
Abstract
Cellular senescence is characterized by silencing of genes involved in DNA replication and cell cycle progression. Stable repression is crucial for preventing inappropriate DNA synthesis and the maintenance of a prolonged senescent state. Many of these genes are targets for E2F transcription factors. The pRB pathway plays a major role in senescence by directly repressing E2Fs and also by regulating chromatin at the promoters of E2F target genes using its LXCXE cleft-dependent interactions. In this study, we sought to investigate the mechanisms by which pRB stably silences E2F target gene transcription during cellular senescence. We report that in mouse embryonic fibroblasts, endogenous promyelocytic leukemia protein (PML) associates with E2F target genes in a pRB LXCXE-dependent manner during HrasV12-induced senescence. Furthermore, using a PML-IV-induced senescence model, we show that the pRB LXCXE binding cleft is essential for PML association with gene promoters, silencing of E2F target genes, and stable cell cycle exit. Binding assays show that pRB can interact with PML specifically during senescence, suggesting that signaling events in senescence regulate assembly of PML and pRB to establish heterochromatin and create a permanent cell cycle arrest.
Introduction
Cellular senescence is a stable cell cycle exit that protects cells from transformation and prevents malignancy.Citation1 Senescence is characterized by cell cycle arrest, repression of proliferative genes, and activation of growth-suppressing genes.Citation2 Senescence-inducing stimuli such as telomere shortening, expression of activated oncogenes, and reactive oxygen species have been shown to cause DNA breaks and subsequent activation of the DNA damage response.Citation3-Citation5 This engages key tumor suppressor pathways regulated by p53 and pRB proteins that act as the effectors of senescence by inducing cell cycle arrest.Citation1
One of the defining characteristics of senescence is its permanence, as senescent cells are highly refractory to growth-promoting signals.Citation6 Maintenance of this stable cell cycle arrest is considered to be critical to the tumor-suppressive role of senescence in vivo.Citation7 Key to the maintenance of a permanent cell cycle arrest is stable repression of proliferative genes involved in DNA replication and cell cycle progression.Citation2 Senescence is often associated with heterochromatin assembly, and this is thought to contribute to stable gene silencing.Citation8 In general, there is enrichment of transcriptionally repressive histone modifications such as H3K9me3 and H3K27me3 and a decrease in activating marks such as H3K4me3 on proliferative gene promoters.Citation9-Citation11 Furthermore, in some cell types, such as IMR90 fibroblasts, these chromatin changes are accompanied by pronounced compaction of whole chromosomes into structures that are called SAHFs (senescence associated heterochromatic foci).Citation8,Citation12,Citation13 Although the precise contribution of each of these chromatin changes to the senescent state is not fully understood, they are proposed to contribute to the permanence of senescent arrest by stably silencing proliferative genes and preventing cell cycle entry.Citation14 There is some in vivo evidence supporting this, as mice lacking the enzymes responsible for these repressive histone modifications show defective chromatin assembly and increased susceptibility to cancer, suggesting that chromatin changes contribute to the tumor-suppressive role of senescence.Citation7
The pRB–E2F pathway is a key tumor suppressor pathway that regulates the expression of a number of genes involved in DNA synthesis and cell cycle advancement in response to growth factor stimuli.Citation15 Many of these proliferative genes are direct targets of E2F transcription factors, which, in turn, are negatively regulated by pRB family proteins. This places the pRB–E2F pathway at the core of the cellular senescence response. Genetic models have confirmed this hypothesis, wherein MEFs lacking all the pRB family proteins (TKO MEFs) fail to senesce and immortalize spontaneously in culture.Citation16,Citation17 Among the pocket protein family, pRB has been shown to have a unique role in senescence. pRB is required for repression of key cell cycle genes and preventing DNA synthesis in response to oncogene expression.Citation2 Furthermore, acute knockdown of pRB alone is sufficient to induce DNA synthesis and cell cycle re-entry in senescent MEFs, suggesting a crucial role for pRb in the maintenance of a stable senescent state.Citation18 This unique role for pRB can be attributed, at least in part, to its ability to regulate the heterochromatinization of cell cycle gene promoters and stable silencing of these genes.Citation10 However, the mechanistic role of pRB in establishing stable senescence is not understood. Taken together, heterochromatin changes accompany senescence-induced cell cycle arrest; however, it is unclear if these are a direct effect of pRB, or an indirect consequence of its other functions.
The pro-myelocytic leukemia protein (PML) has been shown to be essential for senescence.Citation19-Citation21 PML is the primary component of PML nuclear bodies, the sub-nuclear structures that increase in abundance in response to a variety of cellular stresses. Expression of oncogenic HrasV12 in fibroblasts results in a dramatic increase in the number and size of PML nuclear bodies.Citation19 The essential role for PML in senescence comes from the observation that fibroblasts from Pml−/− embryos fail to senesce and continue proliferating in response to HrasV12.Citation21 Furthermore, forced expression of PML is sufficient to induce senescence in primary fibroblasts. The Pml gene is subject to extensive alternate splicing, resulting in at least 7 major isoforms (called PML-I through PML-VII) that differ mainly in their C-terminal region.Citation22 PML-IV, among the major isoforms, is the only one shown to be able to induce senescence when overexpressed, suggesting an important role for this isoform.Citation21 However, PML-IV fails to induce senescence when expressed in Pml−/− MEFs, suggesting that other isoforms are also required for efficient induction of senescence.Citation21
The precise role of PML and its constituent nuclear bodies during senescence is an area of intense research. A functional co-operation between PML and pRB–E2F pathways during senescence was recently reported.Citation23 pRB and E2Fs were shown to localize to the PML nuclear bodies during senescence, and disruption of pRB–E2F interactions, or degradation of RB family proteins by expression of human papilloma virus E7, was sufficient to compromise PML-IV-induced senescence. This association between PML and pRB-E2F is proposed to be responsible for repression of E2Fs and their target gene expression. However, since HPV-E7 inhibits RB family proteins and PML alike, the precise aspects of pRB or PML function that is required for senescence remain unknown.
We previously showed that MEFs from a gene-targeted mouse carrying a mutant pRB that is specifically defective for LXCXE-type interactions (called Rb1∆L) are defective for stable repression of E2F target genes during oncogene-induced senescence.Citation10 This mutation also compromises the stability of senescence arrest and enables escape. In the current study, we explored the mechanism of pRB-mediated silencing and heterochromatinization of E2F-responsive genes using 2 different senescence contexts: oncogene-induced senescence (HrasV12) and PML-induced senescence (PML-IV). Here we show that endogenous PML is enriched at the promoters of E2F target genes in a pRB-LXCXE-dependent manner during both forms of senescence. The same E2F target genes fail to be repressed in Rb1∆L/∆L MEFs overexpressing either HrasV12 or PML-IV. Interestingly, Rb1∆L/∆L MEFs overexpressing PML-IV fail to enrich the repressive histone mark H3K9me3 at Ccne1 and Mcm3 gene promoters. This suggests a requirement for PML recruitment by pRB–LXCXE-type interactions to induce heterochromatinization and gene silencing. Furthermore, using co-immunoprecipitation and GST pull-down experiments, we show that PML is only capable of binding pRB under senescent growth conditions, and these interactions are disrupted by mutations in the pRB–LXCXE binding cleft. Our experiments support a model in which pRb interacts with PML in a LXCXE cleft-dependent manner, and this complex mediates heterochromatinization and silencing of E2F genes during senescence. Taken together, our data demonstrates that the interaction between pRB and PML is critical to switching the chromatin state at cell cycle promoters to create a permanent arrest.
