue that greatly alters the local and systemic secretion of biologically active polypeptides, known as adipocytokines.Citation1 Currently more than 50 different adipocytokines have been identified, which mainly regulate energy metabolism, but also have several pathophysiological functions.Citation2 Of these, adiponectin is a 244 amino acid-long polypeptide that represents the most abundant adipose tissue-derived hormone. It has a protective role against obesity-related disorders, including metabolic syndrome, type-2 diabetes, and cardiovascular disease.Citation3 Unlike most of the other adipokines, serum adiponectin is inversely correlated with body mass index (BMI).Citation4,Citation5 Circulating adiponectin levels are reduced in obesity and type 2 diabetes,Citation6 and mice lacking adiponectin develop insulin resistance, glucose intolerance, hyperglycemia, and hypertension, all characteristics of metabolic syndrome.Citation7,Citation8 A speculative explanation of the reduced adiponectin levels in obesity may be sustained by the enhanced production of cytokines that occurred in such condition, and may contribute to the downregulatory effect on adiponectin secretion by adipose tissue.Citation9 Another potential mechanism indicates a negative feedback of adiponectin on its own production and probably on the expression of its receptors during the development of obesity.Citation10 Adiponectin is synthesized as a single subunit that undergoes oligomerization to form trimers, hexamers, and multimers before secretion. In serum, adiponectin exists in its full-length form (fAdiponectin) or as a proteolytic fragment that corresponds to the globular domain of the protein (gAdiponectin) with enhanced potency.Citation11 The cellular functions of adiponectin are mediated through the adiponectin receptor 1 (AdipoR1) and 2 (AdipoR2) and T-cadherin.Citation3 AdipoR1 and AdipoR2 are integral membrane proteins containing 7 transmembrane domains but are structurally and functionally distinct from G protein-coupled receptors.Citation12 AdipoR1 presents high affinity for gAdiponectin and low affinity for fAdiponectin, and it is expressed ubiquitously but abundantly in skeletal muscle and endothelial cells. AdipoR2 has intermediate affinity for both forms of adiponectin and is predominantly expressed in the liver.Citation13 It has been demonstrated that the globular form of adiponectin binds to AdipoR1,Citation12 which, in turn, through the adaptor protein APPL1 interacting with the intracellular N terminus of AdipoR1, induces activation of MAPK through Src pathway.Citation14,Citation15 APPL1, which contains a pleckstrin homology domain, a phosphotyrosine-binding domain, and a leucin zipper motif, has emerged as an important element in AdipoR1/R2 signaling.Citation14
Recent experimental and clinical investigations suggested that low levels of plasma adiponectin are associated with an increased risk for obesity-related cancers, such as colon, prostate, endometrial, and breast cancer.Citation10,Citation16,Citation17 The pathogenesis of breast cancer is largely dependent on interactions between malignant cells and components of the breast microenvironment. The malignant cell phenotype is regulated not only by autonomous signals originating from cancer cells, but also by the effects of the surrounding stromal cells, which influence mammary epithelial cell growth and differentiation.Citation2 Most important, some studies have suggested that breast tumors arising in patients with hypoadiponectinemia may have a more aggressive phenotype (large tumor size, high histological grade, estrogen receptor negativity, and increased metastasis).Citation17,Citation18 Particularly, in the mammary gland, epithelial cells are exposed to both circulating and locally produced adiponectin from adjacent adipocytes. The close association between mammary epithelial cells and adipocytes may favor a more direct action of adipokines on that tissue.Citation19 Recently it has been reported that low circulating adiponectin levels are associated with a higher risk of breast cancer development, and that this association is independent of age, BMI, and hormonal and reproductive factors.Citation19 Consistent with these evidences, it has been demonstrated that adiponectin mediates an antiproliferative effect in human ERα-negative MDA-MB-231 breast cancer cells through the regulation of genes involved in cell cycle, such as p53, Bax, Bcl-2, c-myc, and cyclin D1.Citation20-Citation22 On the other hand, conflicting observations have been reported on the effects elicited by adiponectin in ERα-positive MCF-7 cells. For instance, some authors evidenced significant anti-proliferative effects;Citation19,Citation22-Citation24 other studies didn’t show any noticeable effect;Citation20,Citation25 while some other studies demonstrated a stimulatory effect on the growth of this cell line.Citation26,Citation27
Thus, stemming from these controversial data, the aim of the present study was to investigate whether the ERα expression influences the response to adiponectin in breast cancer growth.
Our data demonstrated that in ERα-positive breast cancer cells, adiponectin is able to activate ERα at both non-genomic and genomic level, stimulating breast cancer growth.
Results
Adiponectin induces divergent effects on breast cancer cell proliferation
We first investigated the effect of adiponectin (1 and 5 µg/ml) on cell proliferation in both estrogen receptor α-negative (ERα−) as well as positive (ERα+) breast cancer cell lines, by MTT growth assays. After 3 d treatment, adiponectin inhibited cell proliferation of ERα− MDA-MB-231 and SKBR-3 cells, whereas it induced growth in MCF-7 and T47D cells (), which express high levels of ERα.Citation28
Figure 1. Effects of adiponectin on breast cancer cell growth. (A) MTT growth assays in MCF-7, T47D, MDA-MB-231, and SKBR-3 cells treated with vehicle (C) or adiponectin 1 (A1) or 5 μg/ml (A5) for 72 h. Cell proliferation is expressed as fold change ± SD relative to vehicle-treated cells and is representative of 3 different experiments each performed in triplicate. (B) Soft agar growth assay in MCF-7 and MDA-MB-231 cells plated in 0.35% agarose and treated as indicated above. After 14 d of growth colonies >50 µm diameter were counted. (C) MCF-7 3-dimensional cultures were untreated or treated as indicated for 48 h and then photographed under phase-contrast microscopy. (D) Cell numbers obtained from 3-dimensional spheroids in MCF-7 cells treated as indicated for 48 h. (E) MCF-7 cells exposed to ICI 182 780, or transfected in suspension with 30 nM siRNAs/well (ERα siRNA or a scrambled siRNA for control samples), were treated with vehicle (C) or A1 or A5 for 72 h before testing cell viability using MTT assay. Results are expressed as fold change ± SD relative to vehicle-treated cells and are representative of 3 different experiments, each performed in triplicate. *P < 0.05 compared with vehicle.
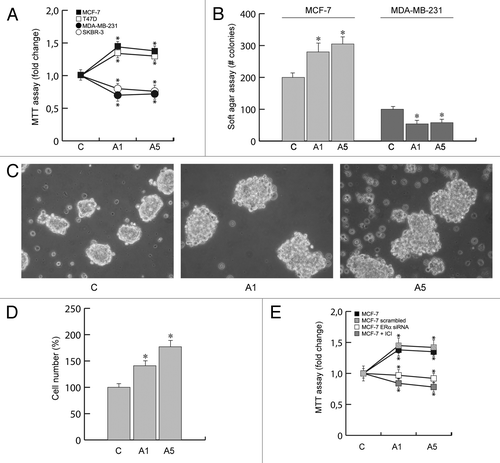
The same dichotomic pattern has been reproduced in anchorage-independent growth assays, using both ERα+ and ERα− breast cancer cells ().
We then performed 3-dimensional MCF-7 cell cultures, which closely mimic some in vivo biologic features of tumors.Citation29 Our results demonstrated that adiponectin treatment enhanced cell–cell adhesion () as well as increased cell growth () compared with untreated cells.
