
Abstract
As a member of imitation switch (ISWI) family in ATP-dependent chromatin remodeling factors, RSF complex consists of SNF2h ATPase and Rsf-1. Although it has been reported that SNF2h ATPase is recruited to DNA damage sites (DSBs) in a poly(ADP-ribosyl) polymerase 1 (PARP1)-dependent manner in DNA damage response (DDR), the function of Rsf-1 is still elusive. Here we show that Rsf-1 is recruited to DSBs confirmed by various cellular analyses. Moreover, the initial recruitment of Rsf-1 and SNF2h to DSBs shows faster kinetics than that of γH2AX after micro-irradiation. Signals of Rsf-1 and SNF2h are retained over 30 min after micro-irradiation, whereas γH2AX signals are gradually reduced at 10 min. In addition, Rsf-1 is accumulated at DSBs in ATM-dependent manner, and the putative pSQ motifs of Rsf-1 by ATM are required for its accumulation at DSBs. Furtheremore, depletion of Rsf-1 attenuates the activation of DNA damage checkpoint signals and cell survival upon DNA damage. Finally, we demonstrate that Rsf-1 promotes homologous recombination repair (HRR) by recruiting resection factors RPA32 and Rad51. Thus, these findings reveal a new function of chromatin remodeler Rsf-1 as a guard in DNA damage checkpoints and homologous recombination repair.
Introduction
Upon DNA damage, cells strive to sustain genomic integrity by coordinating with activation of DNA damage checkpoint signals and the proper repair machineries. DNA double-strand breaks (DSBs) repair is initiated by ataxia-telangiectasia mutated (ATM), which phosphorylates H2AX (γH2AX), as known for histone marker in DNA damage response (DDR). In response to DNA damage, γH2AX spreads immediately for mega-bases away from DNA lesions.Citation1,Citation2 In turn, γH2AX offers a platform to propagate and sustain DNA damage signaling cascades, such as mediator of DNA damage checkpoint protein 1 (MDC1), breast cancer susceptibility 1 (BRCA1), or p53-binding protein 1 (53BP1) and damage repair machineries.Citation3-Citation5 Next, these signals serve to recruit DNA damage repair factors. There are 2 different major DNA repair pathways: non-homologous end-joining (NHEJ), which is error-prone and homologous recombination (HR), which is error-free.Citation6,Citation7 NHEJ occurs in all phases of the cell cycle, but mainly presents in G1 phase, because this repair pathway does not need sister chromatids or homologous templates. On the other hand, HR is dependent on the presence of homologous DNA templates or sister chromatids, which is the reason why HR is limitedly involved from S to G2 phase.Citation8 Since HR-deficient cells show hypersensitivity to DNA-damaging agents such as methyl methanesufonate (MMS) and radio-mimetic drug, phleomycin, defects in HR lead to accumulation of unrepaired chromatin breaks.Citation9 Furthermore, replication protein A (RPA) and Rad51 are important to intermediate the DNA template switch, and their reduced accumulation to DSBs and inactivation significantly reduce HR efficiency.Citation10
Recently, several studies show that chromatin remodeling factors are important for recruiting DNA repair proteins to access into DNA in DDR.Citation8,Citation11 In eukaryotes, 4 families of chromatin remodeling factors have been identified: INO80, SWI/SNF, CHD4, and ISWI.Citation12 Most of them have been reported to be recruited at DNA lesions to mobilize the nucleosome and remodel the chromatin structure. INO80 was initially found to be required for DNA repair in yeast.Citation13-Citation15 Later, it was reported that mammalian INO80 is also involved in DSB repair through its role in DNA end resection.Citation16 SWI/SNF complexes are shown to promote the DSB repairs by facilitating γH2AX induction.Citation17,Citation18 CHD4 also regulates both DNA damage signaling and repair pathways.Citation19-Citation21 In the ISWI family, ACF1 is reported to be required for DNA repair by interacting with KU70/80 complex.Citation22 In addition, SNF2h ATPase, the catalytic subunit in ISWI family, is also known to be recruited at DSBs in a PARP-dependent manner.Citation23
RSF complex, consisting of Rsf-1 (remodeling and spacing factor-1) and SNF2h ATPase, was initially identified as a chromatin assembly factor that deposits nucleosomes and regulates the spaced nucleosome.Citation24 As a subunit of RSF complex, Rsf-1 has been studied in clinical research, and its overexpression has been found in many types of cancer patients.Citation25-Citation29 Recent studies about Rsf-1 showed that Rsf-1 overexpression induces DNA damage response through ATM-Chk2-p53-p21 pathway and inhibits cell cycle progression followed by apoptosis and chromosome instability.Citation30-Citation32
Even if Rsf-1 has been implicated in response to DNA damage, the biological function of Rsf-1 still remains unclear. Here, we show that the accumulation of Rsf-1 by ATM to DSBs facilitates DNA damage signaling pathway and promotes homologous recombination repair.