Results
Defective enrichment of PML on E2F target gene promoters in Rb1∆L/∆L cells during senescence
In a previous study we investigated the role of pRb–LXCXE interactions in cellular senescence using MEFs derived from Rb1∆L/∆L mice.Citation10 We reported that Rb1∆L/∆L MEFs enter a state of partial senescence in response to oncogenic HrasV12 expression, in which they take on many of the morphological features of senescent cells but fail to stably repress E2F target genes, and these genes remain susceptible to activation by ectopic stimuli. Importantly, the Rb1∆L mutation allows partially senescent Rb1∆L/∆L MEFs to initiate DNA synthesis, re-enter the cell cycle, and resume proliferation. We found defective enrichment of the repressive histone modification H3K9me3 on E2F target gene promoters in Rb1∆L/∆L MEFs during senescenceCitation10 and . Our goal in this study was to use Rb1∆L/∆L cells to identify components of the mechanism that converts reversible growth arrest into permanent withdrawal from the cell cycle through heterochromatin formation at E2F promoters.
Figure 1. Defective enrichment of the PML (pro-myelocytic leukemia) protein on E2F-responsive cell cycle gene promoters in senescent Rb1∆L/∆L cells. Asynchronously growing wild-type and Rb1∆L/∆L MEFs were induced to senesce by retroviral-mediated expression of oncogenic HrasV12. Chromatin from proliferating and senescent cells was used for chromatin immunoprecipitation. Real-time PCR was used to amplify the immunoprecipitated DNA using primers specific to Cyclin E1 (left) and Mcm3 (right). The quantity of precipitated DNA is represented as percent of input chromatin. (A) ChIP of proliferating and senescent cells of the indicated genotypes using a α-H3K9me3 antibody or an IgG control. (B) ChIP of chromatin from proliferating and senescent wild-type and Rb1∆L/∆L cells using a α-PML antibody and an IgG control. (C) ChIP on senescent wild-type and Rb1∆L/∆L cells using a α-pRB antibody and an IgG control. All experiments were reproduced in at least 3 independent pairs of MEFs. Error bars indicate one standard deviation from the mean of at least 3 biological replicates. An asterisk indicates a statistically significant difference (t test, P < 0.05).
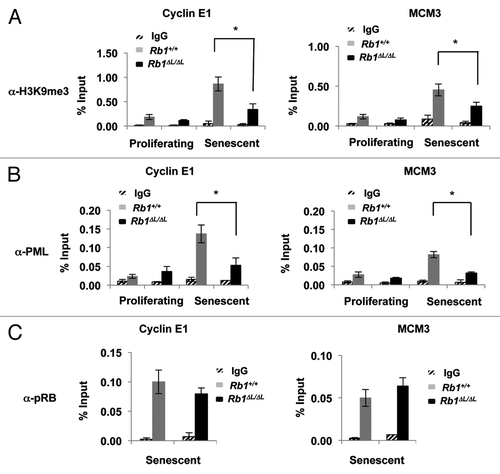
We searched for proteins whose association with E2F-responsive promoters in chromatin immunoprecipitation assays (ChIP) was sensitive to the Rb1∆L mutation. We examined the Ccne1 and Mcm3 gene promoters, as these genes are key targets of pRB in proliferative control during senescence.Citation2 As shown in endogenous PML is enriched at both these promoters during senescence in Rb1+/+ cells. Strikingly, this enrichment is eliminated in Rb1∆L/∆L cells, suggesting that PML requires pRb–LXCXE binding cleft-mediated interactions for recruitment. Furthermore, pRB is equally enriched at these promoters in both Rb1+/+ and Rb1∆L/∆L cells (). This shows that while pRb∆L is capable of binding to E2F target genes during senescence, this mutation specifically disrupts PML association with these promoters, suggesting that it participates in the switch from short-term to long-term growth arrest.
Early events during senescence induction occur normally in Rb1∆L/∆L MEFs
Expression of oncogenic ras in primary fibroblasts induces hyper-proliferation resulting in replicative stress and DNA damage.Citation3-Citation5 This leads to the activation of DNA damage signaling and activation of p53 and pRB pathways. Ras expression also leads to induction of PML and PML nuclear body formation in a p53-dependent manner.Citation24 Activation of the DNA damage response has also been shown to be important for the maintenance of senescence arrest.Citation4 We wanted to investigate whether these signaling events, which are required for senescence induction, are intact in Rb1∆L/∆L MEFs and could explain failure to recruit PML to E2F-regulated promoters in senescent Rb1∆L/∆L cells.
First, we tested if DNA damage signaling is intact in Rb1∆L/∆L MEFs, and if it is activated in response to ras similar to wild-type controls. Rb1+/+ and Rb1∆L/∆L MEFs induced to senesce by expression of oncogenic HrasV12 were stained with antibodies against γH2AX, a marker of DNA double-strand breaks. As a control, we also assessed DNA damage in low passage, proliferating MEFs. As shown in , HrasV12 expression induces a significant increase in the number of γH2AX foci both in wild-type and Rb1∆L/∆L MEFs. The damage foci can be seen very early after the expression of HrasV12, and this damage also persists during senescence in both genotypes. Furthermore, western blotting for this DNA damage marker in proliferating and senescent cell extracts showed a similar increase in the levels of γH2AX in both wild-type and Rb1∆L/∆L MEFs under senescent culture conditions ().
Figure 2. Oncogenic Ras induces DNA damage and accumulation of PML nuclear bodies in both wild-type and Rb1∆L/∆L cells. Asynchronously growing wild-type and Rb1∆L/∆L MEFs were induced to senesce by retroviral-mediated expression of oncogenic HrasV12. After 3 d of selection, cells were re-plated and cultured for the indicated amount of time. (A) Immunofluorescent (IF) staining of wild-type and Rb1∆L/∆L cells with γH2AX antibody (red) to detect double strand breaks at different times after induction of senescence. Nuclei were counter stained with DAPI (blue). (B) Quantification of DNA damage foci in (A). The percent of nuclei with >3 γH2AX foci were compared between genotypes. (C) Whole-cell extracts from asynchronously growing or oncogenic HrasV12 expressing senescent wild-type and Rb1∆L/∆L MEFs were probed with a γH2AX antibody. β-actin levels are used as loading control. (D) IF staining for PML nuclear bodies using a α-PML antibody (green) and DNA counterstaining with DAPI (blue). Inset images show detailed PML staining of individual nuclei. (E) Quantification of the number of PML bodies per nucleus in (D). The proportion of cells with fewer than 10, 10 to 25, or more than 25 PML bodies per nucleus are displayed in graphical format. All experiments were reproduced in at least 3 independent pairs of MEFs. Error bars represent one standard deviation from the mean of at least 3 biological replicates. Scale bars are 50 μM.