In order to investigate the role of ERα in modulating the effect of adiponectin on cell proliferation, in MCF-7 cells ERα was knocked down by siRNA or abrogated by the potent and specific antiestrogen ICI 182 780. In these conditions the adiponectin-induced cell proliferation was completely reversed ().
These results address how adiponectin may affect breast cancer growth through the involvement of ERα.
Effect of adiponectin on ERα non-genomic signal
The biological actions of ERα are mediated by non-genomic action outside of the nuclear compartment and by genomic effects via nuclear ERs.Citation30 The non-genomic effects lead to the rapid activation of the MAPK signaling pathway. Both the genomic and non-genomic actions of ERα play pivotal roles in ERα-induced cancer cell proliferation and survival.
Thus, we investigated the possible cross-talk existing between membrane ERα transductional pathways and adiponectin receptor, since both signaling appear to converge to MAPK cascade. Indeed, it has been demonstrated that AdipoR1 binds to the adaptor protein APPL1Citation14 and induces activation of MAPK through Src pathway.Citation15 For instance, c-Src is also an initial and integral crossroad of different membrane signaling events mediated by the ERα as well as by its cross-talk with growth factors. Thus, we found it reasonable to verify whether AdipoR1/APPL1 may interact with other membrane signaling involved in breast cancer cell growth and progression. We demonstrated for the first time how, in basal conditions, APPL1 coimmunoprecipitated with AdipoR1, ERα, IGF-IR, and c-Src, but not with EGF-R at both 15 min () and 48 h (). These interactions appeared enhanced by adiponectin exposure ().
Figure 2. Adiponectin induces MAPK activation through the formation of a multimeric protein complex. Five hundred micrograms of protein lysates from MCF-7 cells, untreated (C) or treated with A1 or A5 for 15 min (A) or 48 h (B), were immunoprecipitated with an APPL1 antibody and then blotted with the indicated antibodies on the right. To verify equal loading, the membrane was probed with anti-APPL1 antibody. Normal rabbit IgG was used as a negative control to precipitate A5-treated samples. Numbers on top of the blots represent the average fold change vs. untreated cells. One of 3 similar experiments is presented. (C) MCF-7 cells, treated with adiponectin for 15 min, were lysed and immunoprecipitated with an anti-APPL1 antibody/protein A/G complex and assayed for c-Src-kinase activity using acid-treated enolase as described in “Materials and Methods”. These results are representative of 3 independent experiments. c-Src and enolase position is indicated. (D) pIGF-IRTyr1131 levels in MCF-7 cells treated with vehicle (−) or A1 and A5 as reported in absence or presence of ERα siRNA. (E) Total cellular proteins were isolated from MCF-7 cells in the absence or presence of ERα, IGF-IR, or APPL1 siRNA or pre-treated with PP2 or H89 and treated with A1 and A5. pMAPK, levels were evaluated by immunoblotting. Total MAPK was used as a loading control. (F) ERα, IGF-IR, and APPL1 levels were shown as control of silencing. Western blotting showed are representative of 3 different experiments.
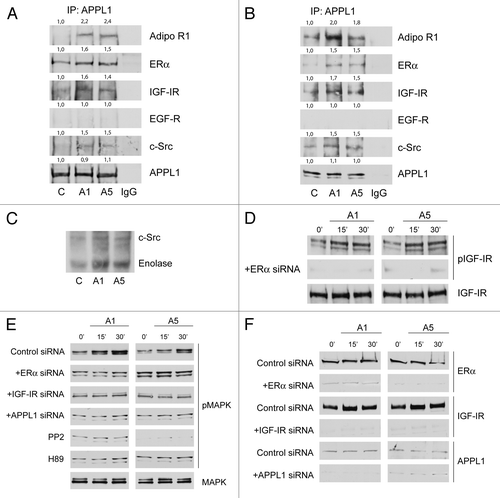
shows the in vitro effect of the protein complex on Src kinase activity. MCF-7 cell lysates were immunoprecipitated with an anti-APPL1 antibody, and the Src kinase activity was measured using the exogenous acid-treated enolase as substrate in the absence and in the presence of adiponectin after 15 min of exposure. The results provide evidence that adiponectin activates c-Src, as evidenced by both the autophosphorylation of c-Src and the concomitant phosphorylation of enolase. Furthermore, we observed an increase of IGF-IR phosphorylation after 15 and 30 min of adiponectin treatment, which was reduced after knockdown of ERα ().
In MCF-7 cells we observed a rapid activation of MAPK by adiponectin (). To determine the mechanism through which AdipoR1 stimulates MAPK, we employed a panel of RNA silencers targeting ERα, IGF-IR, and APPL1 (), or selective inhibitor targeting Src family kinase or PKA. We observed that MAPK activity was abrogated in the presence of all these agents (), suggesting that MAPK cascade upon adiponectin exposure requires APPL1, ERα, IGF-IR, c-Src, and PKA activity.
Finally, in the presence of APPL1 silencer, AdipoR1 was no longer associated with ERα, IGF-IR, and c-Src (data not shown).
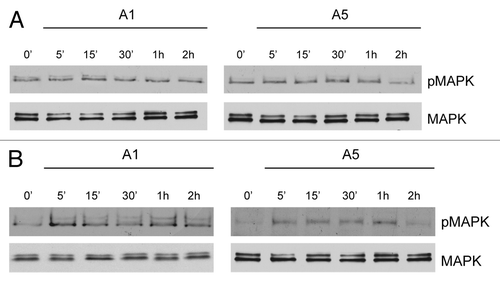
The role of ERα membrane signal in the above reported membrane complex emerges from the evidence that in ERα− MDA-MB-231 cells, adiponectin was no longer able to induce MAPK phosphorylation (). However, when these cells were transfected with a membrane ERα (mERα) construct, which consists solely of the AF-2/ligand binding domain of ERα, adiponectin short exposure was able to upregulate MAPK activity ().
Figure 3. Time course of MAPK activation upon adiponectin exposure in MDA-MB-231 cells ectopically expressing membrane ERα. MDA-MB-231 (A) and MDA-MB-231 transfected with a plasmid codifying for membrane ERα (B) were serum-starved for 24 h followed by treatment with adiponectin 1 or 5 μg/ml for the indicated times. Western blots show the phosphorylation status of MAPK. Total MAPK is used as a loading control. One of 3 similar experiments is presented
Adiponectin transactivates ERα through MAPK signaling
Several studies in recent years suggest that membrane-initiated signaling can converge into genomic events, leading to more long-term consequences. For instance, in MCF-7 breast cancer cells adiponectin revealed an induced MAPK activation still after 48 h treatment (). Since MAPK is involved in enhancing ERα functional activation in a ligand-independent manner, we wondered whether adiponectin was able to modulate ERα transactivation.
Figure 4. Effects of adiponectin on ERα transactivation. (A) Total cellular proteins were isolated from MCF-7 cells treated with A1 and A5 for 48 h. pMAPK, levels were evaluated by immunoblotting. Total MAPK was used as a loading control. (B) MCF-7 cells were transfected with the luciferase reporter plasmid XETL. HeLa cells were cotransfected with XETL and HEGO plasmids. The cells were untreated or treated for 48 h with A1 and A5 or 100 nM E2, used as positive control. (C) MCF-7 cells were transfected with the luciferase reporter plasmid XETL. The cells were untreated or treated for 48 h with A1 and A5 or in combination with Compound C (CC), H89, PP2, or PD98059. *P < 0.05 compared with control (−); ●P < 0.05 compared with A1; ○P < 0.05 compared with A5. (D) MCF-7 cells were transfected with XETL plasmid in the absence or presence of IGF-IR siRNA and treated with adiponectin for 48 h. *P < 0.05 compared with control (−). The values represent the means ± SD of 3 different experiments. In each experiment, the activities of the transfected plasmids were assayed in triplicate transfections.