Results
Rsf-1 is recruited at DSB(s)
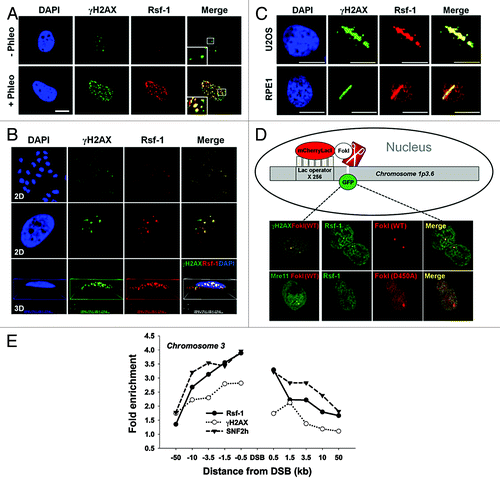
To understand the role of Rsf-1 in DNA damage response (DDR), we first examined whether Rsf-1 is recruited at DNA double-strand breaks (DSBs) upon DNA damage. Rsf-1 formed DNA damage foci with DNA damaging agent phleomycin (radio-mimetic drug making DSBs), which is co-localized with γH2AX (pS139) and pATM (pS1981), as the major hallmarks of DSBs. Furthermore, to demonstrate whether its localization is specific to DSBs, we analyzed 2-dimentional (2D) and 3-dimentional (3D) images by workstation program (Nikon). As expected, Rsf-1 and γH2AX or Rsf-1 and pATM are co-localized at DSBs (; Fig. S1). Previously, it has been shown that Rsf-1 is overexpressed in cancer cells rather than in normal cells.Citation25-Citation29 To confirm clearly if the recruitment of Rsf-1 at DSBs is not specific in cancer cells, we immunostained the endogenous Rsf-1 with γH2AX at 10 min after micro-irradiation in normal human RPE1 and U2OS cancer cells, and found that Rsf-1 is indeed co-localized with γH2AX at the micro-irradiated sites in both cell lines (). Again, in order to exclude the possibility that the recruitment of Rsf-1 at micro-irradiated sites is caused by non-specific damaging effects other than by DNA double-strand breaks, we validated the recruitment of Rsf-1 in the stably integrated LacO–LacI nuclease system.Citation33 Briefly, the fused mCherry-LacI with FokI endonuclease (mCherry-LacI-FokI) generates a single double-strand break on chromosome 1 stably integrated with LacO repeats (, diagram). To detect a single focus at DSB in a single cell, H2AX-GFP, Mre11-YFP, or Rsf-1-GFP was transfected with mCherry-LacI-FokI (WT or inactive nuclease [D450A]). As expected, H2AX-GFP and Mre11-YFP were co-localized with FokI wild-type nuclease to FokI-induced single break site. Furthermore, Rsf-1-GFP was co-localized with FokI wild-type at this site, whereas the nuclease-deficient mutant form of FokI (D450A) did not induce a focus of Rsf-1-GFP at the site (). The recruitment of Rsf-1 at DSB was again confirmed using K230-ZFN, which is zinc finger nuclease generating unique DSB on chromosome 3 specifically (see the previous reference to detail).Citation34 By chromatin immunoprecipitation (ChIP) assay, we demonstrated the accumulation of Rsf-1 at DSB together with γH2AX (). Furthermore, SNF2h ATPase, given that SNF2h is recruited in a poly(ADP-ribosyl) polymerase 1 (PARP1)-dependent manner,Citation23 was also recruited at DSB on chromosome 3. Thus, these results strongly indicate that Rsf-1 is recruited at DSBs upon DNA damage and may function in DDR.