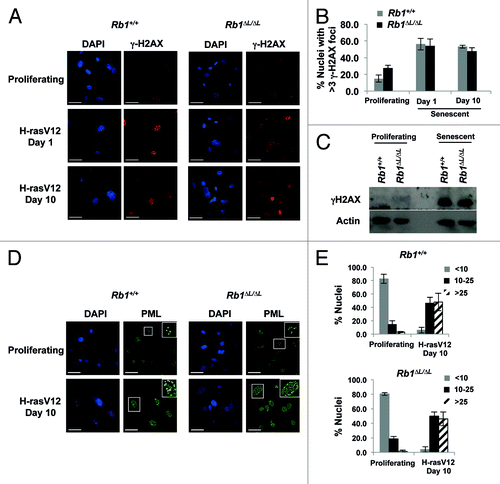
We next determined if PML bodies are formed normally in our mutant background. We used immunofluorescence staining with an α-PML antibody in Rb1+/+ and Rb1∆L/∆L MEFs expressing oncogenic HrasV12. We observed a clear increase in the number of PML nuclear bodies in senescent cells compared with asynchronously proliferating MEFs (). Importantly, Rb1∆L/∆L MEFs showed accumulation of PML bodies similar to wild-type cells (). We observed a significant shift toward more PML bodies per nucleus (> 10) in MEFs induced to senesce by HrasV12 expression ().
Taken together the above experiments suggest that the early events in senescence leading up to PML body assembly occur normally in Rb1∆L/∆L cells compared with wild type. This suggests that the defective enrichment of PML on E2F target gene promoters we observed is not due to decreased PML accumulation in Rb1∆L/∆L MEFs or diminished signals that induce senescence.
Defective senescence arrest in Rb1∆L/∆L MEFs expressing PML-IV
We next sought to determine if ectopic PML expression could rescue defective association with E2F-regulated promoters in Rb1∆L/∆L fibroblasts undergoing senescence. We took advantage of the ability of PML-IV to induce senescence when overexpressed in MEFs.Citation19,Citation21 We induced senescence in Rb1+/+ and Rb1∆L/∆L MEFs by expressing FLAG-tagged PML-IV by retroviral transduction and followed the cells over a 10-d time course as above. Following selection in puromycin, cells were re-plated at low density and cultured for 10 more days to investigate the induction of senescence. Since PML induced senescence occurs earlier than HrasV12 (data not shown), we have focused on day 8 as an equivalent endpoint for these experiments. We analyzed PML-IV expressing cells for DNA synthesis, senescence-associated β-galactosidase expression, and E2F target gene expression. As shown in , FLAG-PML-IV is expressed in most cells in both genotypes tested. Eight days post-re-plating most cells had stopped proliferating, as determined by BrdU and senescence-associated β-galactosidase (SA-β-gal) staining. However, we noticed that the Rb1∆L/∆L cultures are more densely packed compared with Rb1+/+ MEFs at the same time points, suggesting more cell growth during this time course. To determine if Rb1∆L/∆L cells continue to proliferate faster following PML-IV expression before eventually exiting the cell cycle, we performed BrdU labeling at different time points after initial selection and re-plating. In response to PML-IV expression, wild-type MEFs arrest as early as day 1 after re-plating and remain arrested throughout the experiment (). In contrast, Rb1∆L/∆L cells showed elevated DNA synthesis at earlier time points, as indicated by higher BrdU incorporation relative to wild type (). However, 8 d post-re-plating Rb1∆L/∆L cells reduce DNA synthesis to control levels (). Furthermore, both genotypes displayed features of senescent cells at this time point, as they were flat and ubiquitously positive for SA-β-gal expression (). This suggests that pRB-LXCXE interactions are essential for efficient arrest of DNA synthesis and proper cell cycle exit in response to PML-IV expression; however, mutant cells still possess features of senescence in response to PML-IV.
Figure 3. Defective senescent arrest in Rb1∆L/∆L MEFs expressing PML-IV. Asynchronously growing wild-type and Rb1∆L/∆L MEFs were transduced with retroviruses expressing pBABE-FLAG-PML-IV. After 3 d of drug selection, cells were plated in medium and cultured for the indicated amount of time. (A) Immunofluorescent (IF) staining was performed with a α-FLAG antibody (green) to detect PML-IV. Nuclei were counter stained with DAPI (blue). The inset image shows higher magnification of individual nuclei. (B) Cells of the indicated genotypes were pulsed with BrdU for 4 h, followed by fixation and staining with α-BrdU antibodies. The percentage of BrdU-positive nuclei at the indicated time points following FLAG-PML-IV expression are plotted. (C) PML-IV-expressing cells were stained for senescence-associated β-galactosidase (SA-β-gal) expression 8 d after the expression of PML-IV. (D) The number of SA-β-gal-positive cells in each genotype were quantified and plotted. (E) Quantification of E2F target gene mRNA from wild-type and Rb1∆L/∆L MEFs either from proliferating (left), or PML-IV-expressing senescent cells (right). Samples were normalized to expression of the ribosomal protein gene Rplp0. (F) Western blots to determine the expression of protein products of E2F target genes (p107 and cyclin E) following PML expression are shown. (G) DNA synthesis in response to ectopic E2F1 expression was measured by BrdU incorporation. Two days following Ad-E2F1 infection, cells were pulse labeled with BrdU for 16hrs and positive cells were identified by immunofluorescence microscopy and shown in the graph (bottom left). The fold increase in BrdU incorporation between control and E2F1 infected cells was calculated for both the genotypes and is shown in the graph on the bottom right. The mean fold increase was compared by a t test. The expression level of ectopic E2F1 in both the wild-type and Rb1∆L/∆L MEFs is shown by western blotting along with β-actin as a loading control. An asterisk on a graph indicates a statistically significant difference (t test, P < 0.05), an asterisk on a blot represents a non-specific band. All experiments were reproduced in at least three independent pairs of MEFs. Error bars represent one standard deviation from the mean of at least 3 biological replicates. Scale bars are 50 μM.
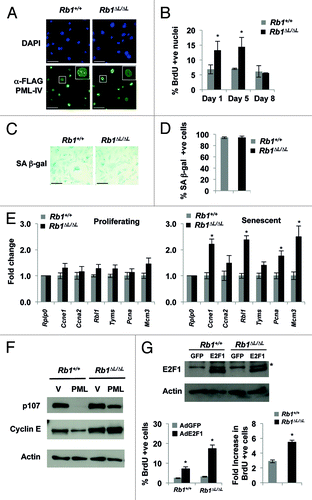
One of the major roles of pRB in senescence is repression of E2F target genes involved in DNA replication and cell cycle advancement.Citation2 We next investigated if the E2F target genes are silenced in Rb1∆L/∆L MEFs in response to PML-IV expression and senescence induction. Based on our results from we used 8 d post re-plating as our time point for assaying E2F target gene message levels, as both Rb1+/+ and Rb1∆L/∆L MEFs showed similar inhibition of DNA synthesis and equal SA-β-gal expression at this time point. We quantified the mRNA levels of 6 known E2F target genes: Ccne1 (cyclin E1), Ccna2 (Cyclin A2), Rbl1 (p107), Tyms (thymidylate synthase), Pcna (proliferating cell nuclear antigen), and Mcm3 (minichromosome maintenance deficient 3), along with Rplp0 (60S acidic ribosomal protein P0) as a control (). In proliferating cultures, the expression levels of E2F target genes are similar in Rb1∆L/∆L MEFs compared with wild type (, left). However, in senescent cultures expressing PML-IV, 8 d post-re-plating, we observed elevated expression of E2F target genes in Rb1∆L/∆L MEFs relative to wild-type controls (, right). Moreover, western blotting further confirmed the failure of Rb1∆L/∆L MEFs to properly repress E2F target gene expression in response to PML-IV as p107 and Cyclin E protein levels are elevated compared with controls ().