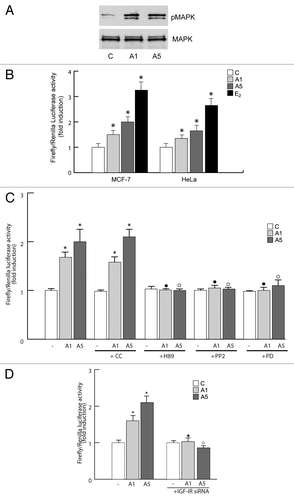
In MCF-7 cells exposed to adiponectin for 48 h, a significant activation of the estrogen-responsive gene XETL was observed (). Similar results were reproduced in ER-negative HeLa cells ectopically expressing ERα and transfected with XETL (). It is worth noting that the specific anti-estrogen ICI 182 780 efficiently antagonized the stimulatory effect of adiponectin on ERα transactivation in both cell types (data not shown).
In the presence of the MAPK inhibitor PD98059, the direct capability of adiponectin in transactivating ERα was completely lost. When we tested different inhibitors of signaling converging on MAPK activation, we observed how PKA and c-Src inhibitors, H89 and PP2, respectively, were able to prevent adiponectin-induced ERα transactivation (). Moreover, knocking down of IGF-IR exerted similar effects (). These results address MAPK signaling as crucial in modulating ERα transactivation upon adiponectin exposure.
In addition, we also studied which functional domain was involved in ERα transactivation by adiponectin. To this aim, HeLa cells were cotransfected with the XETL reporter gene and plasmids codifying for AF-1 or AF-2 domain (). The treatment with adiponectin induced an increased transcriptional activation only in transfected cells bearing the plasmid codifying for AF-1 domain (). These data demonstrate that the N-terminal AF-1 domain is essential in mediating the adiponectin response. Moreover, the activation of ERα occurred at the genomic level, as demonstrated by trasfecting HeLa cells with the plasmid HE241G, encoding ERα lacking the nuclear localization sequence ().
Figure 5. Effects of adiponectin on ERα functional domains. (A and C) Schematic illustration of ER constructs used for the experiments. HEGO is a wild-type ER-expressing vector that encodes a 595-amino acid protein. HE15-(1–282) contains AF-1 and the DNA binding domain (DBD). HE19-(179–95) contains DBD, and AF-2 domains. HE241G encodes a mutated ERα, which has the nuclear localization sequence deleted (250–303). ER plasmids mutated in serine residues 104, 106, 118, and 167 to Ala. (B) HeLa cells were transiently cotransfected with XETL and either HEGO or PSG5/HE15 (AF-1) or PSG5/HE19 (AF-2) or HE241G plasmids. (D) HeLa cells were transiently cotransfected with XETL and either HEGO or Ser-104/106/118A-ER or Ser-118A-ER or Ser-167A-ER. The cells were treated for 48 h in the absence (C) or in the presence of A1 and A5 or E2 (100 nM) used as positive control. The values represent the means ± SD of 3 different experiments. In each experiment, the activities of the transfected plasmids were assayed in triplicate transfections. *P < 0.05, **P < 0.01 compared with (C). (E) Total extracts from cells treated with A1 or A5 for 30 min were analyzed for phosphorylation of serines 118 and 167 (pS118 and pS167) and expression of ERα by immunoblot analysis. GAPDH was used as a control for equal loading and transfer.
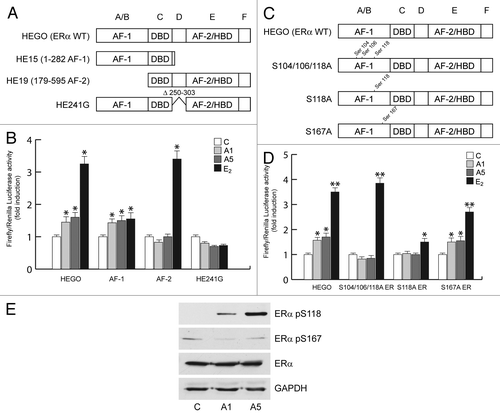
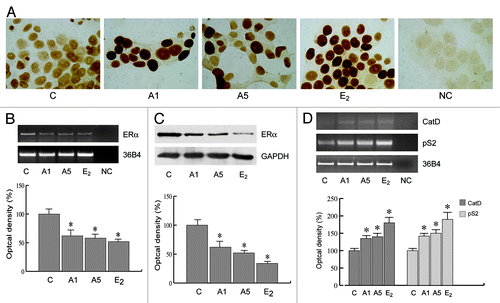
The involvement of AF-1 domain in the activation of ERα by adiponectin was confirmed cotransfecting HeLa cells with XETL and with either HEGO or different plasmids in which ERα was mutated in specific phosphorylation sites, such as Ser-104/106/118A-ER or S118A-ER or S167A-ER (), which are effectors of phosphorylative signaling. As shown in , in transfectants with either Ser-104/106/118A-ER or S118 mutants, adiponectin was no longer able to elicit any substantial activation on ERE luciferase signal as compared with the cells bearing wild-type ERα. In contrast, the activation still persisted in cells transfected with S167 mutant (). Western blotting analysis confirmed how adiponectin enhanced the phosphorylation of ERα at Ser118, while it was unable to affect activation at Ser167 (). The biological correlates of these events reproduce the classic features of ERα transactivation in breast cancer cells. In MCF-7 cells cultured in serum deprivation conditions for 96 h, ERα immunoreactivity was no longer detectable in the control, whereas treatment with adiponectin for 24 h induced a strong ERα immunoreactivity in the nuclear compartment (), which was abrogated in the presence of MAPK inhibitor PD98059 (data not shown). These results show that adiponectin mimics the effect of estradiol on ERα compartmentalization in breast cancer cells. In addition, in MCF-7 cells, we observed a significant downregulation of ERα mRNA and protein levels after adiponectin treatment (), which is a typical hallmark of the receptor activation by an agonist. Moreover, adiponectin was able to increase the expression of classic estrogen-dependent genes, such as Catepsin D and pS2 (), providing further evidence for the ability of this adipokine to activate ERα.
Figure 6. Adiponectin actions on cellular localization and expression of ERα. (A) MCF-7 cells were incubated in serum-free medium for 96 h and then treated with vehicle (C), A1, A5, or 100 nM E2, used as positive control, for 24 h. No immunodetection was observed replacing the anti-ER antibody with an irrelevant mouse IgG (NC). Each experiment is representative of at least 10 tests. (B) RT-PCR of ERα mRNA. MCF-7 cells were stimulated for 48 h with A1 and A5 or E2 (100 nM); 36B4 mRNA levels were determined as a control. (C) Immunoblot of ERα from MCF-7 cells treated as above; GAPDH serves as loading control. (D) RT-PCR of Catepsin D (CatD) and pS2 mRNA. MCF-7 cells were treated as reported. The histograms represent the mean ± SD of 3 separate experiments, in which band intensities were evaluated in terms of optical density arbitrary units and expressed as the percentage of the control assumed as 100%. *P < 0.05 vs. control.