Figure 1. Rsf-1 is recruited at DNA double-strand break site(s). (A and B) U2OS cells were immunostained with anti-Rsf-1 and anti-γH2AX at 2 h after treatment of phleomycin (+Phleo) (50 μg/ml) or untreatment of phleomycin (-Phleo). Scale bar, 10 μm. The damage foci were analyzed in 2-dimentional (2D) and 3-dimentional (3D) image with Workstation (Nikon). (C) U2OS and RPE1 cells were pre-sensitized with BrdU (10 μM) for 30 h, followed by laser micro-irradiation. Both cell lines were immunostained with anti-Rsf-1 and anti-γH2AX at 10 min after micro-irradiation. (D) Diagram of the recruitment of GFP-fused proteins and mCherry-LacI-FokI (wild-type and D450A mutant) on chromosome 1p3.6, stably integrated with LacO repeats, and co-localization of GFP-fused proteins and FokI wild-type (WT) or FokI mutant (D450A) in live cells. H2AX-GFP and Mre11-YFP used as controls. (E) Enrichment of Rsf-1, SNF2h, and γH2AX on site-specific DSB on chromosome 3. DSB was induced by transfection with K230-ZFN in U2OS cells, and fold enrichment by ChIP was divided by input.
Rsf-1 is rapidly recruited and accumulated at DSBs
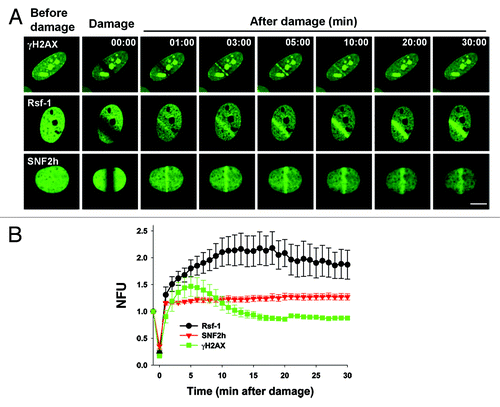
To investigate how Rsf-1 is involved in DDR, we further examined the kinetics of Rsf-1, γH2AX, and SNF2h after micro-irradiation. The initial accumulation of Rsf-1-GFP to DSBs was observed at 1 min after micro-irradiation, and this signal was continuously accumulated to DSBs over 30 min (; Video S2). However, the initial accumulation of H2AX-GFP to DSBs was apparent at 3 min and signal of H2AX-GFP was disappeared at 10 min after micro-irradiation (; Video S1). Previous studies showed that SNF2h, the catalytic subunit of RSF complex (Rsf-1 and SNF2h), is accumulated at DSBs in a PARP-dependent manner.Citation23 We next monitored SNF2h accumulation to DSBs after micro-irradiation. Consistent with the previous data, SNF2h-GFP was initially accumulated at 1 min and remained in a steady level over 30 min as similar in Rsf-1 signal (; Video S3). Unexpectedly, the accumulation of Rsf-1-GFP to DSBs was more intensive compared with that of SNF2h-GFP after micro-irradiation. Thus, these results suggest that Rsf-1 is not only required in response to DNA damage, but it might be also function in a different way than SNF2h.
Figure 2. The kinetics of accumulation of Rsf-1, SNF2h, and γH2AX at DSBs. (A) U2OS cells were transfected with Rsf-1-GFP, SNF2h-GFP, or H2AX-GFP, and treated with BrdU (10 μM) for 30 h, followed by laser micro-irradiation. Scale bar, 10 μm. (B) Quantitative analysis of the fluorescence intensity in result (A). The fluorescence intensity at micro-irradiated sites was normalized by the intensity around the background. The normalized fluorescence units were plotted against time. The plotted values are mean ± SEM of more than 10 individual cells.