We next wanted to determine the stability of PML-IV-induced senescence arrest in Rb1∆L/∆L MEFs relative to wild-type controls. In order to do this, we induced senescence by expressing PML-IV and ectopically expressed human E2F1 by subsequent adenoviral infection. In this way we were able to test the propensity of the Rb1∆L/∆L MEF cells to reinitiate DNA synthesis. As shown in , ectopic E2F1 induced higher levels of BrdU incorporation in Rb1∆L/∆L mutant cells compared with wild type. The fold induction of BrdU incorporation in response to ectopic E2F1 expression was also significantly higher in mutant cells. This suggests that the PML-induced senescent cell cycle arrest in Rb1∆L/∆L cells is less stable and is more susceptible than wild-type cells to ectopic proliferative signals.
Taken together, BrdU incorporation, E2F target gene expression analysis, and ectopic E2F1 expression suggest that pRB-LXCXE interactions are required for proper repression of proliferative genes and efficient exit from the cell cycle in response to PML-IV expression. Robust induction of SA-β-gal suggests that Rb1∆L/∆L MEFs respond normally to other aspects of PML-IV-induced senescence. These data suggest that PML-IV induces an incomplete state of senescence, similar to Hras-V12, as we have reported previously.Citation10 This suggests that PML function is critical to the switch that creates a permanent barrier to proliferation in senescence.
Induction of senescence signals the assembly of PML–pRB complexes that are essential for heterochromatin formation in senescence
Senescence is associated with a number of chromatin changes, and heterochromatin assembly has been suggested to play an important role.Citation8,Citation12,Citation13,Citation25-Citation27 Both pRB and PML have been shown to be involved in heterochromatin formation during senescence.Citation8,Citation13 Consequently, we hypothesized that the defective repression of E2F target genes observed in Rb1∆L/∆L MEFs might be due to the inability of PML to assemble with pRB and regulate heterochromatin at these promoters.
Our analysis of PML bodies in proliferating and senescent cells in indicates that PML bodies exist under both growth conditions. Previously, pRB has been shown to bind to PML in interaction assays when overexpressed in cancer cell lines.Citation28 To distinguish if PML–pRB interactions are simply driven by abundance, or whether there is a regulated assembly process, we tested PML binding to the large pocket fragment of pRB (amino acids 379–928) fused to GST. We performed pull-down experiments with GST-RB or GST-RB∆L using nuclear extracts prepared either from wild-type proliferating MEFs or those made senescent by expressing oncogenic HrasV12. As shown in GST-RB is able to pull-down PML protein from senescent nuclear extracts, but not from proliferating nuclear extracts, even with relatively equal input of PML proteins. In contrast GST-RB∆L is unable to pull-down PML from the same extract (). Furthermore, GST-p107 is incapable of pulling down PML from the same extracts (). As a control to show that the GST-RB∆L and GST-p107 proteins are functional, and that equivalent amounts of extract were used in each, we stripped and re-probed the membranes with either E2F3 or E2F4 antibodies, respectively. As shown in , GST-RB∆L is able to pull-down E2F3 as efficiently as wild-type, and GST-p107 is capable of pulling down E2F4 from the nuclear extracts (). This indicates that GST-RB is specifically capable of interacting with PML from senescent nuclear extracts, it is dependent on LXCXE cleft interactions, and this ability is unique to pRB.
Figure 4. The pRB∆L mutation disrupts PML-pRB interactions during senescence. GST pull-down experiments were performed using nuclear extracts from proliferating or senescent MEFs induced to senesce by expression of oncogenic HrasV12. (A) GST pull-down using GST tagged pRB large pocket or pRB large pocket with ∆LXCXE mutations (∆L). GST alone is used as a negative control. Pull-down fractions were probed with antibodies specific to murine PML and E2F3. (B) GST pull-down as in (A) using GST tagged p107 large pocket. Pull-down fractions were probed with antibodies specific to either murine PML or E2F4. (C) Nuclear extracts from wild-type and Rb1∆L/∆L MEFs that were induced to senesce by expressing oncogenic HrasV12 were used for co-immunoprecipitation using a sheep α-RB antibody or a non-specific IgG control. The immunoprecipitated fractions were probed with a monoclonal antibody against PML (Millipore). The blots were then stripped and reprobed for E2F3. An asterisk indicates a non-specific band. (D) Nuclear extracts from proliferating and senescent cells were analyzed by SDS-PAGE and western blotting with a pan PML antibody that recognizes numerous isoforms (SC, shown at left). The same extracts were probed with a murine specific antibody to PML as above (MP, shown to the right). The arrows indicate differentially expressed bands. (E) GST pull-downs were performed as in (A), except the blot was probed with the pan PML antibody that recognizes many PML isoforms. Arrows indicate different PML species that are sensitive to ∆L mutations in pRB. Stars indicate cross reactivity with the GST-RB protein. (F) MEFs were transfected with expression constructs for each of the indicated PML isoforms. Following SDS-PAGE and western blotting, membranes were probed with the same pan PML antibody as in (E) to identify the migration pattern of different PML isoforms.
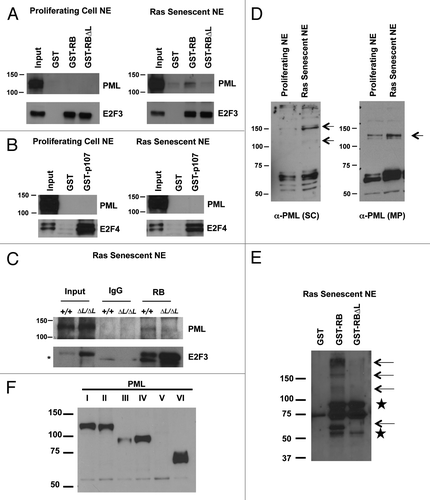
We next wondered whether we could detect endogenous pRB-PML complexes in cells undergoing senescence, and if they are affected by the Rb1∆L mutation. In order to test this, we performed co-immunoprecipitation assays with a pRB antibody using nuclear extracts from wild-type and Rb1∆L/∆L cells that are induced to senescence by oncogenic HrasV12 expression. As shown in , endogenous pRB associates with PML in wild-type senescent nuclear extracts. This interaction is clearly diminished in Rb1∆L/∆L cells, further supporting our GST pull-down experiments (). As a control for our immunoprecipitations, we also tested the ability of pRB∆L to pull-down E2F proteins by re-probing the same blot with an antibody against E2F3. As shown in (bottom) pRB∆L is able to immunoprecipitate E2F3 efficiently from the nuclear extracts. It is interesting to note that E2F3 is itself an E2F target gene, and we have previously reported de-regulation of the E2F target genes in Rb1∆L/∆L cells undergoing senescence. This explains the relatively higher level of E2F3 expression in Rb1∆L/∆L extracts (input lanes) and a corresponding increase in the E2F3 immunoprecipitated from these extracts. Taken together, our GST pull-downs and co-immunoprecipitation experiments suggest that senescence-inducing stimuli, such as HrasV12, signal the generation of a distinct PML body that can assemble with pRB through its LXCXE binding cleft.
To further expand this analysis and better understand the signal that initiates pRB–PML interactions in senescence, we used polyclonal antibodies that recognize most isoforms of PML. First we examined PML protein expression in nuclear extracts from proliferating and HrasV12 senescent fibroblasts (). This demonstrates the senescent-dependent appearance of bands that react with PML antibodies, most notably at 150 kD molecular weight (, left panel, upper arrow). Likewise, the monoclonal antibody to PML used previously preferentially recognizes the increase of a 125-kD form of PML (, arrow by right panel). In GST–RB pulldown assays we observed binding of multiple isoforms of PML with pRB in a LXCXE-dependent manner (, marked by arrows). To clarify the identity of PML proteins in this pulldown assay, we expressed FLAG-tagged versions of PML I-VI individually by transfection and resolved nuclear extracts by SDS-PAGE and identified PML by western blotting with pan PML antibodies (). In agreement with previous publications, PML isoforms range from approximately 50 kD to 120 kD. Our pulldown assays reveal that some PML bands correspond to individual isoforms (e.g., at 60 kD). However, it is notable that others at 150 kD and higher do not. PML has been shown to be extensively modified post translationally by Sumo, among others in response to stress, which could alter their electrophoretic mobility.Citation22 We hypothesize that pRB-PML interactions in senescence rely on post-translational modification of different PML isoforms, to stimulate their interaction.