Evidence for the contribution of the multiprotein complex in adiponectin-induced MCF-7 cell growth
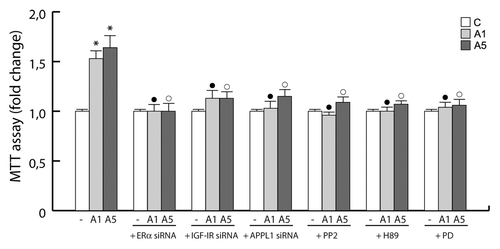
Finally, we evaluated the involvement of the multiprotein complex in mediating adiponectin-induced MCF-7 cell growth. When we knocked down the components of the multiprotein complex (ERα, IGF-IR, and APPL1) or inhibited its signaling cascade, through PP2, H89, and PD98059 (inhibitors of c-Src, PKA, and MAPK, respectively), the stimulatory role of adiponectin on MCF-7 cell growth was completely abrogated ().
Figure 7. Involvement of the protein complex in the adiponectin-induced MCF-7 cell growth. MTT growth assays in MCF-7 cells untreated or treated for 72 h with A1 and A5 in the absence or presence of ERα, IGF-IR, or APPL1 siRNA, or in combination with PP2, H89, or PD98059 inhibitor of c-Src, PKA, and MAPK, respectively. Cell proliferation is expressed as fold change ± SD relative to vehicle treated cells, and is representative of 3 different experiments each performed in triplicate. *P < 0.05 compared with control (−); ●P < 0.05 compared with A1; ○P < 0.05 compared with A5.
Discussion
Accumulating evidences have addressed how adipose tissue can act as an effector organ that influences breast cancer risk, tumor behavior, and clinical outcome.Citation31
The main underlying mechanisms that link obesity to breast cancer development and progression include: (1) abnormalities of insulin resistance and the IGF-I system deregulation; (2) the impact of adiposity on the biosynthesis and bioavailability of endogenous sex hormones; (3) obesity-induced low-grade chronic systemic inflammation; and (4) alterations in the levels of adipocyte-derived factors.Citation32 Indeed, adipose tissue is now widely considered to be an active endocrine organ, secreting several bioactive adipokines, including adiponectin, that exert distinct metabolic functions.Citation33 Adiponectin plays a protective function against obesity-related disorders and the metabolic syndrome, particularly in the pathogenesis of type II diabetes and cardiovascular disease.Citation34 Beyond these metabolic effects, adiponectin deficiency, commonly observed in obesity, may contribute to the natural history of several malignancies, such as breast cancer.Citation18 In particular, low adiponectin levels have been strongly associated with an increased breast cancer risk, and patients with reduced adiponectin levels develop a biologically aggressive phenotype independently of estrogen receptor status,Citation16 although inconclusive results have been published in the latter concern. For instance, several data point toward cell line-dependent effects.Citation35 Adiponectin has been shown to inhibit the growth of ERα− normal MCF-10A human mammary epithelial cells,Citation25 as well as cancerous MDA-MB-231Citation20,Citation27,Citation36 and SKBR-3Citation22 cells. This anti-proliferative effect involves an enhanced expression of Bax, Bcl-2, p53, c-myc, and reduced level of cyclin D1.Citation37
However, in ERα+ MCF-7 and T47D cells, adiponectin appears to stimulateCitation26,Citation27 or to inhibit cell growth,Citation19,Citation22-Citation24 or to play no noticeable effect.Citation20,Citation25 Possible explanations of the different cellular responses may be different content of ERα, differences in culture conditions, specific adiponectin isoform used, incubation time, or dosage.
Here, we elucidated the complex mechanisms involved in adiponectin response in breast cancer growth in dependency on ERα status. Particularly, we demonstrated that MAPK activation, induced by adiponectin/ERα-mediated effect, produces MCF-7 cell proliferation, and it represents the discriminator factor determining the opposite effect induced by adiponectin in ERα+ and ERα− breast cancer cells.
For instance, adiponectin inhibits the growth of ERα− breast cancer cells, whereas it induces proliferation in ERα+ cells. The evidence that this dichotomic effect is dependent on the expression of ERα emerges from the observation that in the presence of its knocking down, adiponectin induced inhibition of MCF-7 cell growth. Thus, we asked through which molecular mechanisms ERα may interfere with adiponectin effect on breast cancer cell growth.
It has been described that a fraction of ERα is extranuclear and exists as cytoplasmatic/membrane-bound population that mediates rapid “non-genomic” events, leading to cell morphologic changes and inhibition of apoptosis.Citation30,Citation38 According to this function, cytoplasmatic ERα can physically interact with different signaling molecules, including IGF-IRCitation38 and c-Src.Citation39 Recent findings demonstrated that AdipoR1, through its most important adaptor protein APPL1, activated c-Src/MAPK pathway as specific effector of adiponectin signaling.Citation15
Thus, APPL1 appears to be a key intracellular mediator of most of adiponectin responses reported to date. Activation of MAPK cascade is essential for cell cycle initiation and plays a key role in the control of cell proliferation, differentiation, and survival,Citation40 hypothesizing that adiponectin might act as a growth factor. MAPK serves as a point of convergence for diverse membrane signal inputs. All this has given the rationale to ascertain the existence of a mechanistic link existing between AdipoR1/APPL1 and other ERα driven membrane signaling. To date, nearly 14 proteins have been reported to associate with APPL1 in various types of cells, and they could be categorized into 3 different groups: membrane receptors, signaling proteins, and others.Citation41 On the basis of coimmunoprecipitation assays, we demonstrated that AdipoR1 and APPL1 interact with ERα, c-Src, and IGF-IR, all converging into MAPK activation. Indeed, the latter event was completely abrogated in the presence of ERα, IGF-IR, and APPL1 siRNA or pharmacological inhibitor of c-Src, PP2. The role of c-Src in maintaining protein complex formation is sustained by evidence demonstrating that, in MCF-7 cells, it allows ERα interaction with IGF-IR, exhibiting a docking site for ERα membrane association.Citation38,Citation39 Moreover, our results demonstrated that short exposure to adiponectin was able to rapidly enhance tyrosine phosphorylation of IGF-IR, which was no longer noticeable in the presence of ERα siRNA. On the basis of these findings, we hypothesize that adiponectin induces a linear pathway involving AdipoR1, ERα/IGF-IR/c-Src/MAPK.
The role of ERα in mediating the adiponectin-induced activation of MAPK in breast cancer cells emerges from the evidence that ectopical expression of membrane ERα in MDA-MB-231 cells was able to restore MAPK phosphorylation by adiponectin. This addresses how the presence of rapid ERα membrane signal is a prerequisite for adiponectin-induced MAPK activation.
In addition, membrane ERα, driving membrane signaling, may, in the long-term, induce a cascade of events converging in MAPK activation, which, in turn, transactivates ERα at genomic level.Citation30 In our study, we have provided the first evidence that adiponectin induces transactivation of ERα in MCF-7 cells. Indeed, it is worth observing that this occurred according to the classical features of ERα transactivation: nuclear localization, downregulation of mRNA and protein levels, and upregulation of estrogen-dependent genes.
Adiponectin was able to transactivate ERα in a ligand-independent manner, since only the AF-1 domain is essential in mediating the specific response through the phosphorylation of Ser118. Serine118 in the N terminus of human ERα is a well-studied phosphorylation site, and both rapid estrogens action and growth factors such as EGF and IGF-I, all converging in MAPK activation, result in phosphorylation of Ser118.Citation42 Indeed, our results demonstrated that upon adiponectin exposure, ERα transactivation was abrogated in the presence of MAPK inhibitor PD98059. Also, PKA appears to play a role in the latter concern, since its inhibition abolishes adiponectin-stimulated MAPK activation.
In our model, we demonstrated that adiponectin via PKA signal influences MAPK pathway, thus inducing ERα transactivation. Indeed, both adiponectin-induced MAPK phosphorylation and ERα transactivation are completely abrogated in the presence of a specific PKA inhibitor, H89.