ATM-dependent accumulation of Rsf-1 at DSBs
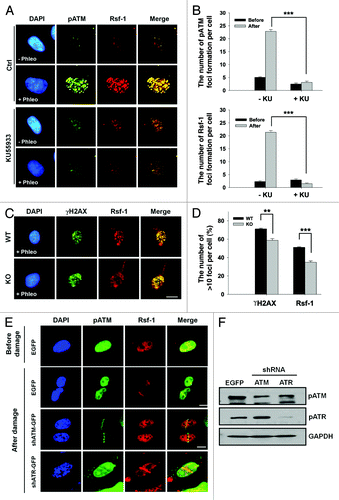
Based on the large-scale proteomic analysis of ATM and ATR substrates and mass spectrometry-based quantitative proteomics, Rsf-1 was found to be one of the potential substrates for ATM/ATR and its phosphorylation is upregulated upon DNA damage.Citation35,Citation36 Since ATM kinase is involved in the earliest step in DDR and the recruitment of Rsf-1 was rapidly occurred to DSBs after micro-irradiation, we focused on the effect of ATM on the function of Rsf-1 upon DNA damage. We examined the foci formation of Rsf-1 and pATM after treatment with ATM inhibitor KU55933 prior to adding phleomycin. The average number of foci formation of Rsf-1 and pATM in cells pre-incubated with KU55933 was compared with the average number of foci in control cells without KU55933 (). These results suggest that ATM inhibition diminished the foci formation of Rsf-1 upon DNA damage. In order to validate again whether the foci formation of Rsf-1 depends on ATM upon DNA damage, we treated human A-T cells (ATM-knockout fibroblasts) with phleomycin and counted foci formation (>10 foci) of γH2AX or Rsf-1 in ATM wild-type and ATM-knockout cells. As expected, since it had been reported that DNA damage can induce the foci formation of γH2AX by all 3 PI 3-kinases (ATM, ATR, and DNA-PK),Citation37 the foci formation of γH2AX was not reduced dramatically in ATM wild-type cells vs. ATM-deficient cells upon DNA damage. In contrast to γH2AX, the foci formation of Rsf-1 was attenuated significantly in ATM-KO cells, compared with ATM wild-type cells (). Thus, we speculated that Rsf-1 could be affected by prolonged loss of ATM in ATM-KO cells, altering the chromatin structure of these cells through altered protein modification upon DNA damage. Next, we performed the micro-irradiation in ATM- or ATR-depleted cells using short hairpin RNA (shRNA). The accumulation of Rsf-1 was impaired in ATM-depleted cells, while ATR depletion did not affect the accumulation of Rsf-1 (). These data therefore provide that ATM is required for accumulation of Rsf-1 to DSBs in DDR, rather than ATR.
Figure 3. The accumulation of Rsf-1 at DSBs is dependent on ATM activity. (A) U2OS cells were untreated (Ctrl) or treated with ATM inhibitor (KU55933) (20 μm) prior to phleomycin treatment (+Phleo) or untreatment (−Phleo) and immunostained with anti-pATM (pS1981) and anti-Rsf-1. (B) Quantitative analysis of result in (A). “Before” means untreatment of phleomycin; “after” means treatment of phleomycin. The average number (mean ± S.E.M.) of foci per cell was counted from over 100 cells. ***P < 0.005 by Student t test. (C) Human fibroblast wild-type (WT) or ATM knockout (KO) cell was untreated (−Phleo), or treated with phleomycin (+Phleo) for 2 h and immunostained with anti-γH2AX and anti-Rsf-1. Scale bar, 10μm. (D) Quantitative analysis of result in (C). The percentage of foci-positive cells (>10 foci per cell) was counted. **P < 0.01, ***P < 0.005 by Student t test. (E) U2OS cells were transfected with EGFP, shATM, or shATR and pre-sensitized with BrdU for 30 h, followed by laser micro-irradiation. Cells were immunostained with anti-Rsf-1 and anti-pATM at 1 h after micro-irradiation. Scale bar, 10 μm. (F) The level of pATM (pS1981) and pATR (pS428) was analyzed by western blot.
The putative pSQ motifs of Rsf-1 by ATM are required for its accumulation at DSBs
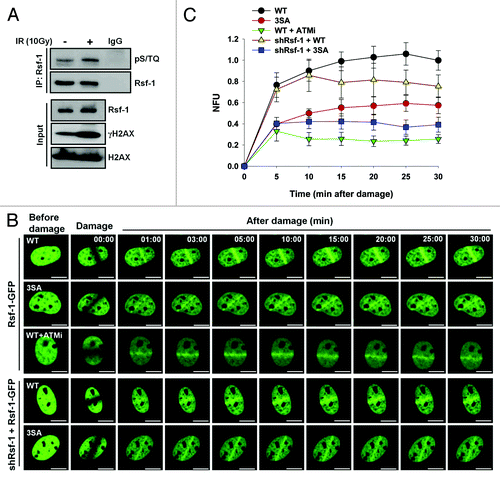
Rsf-1 has the putative motifs (pS524, pS1226, pS1325) for ATM.Citation35,Citation36 We thus focused on whether these motifs are important for accumulation of Rsf-1 at DSBs. To evaluate whether the motifs of Rsf-1 by ATM are important in DDR, we checked its phosphorylation by phspho-specific antibody (α-pSQ/TQ). The phosphorylation level of Rsf-1 was increased in DNA-damaging condition (). Next, we examined the recruitment of Rsf-1 to DSBs after treatment with ATM inhibitor KU55933 prior to micro-irradiation. In the presence of the ATM inhibitor, Rsf-1 recruitment to DSBs was significantly reduced compared with its recruitment in normal condition (). Next, we examined whether the putative motifs of Rsf-1 are important for its recruitment at DSBs. After substituting 3 serine residues (S524, S1226, and S1325) to alanines, we tested the accumulation of Rsf-1 3SA mutant after micro-irradiation. Interestingly, overexpression of the 3SA mutant (3SA-GFP) showed reduced accumulation to DSBs, compared with overexpression of Rsf-1 wild-type (WT-GFP). Again, after Rsf-1-GFP wild-type or 3SA mutant was transfected into stably Rsf-1-depleted cells (HeLa), we found that the accumulation of 3SA mutant was declined at DNA lesions compared with that of wild-type (). Thus, these data suggest that the pSQ motifs of Rsf-1 are phosphorylated by ATM upon DNA damage, and its phosphorylation is important for the accumulation of Rsf-1 to DSBs.