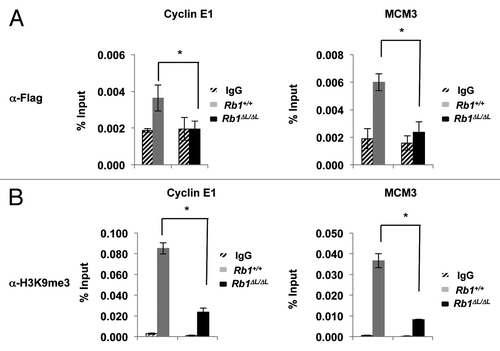
In order to determine the functional relevance of pRB-PML interactions in senescence we performed ChIP using α-FLAG antibodies on chromatin from cells that were induced to senesce by expressing FLAG-PML-IV. While we were able to detect FLAG-PML-IV on both Ccne1 and Mcm3 promoters in wild-type MEFs, we could not detect a signal above background in Rb1∆L/∆L MEFs (). This further suggested that PML–pRB interactions are LXCXE-dependent directly at E2F-responsive gene promoters. We next tested if PML-IV interaction with these gene promoters in Rb1+/+ MEFs is coincident with heterochromatinization by ChIP assay. As shown in , in Rb1+/+ MEFs expressing FLAG-PML-IV, H3K9me3 is enriched at Ccne1 and Mcm3 gene promoters. In contrast, in Rb1∆L/∆L MEFs there is no enrichment of this repressive histone modification following the expression of PML-IV.
Figure 5. Defective enrichment of Flag-PML-IV and heterochromatin formation at E2F target gene promoters in Rb1∆L/∆L MEFs. Asynchronously growing wild-type and Rb1∆L/∆L MEFs were transduced with pBABE-FLAG-PML-IV retrovirus. After 3 d of drug selection, cells were re-plated and cultured for 8 more days before processing for chromatin immunoprecipitation. (A) ChIP on wild-type and Rb1∆L/∆L MEFs expressing PML-IV using a α-FLAG antibody or an IgG control. Real-time PCR was used to amplify the immunoprecipitated DNA using primers specific to the promoter regions of Ccne1 (left) and Mcm3 (right). (B) ChIP on wild-type and Rb1∆L/∆L MEFs expressing PML-IV using a α-H3K9me3 antibody or an IgG control. Real-time PCR was used to amplify the immunoprecipitated DNA using primers specific to the promoter regions of Ccne1 and Mcm3. All experiments were reproduced in at least 3 independent pairs of MEFs. Error bars represent one standard deviation from the mean of at least 3 biological replicates. An asterisk indicates a statistically significant difference (t test, P < 0.05).
The above experiments show that pRB and PML functionally interact to regulate the assembly of repressive heterochromatin at E2F target genes involved in replication and cell cycle progression. Furthermore, this interaction is mediated by the LXCXE binding cleft of pRB and pRB–PML interactions are actively stimulated by senescence.
Discussion
Our study demonstrates the cooperative action of PML and pRB during senescence in silencing of E2F target genes involved in DNA synthesis and cell cycle advancement. This interaction is important for heterochromatinization of these promoters as H3K9me3 deposition is blocked when PML and pRB are unable to assemble together at these promoters. Using a mutant version of pRB that is defective for LXCXE-type interactions, we demonstrated that senescence actively stimulates interactions between PML and pRB through this conserved interaction domain on pRB. The complexity of PML protein isoforms that exist in senescent cells likely contributes to their interaction with pRB. This assembly step is key to understanding the events that commit senescent cells to a permanent cell cycle arrest, and our study adds important new knowledge to ongoing work on this question.
Previous work using Rb1∆L/∆L MEFs and mice has allowed us to probe the circumstances where pRB uses LXCXE-type interactions in cell cycle arrest. Surprisingly, pRB–LXCXE interactions are critical for stress-responsive growth arrest, but not in reversible growth arrest or cell cycle arrest in development, even though each paradigm of proliferative control is pRB-dependent.Citation10,Citation29,Citation30 Notably, senescent Rb1∆L/∆L MEFs can re-initiate DNA synthesis in response to ectopic E2F1 expression, whereas wild-type cells are resistant.Citation10 Furthermore, serially cultured Rb1∆L/∆L MEFs escape senescence more readily than their wild-type counterparts, suggesting that defective silencing of proliferative genes can compromise the long-term stability of senescence arrest.Citation10 For these reasons, we have described Rb1∆L/∆L cells as entering into a state of partial senescence, whereby morphological features of senescence and SA-β-gal activity are typical of senescent cells, but their arrest remains reversible. A role for PML in gene silencing in growth control has been suggested, but has been less clear. First, ectopic expression of PML has been shown to inhibit growth of a number of cancer cell lines.Citation31,Citation32 In addition, PML has been shown to be able to suppress the transformation of fibroblasts by activated oncogenes.Citation33,Citation34 Indeed, Pml−/− mice show increased susceptibility to cancer-promoting agents.Citation35,Citation36 Interestingly, in a recent study by Vernier et al. the authors showed that expression of PML in U2OS osteosarcoma cells results in the association of PML with E2F target genes and repression of their expression.Citation23 However, other studies have suggested that the formation of PML nuclear bodies is dispensable for induction of senescence, and that the individual PML proteins are key.Citation21 Our work reconciles these conflicting observations from 2 perspectives. Demonstration that endogenous PML proteins associate with E2F target promoters during the induction of senescence places PML in the right genomic location at the appropriate time to play an active role in repression of these genes by directing heterochromatin assembly. Furthermore, studies that suggest PML body formation is dispensable for senescence pre-date our description of incomplete senescence.Citation10 Thereby, cells that are incapable of assembling PML bodies, but that still become SA-β-gal positive, may be in a similar state of incomplete senescence, where the arrest is not yet permanent.
In our studies we demonstrate that Hras-V12 expression in Rb1∆L/∆L fibroblasts triggers a similar DNA damage response as in control Rb1+/+ cells. In addition, the quantity of PML bodies that are induced by Hras-V12 in Rb1∆L/∆L and Rb1+/+ cells is similar. Differences only appear when PML fails to associate with E2F target genes in senescing Rb1∆L/∆L fibroblasts. We interpret this defect as resulting from the failure of a regulated PML–pRB assembly step. We describe this event as regulated assembly, because similar quantities of PML protein from proliferating cells fail to bind to GST-RB in our assays. This interaction assay is highly relevant to PML-pRB interactions in vivo, because it is disrupted by the same LXCXE binding cleft mutation as is present in Rb1∆L/∆L fibroblasts. Understanding how PML engages this binding site on pRB is complex. Since PML is not reported to contain an LXCXE motif, it may be that the interaction is indirect and could be mediated by one or more proteins that bind to pRB through its LXCXE binding cleft. HDACs are one such potential candidate, as they have been shown to interact with both pRB and PML.Citation36,Citation37 We don’t favor this interpretation, because our previous studies have indicated that HDAC-containing complexes interact with pRB in a GST-RB pull-down assay irrespective of the growth state of the cells.Citation29 Based on this line of reasoning, we expect that PML–pRB interactions are likely quite direct.