However, our results showed that adiponectin action on ERα transactivation was independent by phosphorylation of Ser167, targeted by PKA signaling, while it failed to induce ERα signaling in cells ectopically expressing ERα mutated in Ser118, usually phosphorylated by MAPK.
All these observations lead to the idea that adiponectin effects on ERα transactivation are based mostly on MAPK signaling, which is dependent on the formation of a multiprotein complex including AdipoR1/APPL1/c-Src/ ERα/IGF-IR. On the other hand, the activation of MAPK by PKA may occur through c-Src/Rap1/B-Raf cascade.Citation43 Thus the adiponectin/AdipoR1/ERα axis, through c-Src, may converge on MAPK activation.
Adiponectin-induced transactivation of ERα appears not to involve another important effector signaling, such as AMPK, since it is not substantially affected by its pharmacological inhibition.
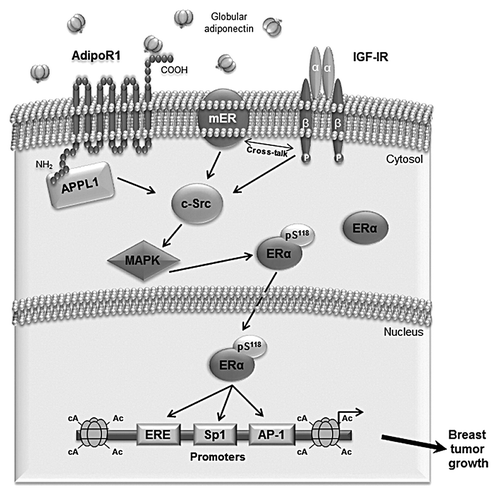
In conclusion, we provided evidence that the effects of adiponectin on breast cancer cell proliferation are dependent on ERα expression through both its non-genomic and genomic actions ().
Figure 8. Proposed model of adiponectin-induced proliferation in ERα-positive breast cancer cells. Globular adiponectin binds to AdipoR1, which interacts with the adaptor protein APPL1, through the intracellular N terminus. Adiponectin induces physical interaction between AdipoR1/APPL1, membrane ERα, IGF-IR, and c-Src, leading to MAPK phosphorylation. This contributes to ERα activation at genomic level through the phosphorylation at Ser118. Consequently, adiponectin, increasing ERα transactivation, positively affects MCF-7 cell growth.
Thus, our results suggest that ERα negatively interferes with the antiproliferative effect induced by adiponectin on breast cancer cell growth.
On the basis of these observations, it is rational to conclude that at the doses tested, adiponectin exerts an antiproliferative role only in ERα-negative breast cancer cells, wherein it may represent a promising pharmacological tool to be implemented in the novel adjuvant strategies adopted in breast cancer.
Materials and Methods
Cell culture
Cells utilized in the studies were obtained from American Type Culture Collection, where they were authenticated, stored according to supplier’s instructions, and used within 4 mo after frozen aliquots resuscitations. Every 4 mo, cells were authenticated by single tandem repeat analysis at our Sequencing Core; morphology, doubling times, estrogen sensitivity, and mycoplasma negativity were tested (MycoAlert, Lonza). MCF-7, T47D, and HeLa cells were cultured as described.Citation44 MDA-MB-231 were maintained in DMEM/F-12 containing 5% fetal bovine serum (Sigma). SKBR-3 breast cancer cells were cultured in RPMI 1640 without phenol red supplemented with 10% fetal bovine serum (FBS). Before each experiment, cells were grown in phenol red-free media, for at least 24 h and then treated with 1 and 5 µg/ml recombinant human gAdiponectin/gAcrp30 (R&D Systems) as described.
Cell proliferation assays
MTT assays
After 3 d of treatment, cell proliferation was assessed by MTT assay as reportedCitation45 and expressed as fold change relative to vehicle-treated cells.
Soft agar growth assays
Anchorage-independent growth assays were conducted as previously described.Citation46 Data represent 3 independent experiments performed in triplicate.
Three-dimensional spheroid culture and cell growth assays
For 3-dimensional cultures, MCF-7 cells plated on 2% agar-coated plates were untreated or treated with adiponectin 1 and 5 μg/ml. After 48 h, 3-dimensional cultures were photographed using a phase-contrast microscope (Olympus), and the cell numbers were evaluated as previously reported.Citation47
RNA silencing
MCF-7 cells were transfected with RNA duplex of stealth siRNA targeted for the human ERα (SI02781401), IGF-IR (SI01074017), or APPL1 (GS26060) mRNA sequence or with a control siRNA (Qiagen, Milan, Italy) that does not match with any human mRNA, used as a control for non-sequence-specific effects. Cells were transfected using RNAiFect Transfection Reagent (Qiagen) as recommended by the manufacturer with minor modifications.Citation48 After 5 h the transfection medium was changed with serum-free medium, and then the cells were exposed to treatments.
Western blotting and immunoprecipitation
Cells were lysed in ice-cold lysis buffer containing 50 mM HEPES (pH 7.5), 150 mM NaCl, 1.5 mM MgCl2, 1 mM EGTA, 10% glycerol, 1% Triton X-100, and a mixture of protease inhibitors (aprotinin, PMSF, and sodium orthovanadate). The protein content was determined using Bradford dye reagent (Bio-Rad). Equal amounts of total protein were resolved on SDS–polyacrilamide gels and transferred onto a nitrocellulose membrane as described.Citation49 Blots were incubated overnight at 4 °C and probed with the specific primary antibodies. Immunoblotting was performed as reported.Citation50 For immunoprecipitation, 500 µg of total protein lysates were precleared for 1 h with protein A/G-agarose (Santa Cruz Biotechnology), incubated with primary Abs at 4 °C for 18 h in HNTG buffer (20 mM HEPES, pH 7.5, 150 mM NaCl, 0.1% Triton X-100, 10% glycerol, and 0.1 mM Na3VO4), and then the antigen-Ab complexes were precipitated with protein A/G agarose for 2 h in HNTG buffer. The immunoprecipitated proteins were washed 3 times with HNTG buffer, separated on SDS-PAGE, and processed by western blotting. The following antibodies were used: AdipoR1, ERα, c-Src, APPL1, EGFR, and GAPDH from Santa Cruz Biotechnology; pERαSer118, pERαSer167, total MAPK and phosphorylated p42/44 MAPK(Thr202/Tyr204), IGF-IR, and pIGF-IRTyr1131 from Cell Signaling Technology.
Kinase activity of c-Src
To assay for c-Src kinase activity MCF-7 cells were grown in PRF-SFM for 24 h and stimulated with 1 or 5 µg/ml adiponectin for 15 min. Cells were then lysed with RIPA lysis buffer (500 mM TRIS-HCl, 150 mM NaCl, 1% Triton X-100) containing 10 mM PMSF, 1.5 mg/ml aprotinin, and 2 mg/ml leupeptin and immunoprecipitated. A Protein G–agarose and an anti-APPL1 antibody complex were prepared to immunopurify the lysates.