There are at least 6 isoforms of PML (I–VI) capable of forming nuclear bodies, and pRB has been shown to bind some isoforms preferentially.Citation28 Oncogene-induced senescence has also been shown to upregulate PML at the transcriptional and translational levels.Citation19,Citation20,Citation24 Thus, expression of PML increases in senescence, but our data indicates that relatively equal quantities of PML obtained from proliferating cells still fail to bind to GST-RB. We hypothesize that the signal to actively form pRB–PML interactions may be coincident with PML body assembly. Examination of the forms of PML present in nuclear extracts of senescent cells compared with proliferating indicates that senescence generates species of PML that are far larger than the predicted molecular weights of the largest PML isoforms. This implies that post-translational modifications, such as sumoylation, may trigger PML body assembly and/or direct the interaction with pRB.
The precise mechanism of how PML–pRB complexes inhibit the expression of E2F target genes is still unclear. Defective enrichment of the repressive histone modification H3K9me3 in Rb1∆L/∆L MEFs provides a clue. PML might recruit and/or facilitate the incorporation of this mark to render the genes transcriptionally inert. Indeed, both pRB and PML are reported to interact with Suv39h1, the enzyme capable of trimethylating histone H3K9.Citation38,Citation39 In addition, Suv39h1 knockout mice are defective for chromatin condensation in senescence.Citation7 Unfortunately, we were unable to reliably detect an interaction between pRB and Suv39h1 either by co-immunoprecipitation or by ChIP in extracts from senescent MEFs. It is possible that this interaction is very transient, and the conditions we used in our experiments were not conducive to detecting this interaction. Alternatively, a different enzyme may be responsible for incorporating this modification at E2F target gene promoters during senescence in a PML–pRB-dependent manner. Nevertheless, ectopic expression of PML-IV in Rb1∆L/∆L MEFs demonstrates that H3K9me3 addition to promoters is dependent on PML–pRB interactions and the regulated assembly processes described above. Previously, Nielsen et al.Citation39 have demonstrated that Suv39h enzymes passively interact with GST-RB in an LXCXE cleft-dependent manner. For this reason, we expect that the actual enzymatic methylation of H3K9 is downstream of a cell’s commitment to enter a permanently arrested state. Our data suggests that PML–pRB interactions are likely closer to the switch that converts reversible arrest to permanent through E2F target gene heterochromatinization. Future work in this area will need to focus on the signals that assemble PML–pRB complexes as they hold the key to understanding how senescent cells become committed to permanent cell cycle arrest.
Material and Methods
Cell culture
Mouse embryonic fibroblasts (MEFs) were generated from E13.5 embryos using standard procedures and cultured as previously described.Citation29 The Rb1∆L allele encodes I746A, N750A, and M754A substitutions, and is detected by PCR genotyping as previously reported.Citation29 All cells were cultured in growth medium that contained Dulbecco modified Eagle medium (DMEM) supplemented with L-glutamine, streptomycin, penicillin, and 10% fetal bovine serum. Cells were maintained in a humidified chamber at 37 °C with 5% CO2. MEFs used for our experiments were all at passage 3 or 4. At these passages, typically only 5–10% of cells in the culture are senescent as determined by senescence-associated β-galactosidase (SA-β-gal) staining, and we generally discard MEF preparations with higher levels of senescent cells. Retroviral transduction with pBABE-H-RasV12, pBABE-flag-PML-IV, or other pBABE-flag-PML constructs was as reported by Serrano et al.,Citation40 and viruses were packaged in Bosc-23 cells. Cells infected with viruses encoding Ras or PML-IV were pre-selected in 4 μg/ml puromycin for at least 3 d before re-plating and further culturing in selection medium for 1, 5, 8, or 10 d depending on the experiment. Senescence associated β-galactosidase (SA-β-gal) staining was performed as described.Citation40 Infections with Ad-GFP and Ad-E2F1 were performed as previously described,Citation10 and cells were cultured for an additional 48 h before labeling with BrdU for 16 h or preparing extracts. PML expression plasmids were a kind gift of Lawrence Banks.Citation41
Immunofluorescence
Cells on coverslips were fixed in 4% PFA for 10 min at room temperature (RT), permeablized with 0.5% triton-X-100 for 5 min at RT, blocked with 3% BSA/PBS for 15 min, followed by incubation with primary antibodies diluted in blocking buffer for 1 h at RT or overnight at 4 °C in a humidified chamber. Cells were washed in the blocking buffer 5 min each, 3 times. Cells were incubated with secondary antibody diluted in blocking buffer for 1 h. at RT. Cells were washed again 3 times in PBS followed by mounting on slides with mounting medium containing DAPI before analyzing on an Olympus Fluoview FV1000 confocal microscope.
GST pull-downs and immunoprecipitations
For GST pull-down assays, nuclear extracts were prepared as described before.Citation42 GST-tagged proteins were expressed in E. coli and purified using glutathione sepharose beads. Nuclear extracts were diluted in low-salt GSE buffer (20 mM Tris [pH 7.5], 200 mM NaCl, 1.5 mM MgCl2, 0.2 mM EDTA, 1 mM dithiothreitol [DTT], and 0.1% NP-40) and incubated with either GST, GST-RB (large pocket, amino acids 379–928), GST-RB∆L (large pocket, with I753A, N757A, and M761A substitutions), or GST-p107 (large pocket, amino acids 385–1069) for 1 h. Protein complexes were collected with 25 µl of glutathione sepharose bead slurry for 1 h. and eluted in 1× Laemmli buffer before using for western blots. For pRB co-immunoprecipitations (IPs), antibodies were cross-linked to Dynabeads using 5 mM Bis [sulfosuccinimidyl] suberate (BS3) (#21580 Thermo scientific). IPs were performed by incubating the nuclear extracts with cross-linked antibodies in the low-salt GSE buffer by rotating at 4 °C for 16 h followed by eluting in 1× Laemmli buffer before using for western blots.
Chromatin immunoprecipitations
Senescent MEFs were fixed in 1% formaldehyde in PBS for 10 min at room temperature. The reaction was stopped with glycine at a final concentration of 0.125 M. Chromatin was extracted as described beforeCitation43 with the following changes. Cells were sonicated for 30 min (30 s on; 30 s off, power high) using a Bioruptor sonicator (Diagenode). Diluted chromatin was pre-cleared at 4 °C for 1 h with 40 µl of a 50% slurry of magnetic Protein G beads (Invitrogen) pre-bound with respective IgG. Pre-cleared chromatin was incubated with 5 µg of the indicated antibody overnight at 4 °C with gentle rotation. DNA was purified by using a PCR purification kit from Invitrogen. Real-time PCR amplification was performed using iQSYBRGreen master mix on a Bio-Rad CFX Connect Real Time System. Primer sequences are: Ccne1 Forward primer: 5′ GAGAACTTGG TAGACCAACT CTAAA 3′, Ccne1 Reverse primer: 5′ GCAGCTGTTC TTAACTCTGT CTAGT 3′, Mcm3 Forward primer: 5′ GAATGCAGTG CTTCCTAGCC 3′, and Mcm3 Reverse primer: 5′ CGGAAGTTTA TGGTGGAGGA 3′.
Antibodies
Anti-H3K9me3 (07–442), anti-γH2AX (Ser139) (05–636), and mouse monoclonal anti-PML (MAB3738) antibodies are from Millipore. Anti-Flag (F1804), and anti-Actin (A2066) antibodies are from Sigma. Anti-pRB (M-153), anti-PML (polyclonal) antibody H-238 (SC-5621), and anti-p107 (SC-318) are from Santa Cruz Biotechnology. The anti-Cyclin E antibody was purchased from Abcam (ab7959). An anti-mouse pRB antibody was raised in sheep against the C-terminal 136 amino acids (Affinity Biologicals). Antibodies were affinity purified against a peptide corresponding to amino acids 867–881 of the mouse pRB protein using previously reported methods.Citation44
| Abbreviations: | ||
| PML | = | pro-myelocytic leukemia |
| RB | = | retinoblastoma |
| SAHF | = | senescence associated heterochromatic foci |
| SA-βgal | = | senescence-associated beta-galactosidase |
| LXCXE | = | leucine, any amino acid, cysteine, any amino acid, glutamate |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
The authors thank colleagues in the Dick Lab, the London Regional Cancer Program, and the Children’s Health Research Institute for advice throughout this study. PML expression vectors were a kind gift of Lawrence Banks (ICGEB, Trieste, Italy). ST was a member of the CaRTT training program during this research. FAD is the Wolfe Senior Fellow in Tumor Suppressor Genes at Western. This work was supported by a CIHR operating grant to FAD (MOP 64253).
Related Research Data
References
- Campisi J. Cellular senescence as a tumor-suppressor mechanism. Trends Cell Biol 2001; 11:S27 - 31; PMID: 11684439
- Chicas A, Wang X, Zhang C, McCurrach M, Zhao Z, Mert O, Dickins RA, Narita M, Zhang M, Lowe SW. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 2010; 17:376 - 87; http://dx.doi.org/10.1016/j.ccr.2010.01.023; PMID: 20385362
- Bartkova J, Rezaei N, Liontos M, Karakaidos P, Kletsas D, Issaeva N, Vassiliou LV, Kolettas E, Niforou K, Zoumpourlis VC, et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006; 444:633 - 7; http://dx.doi.org/10.1038/nature05268; PMID: 17136093
- Di Micco R, Fumagalli M, Cicalese A, Piccinin S, Gasparini P, Luise C, Schurra C, Garre’ M, Nuciforo PG, Bensimon A, et al. Oncogene-induced senescence is a DNA damage response triggered by DNA hyper-replication. Nature 2006; 444:638 - 42; http://dx.doi.org/10.1038/nature05327; PMID: 17136094
- Campisi J, Gaumont-Leclerc MF, Ferbeyre G. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513 - 22; http://dx.doi.org/10.1016/j.cell.2005.02.003; PMID: 15734683
- Campisi J. Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513 - 22; http://dx.doi.org/10.1016/j.cell.2005.02.003; PMID: 15734683
- Braig M, Lee S, Loddenkemper C, Rudolph C, Peters AH, Schlegelberger B, Stein H, Dörken B, Jenuwein T, Schmitt CA. Oncogene-induced senescence as an initial barrier in lymphoma development. Nature 2005; 436:660 - 5; http://dx.doi.org/10.1038/nature03841; PMID: 16079837
- Narita M, Nũnez S, Heard E, Narita M, Lin AW, Hearn SA, Spector DL, Hannon GJ, Lowe SW. Rb-mediated heterochromatin formation and silencing of E2F target genes during cellular senescence. Cell 2003; 113:703 - 16; http://dx.doi.org/10.1016/S0092-8674(03)00401-X; PMID: 12809602
- Chandra T, Kirschner K, Thuret JY, Pope BD, Ryba T, Newman S, Ahmed K, Samarajiwa SA, Salama R, Carroll T, et al. Independence of repressive histone marks and chromatin compaction during senescent heterochromatic layer formation. Mol Cell 2012; 47:203 - 14; http://dx.doi.org/10.1016/j.molcel.2012.06.010; PMID: 22795131
- Talluri S, Isaac CE, Ahmad M, Henley SA, Francis SM, Martens AL, Bremner R, Dick FA. A G1 checkpoint mediated by the retinoblastoma protein that is dispensable in terminal differentiation but essential for senescence. Mol Cell Biol 2010; 30:948 - 60; http://dx.doi.org/10.1128/MCB.01168-09; PMID: 20008551
- Chicas A, Kapoor A, Wang X, Aksoy O, Evertts AG, Zhang MQ, Garcia BA, Bernstein E, Lowe SW. H3K4 demethylation by Jarid1a and Jarid1b contributes to retinoblastoma-mediated gene silencing during cellular senescence. Proc Natl Acad Sci U S A 2012; 109:8971 - 6; http://dx.doi.org/10.1073/pnas.1119836109; PMID: 22615382
- Funayama R, Saito M, Tanobe H, Ishikawa F. Loss of linker histone H1 in cellular senescence. J Cell Biol 2006; 175:869 - 80; http://dx.doi.org/10.1083/jcb.200604005; PMID: 17158953
- Zhang R, Chen W, Adams PD. Molecular dissection of formation of senescence-associated heterochromatin foci. Mol Cell Biol 2007; 27:2343 - 58; http://dx.doi.org/10.1128/MCB.02019-06; PMID: 17242207
- Beauséjour CM, Krtolica A, Galimi F, Narita M, Lowe SW, Yaswen P, Campisi J. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J 2003; 22:4212 - 22; http://dx.doi.org/10.1093/emboj/cdg417; PMID: 12912919
- Dimova DK, Dyson NJ. The E2F transcriptional network: old acquaintances with new faces. Oncogene 2005; 24:2810 - 26; http://dx.doi.org/10.1038/sj.onc.1208612; PMID: 15838517
- Dannenberg J-H, van Rossum A, Schuijff L, te Riele H. Ablation of the retinoblastoma gene family deregulates G(1) control causing immortalization and increased cell turnover under growth-restricting conditions. Genes Dev 2000; 14:3051 - 64; http://dx.doi.org/10.1101/gad.847700; PMID: 11114893
- Sage J, Mulligan GJ, Attardi LD, Miller A, Chen S, Williams B, Theodorou E, Jacks T. Targeted disruption of the three Rb-related genes leads to loss of G(1) control and immortalization. Genes Dev 2000; 14:3037 - 50; http://dx.doi.org/10.1101/gad.843200; PMID: 11114892
- Sage J, Miller AL, Pérez-Mancera PA, Wysocki JM, Jacks T. Acute mutation of retinoblastoma gene function is sufficient for cell cycle re-entry. Nature 2003; 424:223 - 8; http://dx.doi.org/10.1038/nature01764; PMID: 12853964
- Ferbeyre G, de Stanchina E, Querido E, Baptiste N, Prives C, Lowe SW. PML is induced by oncogenic ras and promotes premature senescence. Genes Dev 2000; 14:2015 - 27; PMID: 10950866