A measure of 1 µg of rabbit polyclonal anti-APPL1 antibody and 30 µl of protein G-agarose (Santa Cruz, Biotechnology) were incubated at 4 °C for 1 h in 500 µl of PBS with a tube rotator. The complexes were microfuged and washed with 1 ml of lysis buffer for 3 times. At the end, 1000 µg of each cell lysates were added to the Protein G-agarose/anti-APPL1 antibodies and incubated at 4 °C for 2 h rotating. The proteins/complexes were centrifuged and washed 3 times with the kinase buffer (200 mM PIPES, 100 mM MnCl2). c-Src kinase activity was assayed by a standard in vitro kinase assay using acidified enolase as substrate. The incubation was performed in a total volume of 50 µl composed of the immunoprecipitates and the kinase buffer containing 5 mM ATP, 1 µC of [γ32P]ATP, and 2.5 µg of acid denatured rabbit muscle enolase (Sigma) as exogenous substrate. Samples were incubated at 30 °C for 10 min then reduced with an equal volume of 2× SDS Laemmli sample buffer (Sigma) and aliquots of them (40 µl) were submitted to SDS-PAGE (acrylamide 11%). The dried gel was exposed to X-omat film (Kodak) for 12 h. The gels were stained with Coomassie blue to ensure that an equal amount of enolase was present in all samples.
Plasmids
XETL plasmid, which carries firefly luciferase sequences under the control of an estrogen response element upstream of the thymidine kinase promoter, was provided by Dr Picard, University of Geneva. S104/106/118A-ER, S118A-ER, and S167A-ER plasmids were mutated in serine residues 104, 106, 118, or 167 to Ala, respectively (a gift from Dr DA Lannigan, University of Virginia); HE241G ERα plasmid mutant that lacks a nuclear translocation signal (NLS) (Δ250–303) (kindly provided by Dr P Chambon, CNRS-INSERM, University of Louis Pasteur). mERα plasmid containing the AF-2/ligand binding domain and a signal that targets this portion of the receptor to the plasma membrane (generously provided by Dr ER Levin, University of California).
Transfections and luciferase assays
MCF-7 cells were transfected using the FuGENE 6 reagent as recommended by the manufacturer with the mixture containing 0.5 μg of reporter plasmid XETL. A set of experiments was performed cotransfecting XETL HeLa cells with XETL and HEGO. Another set of experiments was performed by using 0.5 μg/well pSG5/HE15 (AF-1), pSG5/HE19 (AF-2), HE241G, S104/106/118A-ER, S118A-ER, or S167A-ER plasmids. Six hours after transfection, treatments were added, and cells were incubated for 48 h. A concentration 10 μM, of ICI 182,780 (Tocris Bioscience), Compound C (Enzo Life Sciences), H89 and PP2 (Sigma), PD98059 (Calbiochem), was used. TK Renilla luciferase plasmid (25 ng/well) serves to normalize the efficiency of the transfection. Firefly and Renilla luciferase activities were measured using a Dual Luciferase kit. The firefly luciferase data for each sample were normalized on the basis of transfection efficiency measured by Renilla luciferase activity as reported above.Citation51
Immunocytochemical staining
Paraformaldehyde-fixed MCF-7 cells were used for immunocytochemical staining. Endogenous peroxidase activity was inhibited by hydrogen peroxide, and nonspecific sites were blocked by normal horse serum. ERα immunostaining was then performed using as primary antibody a mouse monoclonal antiserum, whereas a biotinylated horse-anti-mouse IgG was utilized as secondary antibody. Avidin–biotin–horseradish peroxidase complex (ABC complex/horseradish peroxidase) was applied, and the chromogen 3,3′-diaminobenzidine tetrachloride dihydrate was used as detection system. TBS-T (0.05 M TRIS-HCl plus 0.15 M NaCl, pH 7.6 containing 0.05% Triton X-100) served as washing buffer. The primary antibody was replaced by normal mouse serum at the same concentration in control experiments on MCF-7 cultured cells.
Reverse transcription-PCR assay
The gene expression of ERα, cathepsin D, pS2, and 36B4 was evaluated by reverse transcription PCR (RT-PCR) method as described.Citation52 Primer sequences include: estrogen receptor α (ERα), forward 5′-TGATTGGTCT CGTCTGGCG-3′ and reverse 5′-CATGCCCTCT ACACATTTTC CC-3′; Cathepsin D (CatD), forward 5′-AACAACAGGG TGGGCTTC-3′and reverse 5′-TTTGAGTAGT CAAAGTCAGA GC-3′; Trefoil factor 1/pS2 (pS2), forward 5′-TTCTATCCTA ATACCATCGA CG-3′ and reverse 5′-TTTGAGTAGT CAAAGTCAGA GC-3′; 36B4, forward 5′-CTCAACATCT CCCCCTTCTC-3′ and reverse 5′- CAAATCCCAT ATCCTCGT -3′. Equal amounts of PCR product were electrophoresed on 1% agarose gels and visualized by ethidium bromide staining.
Statistical analysis
Each datum point represents the mean ± SD of at least 3 independent experiments. Data were analyzed by Student t test using the GraphPad Prism 4 software program. P < 0.05 was considered as statistically significant.
| Abbreviations: | ||
| ERα | = | estrogen receptor alpha |
| AdipoR1 | = | adiponectin receptor 1 |
| IGF-IR | = | insulin-like growth factor I receptor |
| EGFR | = | epidermal growth factor receptor |
| MAPK | = | mitogen activated protein |
| MTT | = | 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide |
| PKA | = | cAMP-dependent protein kinase |
| E2 | = | 17β-estradiol |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by Associazione Italiana Ricerca sul Cancro (AIRC) [MFAG grant number 6180 and IG grant number 11595]. Progetti di Ricerca di Interesse Nazionale (PRIN)-Ministero Istruzione Università e Ricerca (MIUR) [grant number 20085Y7XT5]. European Commission/FSE/Regione Calabria to Emilia Ricchio.
The authors thank Dr Pasquale Cicirelli for his technical assistance.
Related Research Data
References
- MacDougald OA, Burant CF. The rapidly expanding family of adipokines. Cell Metab 2007; 6:159 - 61; http://dx.doi.org/10.1016/j.cmet.2007.08.010; PMID: 17767903
- Andò S, Catalano S. The multifactorial role of leptin in driving the breast cancer microenvironment. Nat Rev Endocrinol 2012; 8:263 - 75; http://dx.doi.org/10.1038/nrendo.2011.184; PMID: 22083089
- Brochu-Gaudreau K, Rehfeldt C, Blouin R, Bordignon V, Murphy BD, Palin MF. Adiponectin action from head to toe. Endocrine 2010; 37:11 - 32; http://dx.doi.org/10.1007/s12020-009-9278-8; PMID: 20963555
- Matsubara M, Maruoka S, Katayose S. Inverse relationship between plasma adiponectin and leptin concentrations in normal-weight and obese women. Eur J Endocrinol 2002; 147:173 - 80; http://dx.doi.org/10.1530/eje.0.1470173; PMID: 12153737
- Cnop M, Havel PJ, Utzschneider KM, Carr DB, Sinha MK, Boyko EJ, Retzlaff BM, Knopp RH, Brunzell JD, Kahn SE. Relationship of adiponectin to body fat distribution, insulin sensitivity and plasma lipoproteins: evidence for independent roles of age and sex. Diabetologia 2003; 46:459 - 69; PMID: 12687327
- Weyer C, Funahashi T, Tanaka S, Hotta K, Matsuzawa Y, Pratley RE, Tataranni PA. Hypoadiponectinemia in obesity and type 2 diabetes: close association with insulin resistance and hyperinsulinemia. J Clin Endocrinol Metab 2001; 86:1930 - 5; http://dx.doi.org/10.1210/jc.86.5.1930; PMID: 11344187
- Kubota N, Terauchi Y, Yamauchi T, Kubota T, Moroi M, Matsui J, Eto K, Yamashita T, Kamon J, Satoh H, et al. Disruption of adiponectin causes insulin resistance and neointimal formation. J Biol Chem 2002; 277:25863 - 6; http://dx.doi.org/10.1074/jbc.C200251200; PMID: 12032136
- Maeda N, Shimomura I, Kishida K, Nishizawa H, Matsuda M, Nagaretani H, Furuyama N, Kondo H, Takahashi M, Arita Y, et al. Diet-induced insulin resistance in mice lacking adiponectin/ACRP30. Nat Med 2002; 8:731 - 7; http://dx.doi.org/10.1038/nm724; PMID: 12068289
- Tilg H, Moschen AR. Adipocytokines: mediators linking adipose tissue, inflammation and immunity. Nat Rev Immunol 2006; 6:772 - 83; http://dx.doi.org/10.1038/nri1937; PMID: 16998510
- Dalamaga M, Diakopoulos KN, Mantzoros CS. The role of adiponectin in cancer: a review of current evidence. Endocr Rev 2012; 33:547 - 94; http://dx.doi.org/10.1210/er.2011-1015; PMID: 22547160
- Fruebis J, Tsao TS, Javorschi S, Ebbets-Reed D, Erickson MR, Yen FT, Bihain BE, Lodish HF. Proteolytic cleavage product of 30-kDa adipocyte complement-related protein increases fatty acid oxidation in muscle and causes weight loss in mice. Proc Natl Acad Sci U S A 2001; 98:2005 - 10; http://dx.doi.org/10.1073/pnas.98.4.2005; PMID: 11172066
- Yamauchi T, Kamon J, Ito Y, Tsuchida A, Yokomizo T, Kita S, Sugiyama T, Miyagishi M, Hara K, Tsunoda M, et al. Cloning of adiponectin receptors that mediate antidiabetic metabolic effects. Nature 2003; 423:762 - 9; http://dx.doi.org/10.1038/nature01705; PMID: 12802337
- Kadowaki T, Yamauchi T. Adiponectin and adiponectin receptors. Endocr Rev 2005; 26:439 - 51; http://dx.doi.org/10.1210/er.2005-0005; PMID: 15897298
- Mao X, Kikani CK, Riojas RA, Langlais P, Wang L, Ramos FJ, Fang Q, Christ-Roberts CY, Hong JY, Kim RY, et al. APPL1 binds to adiponectin receptors and mediates adiponectin signalling and function. Nat Cell Biol 2006; 8:516 - 23; http://dx.doi.org/10.1038/ncb1404; PMID: 16622416
- Lee M-H, Klein RL, El-Shewy HM, Luttrell DK, Luttrell LM. The adiponectin receptors AdipoR1 and AdipoR2 activate ERK1/2 through a Src/Ras-dependent pathway and stimulate cell growth. Biochemistry 2008; 47:11682 - 92; http://dx.doi.org/10.1021/bi801451f; PMID: 18842004
- Kelesidis I, Kelesidis T, Mantzoros CS. Adiponectin and cancer: a systematic review. Br J Cancer 2006; 94:1221 - 5; http://dx.doi.org/10.1038/sj.bjc.6603051; PMID: 16570048
- Miyoshi Y, Funahashi T, Kihara S, Taguchi T, Tamaki Y, Matsuzawa Y, Noguchi S. Association of serum adiponectin levels with breast cancer risk. Clin Cancer Res 2003; 9:5699 - 704; PMID: 14654554
- Mantzoros C, Petridou E, Dessypris N, Chavelas C, Dalamaga M, Alexe DM, Papadiamantis Y, Markopoulos C, Spanos E, Chrousos G, et al. Adiponectin and breast cancer risk. J Clin Endocrinol Metab 2004; 89:1102 - 7; http://dx.doi.org/10.1210/jc.2003-031804; PMID: 15001594
- Körner A, Pazaitou-Panayiotou K, Kelesidis T, Kelesidis I, Williams CJ, Kaprara A, Bullen J, Neuwirth A, Tseleni S, Mitsiades N, et al. Total and high-molecular-weight adiponectin in breast cancer: in vitro and in vivo studies. J Clin Endocrinol Metab 2007; 92:1041 - 8; http://dx.doi.org/10.1210/jc.2006-1858; PMID: 17192291
- Kang JH, Lee YY, Yu BY, Yang BS, Cho KH, Yoon DK, Roh YK. Adiponectin induces growth arrest and apoptosis of MDA-MB-231 breast cancer cell. Arch Pharm Res 2005; 28:1263 - 9; http://dx.doi.org/10.1007/BF02978210; PMID: 16350853
- Dos Santos E, Benaitreau D, Dieudonne MN, Leneveu MC, Serazin V, Giudicelli Y, Pecquery R. Adiponectin mediates an antiproliferative response in human MDA-MB 231 breast cancer cells. Oncol Rep 2008; 20:971 - 7; PMID: 18813842
- Grossmann ME, Nkhata KJ, Mizuno NK, Ray A, Cleary MP. Effects of adiponectin on breast cancer cell growth and signaling. Br J Cancer 2008; 98:370 - 9; http://dx.doi.org/10.1038/sj.bjc.6604166; PMID: 18182989
- Dieudonne MN, Bussiere M, Dos Santos E, Leneveu MC, Giudicelli Y, Pecquery R. Adiponectin mediates antiproliferative and apoptotic responses in human MCF7 breast cancer cells. Biochem Biophys Res Commun 2006; 345:271 - 9; http://dx.doi.org/10.1016/j.bbrc.2006.04.076; PMID: 16678125
- Jardé T, Caldefie-Chézet F, Goncalves-Mendes N, Mishellany F, Buechler C, Penault-Llorca F, Vasson MP. Involvement of adiponectin and leptin in breast cancer: clinical and in vitro studies. Endocr Relat Cancer 2009; 16:1197 - 210; http://dx.doi.org/10.1677/ERC-09-0043; PMID: 19661131
- Treeck O, Lattrich C, Juhasz-Boess I, Buchholz S, Pfeiler G, Ortmann O. Adiponectin differentially affects gene expression in human mammary epithelial and breast cancer cells. Br J Cancer 2008; 99:1246 - 50; http://dx.doi.org/10.1038/sj.bjc.6604692; PMID: 18827813
- Pfeiler GH, Buechler C, Neumeier M, Schäffler A, Schmitz G, Ortmann O, Treeck O. Adiponectin effects on human breast cancer cells are dependent on 17-beta estradiol. Oncol Rep 2008; 19:787 - 93; PMID: 18288417
- Landskroner-Eiger S, Qian B, Muise ES, Nawrocki AR, Berger JP, Fine EJ, Koba W, Deng Y, Pollard JW, Scherer PE. Proangiogenic contribution of adiponectin toward mammary tumor growth in vivo. Clin Cancer Res 2009; 15:3265 - 76; http://dx.doi.org/10.1158/1078-0432.CCR-08-2649; PMID: 19447867