- Pearson M, Carbone R, Sebastiani C, Cioce M, Fagioli M, Saito S, Higashimoto Y, Appella E, Minucci S, Pandolfi PP, et al. PML regulates p53 acetylation and premature senescence induced by oncogenic Ras. Nature 2000; 406:207 - 10; http://dx.doi.org/10.1038/35021000; PMID: 10910364
- Bischof O, Kirsh O, Pearson M, Itahana K, Pelicci PG, Dejean A. Deconstructing PML-induced premature senescence. EMBO J 2002; 21:3358 - 69; http://dx.doi.org/10.1093/emboj/cdf341; PMID: 12093737
- Jensen K, Shiels C, Freemont PS. PML protein isoforms and the RBCC/TRIM motif. Oncogene 2001; 20:7223 - 33; http://dx.doi.org/10.1038/sj.onc.1204765; PMID: 11704850
- Vernier M, Bourdeau V, Gaumont-Leclerc MF, Moiseeva O, Bégin V, Saad F, Mes-Masson AM, Ferbeyre G. Regulation of E2Fs and senescence by PML nuclear bodies. Genes Dev 2011; 25:41 - 50; http://dx.doi.org/10.1101/gad.1975111; PMID: 21205865
- de Stanchina E, Querido E, Narita M, Davuluri RV, Pandolfi PP, Ferbeyre G, Lowe SW. PML is a direct p53 target that modulates p53 effector functions. Mol Cell 2004; 13:523 - 35; http://dx.doi.org/10.1016/S1097-2765(04)00062-0; PMID: 14992722
- Zhang R, Poustovoitov MV, Ye X, Santos HA, Chen W, Daganzo SM, Erzberger JP, Serebriiskii IG, Canutescu AA, Dunbrack RL, et al. Formation of MacroH2A-containing senescence-associated heterochromatin foci and senescence driven by ASF1a and HIRA. Dev Cell 2005; 8:19 - 30; http://dx.doi.org/10.1016/j.devcel.2004.10.019; PMID: 15621527
- Ye X, Zerlanko B, Zhang R, Somaiah N, Lipinski M, Salomoni P, Adams PD. Definition of pRB- and p53-dependent and -independent steps in HIRA/ASF1a-mediated formation of senescence-associated heterochromatin foci. Mol Cell Biol 2007; 27:2452 - 65; http://dx.doi.org/10.1128/MCB.01592-06; PMID: 17242198
- Narita M, Narita M, Krizhanovsky V, Nuñez S, Chicas A, Hearn SA, Myers MP, Lowe SW. A novel role for high-mobility group a proteins in cellular senescence and heterochromatin formation. Cell 2006; 126:503 - 14; http://dx.doi.org/10.1016/j.cell.2006.05.052; PMID: 16901784
- Alcalay M, Tomassoni L, Colombo E, Stoldt S, Grignani F, Fagioli M, Szekely L, Helin K, Pelicci PG. The promyelocytic leukemia gene product (PML) forms stable complexes with the retinoblastoma protein. Mol Cell Biol 1998; 18:1084 - 93; PMID: 9448006
- Isaac CE, Francis SM, Martens AL, Julian LM, Seifried LA, Erdmann N, Binné UK, Harrington L, Sicinski P, Bérubé NG, et al. The retinoblastoma protein regulates pericentric heterochromatin. Mol Cell Biol 2006; 26:3659 - 71; http://dx.doi.org/10.1128/MCB.26.9.3659-3671.2006; PMID: 16612004
- Andrusiak MG, Vandenbosch R, Dick FA, Park DS, Slack RS. LXCXE-independent chromatin remodeling by Rb/E2f mediates neuronal quiescence. Cell Cycle 2013; 12:1416 - 23; http://dx.doi.org/10.4161/cc.24527; PMID: 23574720
- Le XF, Vallian S, Mu ZM, Hung MC, Chang KS. Recombinant PML adenovirus suppresses growth and tumorigenicity of human breast cancer cells by inducing G1 cell cycle arrest and apoptosis. Oncogene 1998; 16:1839 - 49; http://dx.doi.org/10.1038/sj.onc.1201705; PMID: 9583681
- Fagioli M, Alcalay M, Tomassoni L, Ferrucci PF, Mencarelli A, Riganelli D, Grignani F, Pozzan T, Nicoletti I, Grignani F, et al. Cooperation between the RING + B1-B2 and coiled-coil domains of PML is necessary for its effects on cell survival. Oncogene 1998; 16:2905 - 13; http://dx.doi.org/10.1038/sj.onc.1201811; PMID: 9671411
- Liu JH, Mu ZM, Chang KS. PML suppresses oncogenic transformation of NIH/3T3 cells by activated neu. J Exp Med 1995; 181:1965 - 73; http://dx.doi.org/10.1084/jem.181.6.1965; PMID: 7759992
- Mu ZM, Chin KV, Liu JH, Lozano G, Chang KS. PML, a growth suppressor disrupted in acute promyelocytic leukemia. Mol Cell Biol 1994; 14:6858 - 67; PMID: 7935403
- Salomoni P, Pandolfi PP. The role of PML in tumor suppression. Cell 2002; 108:165 - 70; http://dx.doi.org/10.1016/S0092-8674(02)00626-8; PMID: 11832207
- Wang ZG, Delva L, Gaboli M, Rivi R, Giorgio M, Cordon-Cardo C, Grosveld F, Pandolfi PP. Role of PML in cell growth and the retinoic acid pathway. Science 1998; 279:1547 - 51; http://dx.doi.org/10.1126/science.279.5356.1547; PMID: 9488655
- Dick FA. Structure-function analysis of the retinoblastoma tumor suppressor protein - is the whole a sum of its parts?. Cell Div 2007; 2:26; http://dx.doi.org/10.1186/1747-1028-2-26; PMID: 17854503
- Carbone R, Botrugno OA, Ronzoni S, Insinga A, Di Croce L, Pelicci PG, Minucci S. Recruitment of the histone methyltransferase SUV39H1 and its role in the oncogenic properties of the leukemia-associated PML-retinoic acid receptor fusion protein. Mol Cell Biol 2006; 26:1288 - 96; http://dx.doi.org/10.1128/MCB.26.4.1288-1296.2006; PMID: 16449642
- Nielsen SJ, Schneider R, Bauer UM, Bannister AJ, Morrison A, O’Carroll D, Firestein R, Cleary M, Jenuwein T, Herrera RE, et al. Rb targets histone H3 methylation and HP1 to promoters. Nature 2001; 412:561 - 5; http://dx.doi.org/10.1038/35087620; PMID: 11484059
- Serrano M, Lin AW, McCurrach ME, Beach D, Lowe SW. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell 1997; 88:593 - 602; http://dx.doi.org/10.1016/S0092-8674(00)81902-9; PMID: 9054499
- Guccione E, Lethbridge KJ, Killick N, Leppard KN, Banks L. HPV E6 proteins interact with specific PML isoforms and allow distinctions to be made between different POD structures. Oncogene 2004; 23:4662 - 72; http://dx.doi.org/10.1038/sj.onc.1207631; PMID: 15107834
- Cecchini MJ, Dick FA. The biochemical basis of CDK phosphorylation-independent regulation of E2F1 by the retinoblastoma protein. Biochem J 2011; 434:297 - 308; http://dx.doi.org/10.1042/BJ20101210; PMID: 21143199
- Cortázar D, Kunz C, Selfridge J, Lettieri T, Saito Y, MacDougall E, Wirz A, Schuermann D, Jacobs AL, Siegrist F, et al. Embryonic lethal phenotype reveals a function of TDG in maintaining epigenetic stability. Nature 2011; 470:419 - 23; http://dx.doi.org/10.1038/nature09672; PMID: 21278727
- Coschi CH, Martens AL, Ritchie K, Francis SM, Chakrabarti S, Berube NG, Dick FA. Mitotic chromosome condensation mediated by the retinoblastoma protein is tumor-suppressive. Genes Dev 2010; 24:1351 - 63; http://dx.doi.org/10.1101/gad.1917610; PMID: 20551166