- Sflomos G, Brisken C. A new Achilles Heel in breast cancer?. Oncotarget 2013; 4:1126 - 7; PMID: 23907591
- Mauro L, Surmacz E. IGF-I receptor, cell-cell adhesion, tumour development and progression. J Mol Histol 2004; 35:247 - 53; http://dx.doi.org/10.1023/B:HIJO.0000032356.98363.a2; PMID: 15339044
- Simoncini T, Genazzani AR. Non-genomic actions of sex steroid hormones. Eur J Endocrinol 2003; 148:281 - 92; http://dx.doi.org/10.1530/eje.0.1480281; PMID: 12611608
- Schäffler A, Schölmerich J, Buechler C. Mechanisms of disease: adipokines and breast cancer - endocrine and paracrine mechanisms that connect adiposity and breast cancer. Nat Clin Pract Endocrinol Metab 2007; 3:345 - 54; http://dx.doi.org/10.1038/ncpendmet0456; PMID: 17377617
- Park J, Euhus DM, Scherer PE. Paracrine and endocrine effects of adipose tissue on cancer development and progression. Endocr Rev 2011; 32:550 - 70; http://dx.doi.org/10.1210/er.2010-0030; PMID: 21642230
- Kershaw EE, Flier JS. Adipose tissue as an endocrine organ. J Clin Endocrinol Metab 2004; 89:2548 - 56; http://dx.doi.org/10.1210/jc.2004-0395; PMID: 15181022
- Trujillo ME, Scherer PE. Adiponectin--journey from an adipocyte secretory protein to biomarker of the metabolic syndrome. J Intern Med 2005; 257:167 - 75; http://dx.doi.org/10.1111/j.1365-2796.2004.01426.x; PMID: 15656875
- Jardé T, Perrier S, Vasson MP, Caldefie-Chézet F. Molecular mechanisms of leptin and adiponectin in breast cancer. Eur J Cancer 2011; 47:33 - 43; http://dx.doi.org/10.1016/j.ejca.2010.09.005; PMID: 20889333
- Wang Y, Lam JB, Lam KS, Liu J, Lam MC, Hoo RL, Wu D, Cooper GJ, Xu A. Adiponectin modulates the glycogen synthase kinase-3β/β-catenin signaling pathway and attenuates mammary tumorigenesis of MDA-MB-231 cells in nude mice. Cancer Res 2006; 66:11462 - 70; http://dx.doi.org/10.1158/0008-5472.CAN-06-1969; PMID: 17145894
- Chen X, Wang Y. Adiponectin and breast cancer. Med Oncol 2011; 28:1288 - 95; http://dx.doi.org/10.1007/s12032-010-9617-x; PMID: 20625941
- Lanzino M, Morelli C, Garofalo C, Panno ML, Mauro L, Andò S, Sisci D. Interaction between estrogen receptor alpha and insulin/IGF signaling in breast cancer. Curr Cancer Drug Targets 2008; 8:597 - 610; http://dx.doi.org/10.2174/156800908786241104; PMID: 18991569
- Song RX, Chen Y, Zhang Z, Bao Y, Yue W, Wang JP, Fan P, Santen RJ. Estrogen utilization of IGF-1-R and EGF-R to signal in breast cancer cells. J Steroid Biochem Mol Biol 2010; 118:219 - 30; http://dx.doi.org/10.1016/j.jsbmb.2009.09.018; PMID: 19815064
- Pearson G, Robinson F, Beers Gibson T, Xu BE, Karandikar M, Berman K, Cobb MH. Mitogen-activated protein (MAP) kinase pathways: regulation and physiological functions. Endocr Rev 2001; 22:153 - 83; http://dx.doi.org/10.1210/er.22.2.153; PMID: 11294822
- Deepa SS, Dong LQ. APPL1: role in adiponectin signaling and beyond. Am J Physiol Endocrinol Metab 2009; 296:E22 - 36; http://dx.doi.org/10.1152/ajpendo.90731.2008; PMID: 18854421
- Murphy LC, Weitsman GE, Skliris GP, Teh EM, Li L, Peng B, Davie JR, Ung K, Niu YL, Troup S, et al. Potential role of estrogen receptor alpha (ERalpha) phosphorylated at Serine118 in human breast cancer in vivo. J Steroid Biochem Mol Biol 2006; 102:139 - 46; http://dx.doi.org/10.1016/j.jsbmb.2006.09.021; PMID: 17092701
- Luttrell LM. ‘Location, location, location’: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol 2003; 30:117 - 26; http://dx.doi.org/10.1677/jme.0.0300117; PMID: 12683936
- De Amicis F, Russo A, Avena P, Santoro M, Vivacqua A, Bonofiglio D, Mauro L, Aquila S, Tramontano D, Fuqua SA, et al. In vitro mechanism for downregulation of ER-α expression by epigallocatechin gallate in ER+/PR+ human breast cancer cells. Mol Nutr Food Res 2013; 57:840 - 53; http://dx.doi.org/10.1002/mnfr.201200560; PMID: 23322423
- Rovito D, Giordano C, Vizza D, Plastina P, Barone I, Casaburi I, Lanzino M, De Amicis F, Sisci D, Mauro L, et al. Omega-3 PUFA ethanolamides DHEA and EPEA induce autophagy through PPARγ activation in MCF-7 breast cancer cells. J Cell Physiol 2013; 228:1314 - 22; http://dx.doi.org/10.1002/jcp.24288; PMID: 23168911
- Panno ML, Giordano F, Rizza P, Pellegrino M, Zito D, Giordano C, Mauro L, Catalano S, Aquila S, Sisci D, et al. Bergapten induces ER depletion in breast cancer cells through SMAD4-mediated ubiquitination. Breast Cancer Res Treat 2012; 136:443 - 55; http://dx.doi.org/10.1007/s10549-012-2282-3; PMID: 23053665
- Mauro L, Catalano S, Bossi G, Pellegrino M, Barone I, Morales S, Giordano C, Bartella V, Casaburi I, Andò S. Evidences that leptin up-regulates E-cadherin expression in breast cancer: effects on tumor growth and progression. Cancer Res 2007; 67:3412 - 21; http://dx.doi.org/10.1158/0008-5472.CAN-06-2890; PMID: 17409452
- Guido C, Panza S, Santoro M, Avena P, Panno ML, Perrotta I, Giordano F, Casaburi I, Catalano S, De Amicis F, et al. Estrogen receptor beta (ERβ) produces autophagy and necroptosis in human seminoma cell line through the binding of the Sp1 on the phosphatase and tensin homolog deleted from chromosome 10 (PTEN) promoter gene. Cell Cycle 2012; 11:2911 - 21; http://dx.doi.org/10.4161/cc.21336; PMID: 22810004
- Mauro L, Pellegrino M, Lappano R, Vivacqua A, Giordano F, Palma MG, Andò S, Maggiolini M. E-cadherin mediates the aggregation of breast cancer cells induced by tamoxifen and epidermal growth factor. Breast Cancer Res Treat 2010; 121:79 - 89; http://dx.doi.org/10.1007/s10549-009-0456-4; PMID: 19593637
- Guido C, Whitaker-Menezes D, Lin Z, Pestell RG, Howell A, Zimmers TA, Casimiro MC, Aquila S, Ando’ S, Martinez-Outschoorn UE, et al. Mitochondrial fission induces glycolytic reprogramming in cancer-associated myofibroblasts, driving stromal lactate production, and early tumor growth. Oncotarget 2012; 3:798 - 810; PMID: 22878233
- De Amicis F, Zupo S, Panno ML, Malivindi R, Giordano F, Barone I, Mauro L, Fuqua SA, Andò S. Progesterone receptor B recruits a repressor complex to a half-PRE site of the estrogen receptor alpha gene promoter. Mol Endocrinol 2009; 23:454 - 65; http://dx.doi.org/10.1210/me.2008-0267; PMID: 19147702
- Casaburi I, Avena P, Lanzino M, Sisci D, Giordano F, Maris P, Catalano S, Morelli C, Andò S. Chenodeoxycholic acid through a TGR5-dependent CREB signaling activation enhances cyclin D1 expression and promotes human endometrial cancer cell proliferation. Cell Cycle 2012; 11:2699 - 710; http://dx.doi.org/10.4161/cc.21029; PMID: 22751440