Figure 4. The putative pSQ motifs of Rsf-1 by ATM are required for its accumulation at DSBs. (A) U2OS cells were irradiated at 10 Gy and harvested at 1 h after laser micro-irradiation. Whole-cell extracts were lysed and immunoprecipitated with Rsf-1 antibody. The immunoprecipitated was detected with pSQ/TQ antibody. γH2AX antibody used as a DNA damage control. (B) Rsf-1-GFP (WT) or Rsf-1 mutant (3SA) was transfected in U2OS cells and pre-sensitized with BrdU for 30 h, followed by laser micro-irradiation. ATM inhibitor KU55933 was pre-treated for 12 h prior to micro-irradiation. Scale by bar, 10 μm. (C) Quantitative analysis of fluorescence intensity at DSBs. The accumulated fluorescence intensity at micro-irradiated sites was normalized the background around the sites. The values (mean ± S.E.M.) were calculated and plotted against time from at least 10 individual cells.
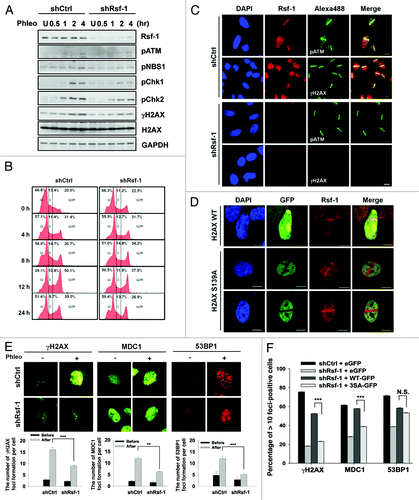
Depletion of Rsf-1 attenuates DNA damage checkpoint signals
In general, DNA damage induces ATM/ATR activation, followed by the phosphorylation of γH2AX and recruitment of the mediators, including MDC1, BRCA1, 53BP1, and others.Citation38 Thus, the accumulation of Rsf-1 in an ATM-dependent manner upon DNA damage led us to examine whether Rsf-1 is involved in DNA damage checkpoint signaling. We used stably Rsf-1-depleted cell lines (HeLa) and checked the DNA damage checkpoint signaling. Surprisingly, the depletion of Rsf-1 attenuated DNA damage checkpoints (pNBS1, pChk1/2), especially in the propagation of γH2AX (pS139) (). This result was confirmed again with a U2OS cell line in which Rsf-1 was transiently knocked-down by siRNA (Fig. S2A). Next, we checked whether the depletion of Rsf-1 affects the cell cycle progression upon DNA damage. In consistent with previous data,Citation39 FACS analysis demonstrated that cell cycle progression remained unchanged in Rsf-1-depleted or control cells at 12 h after the exposure to IR (Fig. S2B). However, in the presence of phleomycin, G2/M arrest at 12 h was weakened in Rsf-1-depleted cells, compared with control cells (). These data indicated that Rsf-1 is required to regulate the DNA damage checkpoint at G2/M in continuously DNA damaged conditions. Next, we monitored the accumulation of γH2AX and pATM at 10 min after micro-irradiation. The accumulation of γH2AX was dramatically abrogated in Rsf-1-depleted cells, whereas pATM signal was unlikely to be reduced in Rsf-1-depleted cells (). Considering that the Rsf-1 is one of the chromatin remodeling factors, Rsf-1 might function on opened chromatin structure, which is mostly reflected on γH2AX propagation. In order to address the possibility that Rsf-1 works upstream of γH2AX, we tested whether H2AX phsphorylation affects the recruitment of Rsf-1 at DSBs. H2AX S139 is known to be the target site of phosphorylation by ATM. We found that Rsf-1 recruitment to DSBs was not affected by the overexpression of H2AX wild-type and S139A mutant (). We next checked the accumulation of the downstream cascades of ATM after DNA damage. The foci formation of MDC1 and 53BP1, including γH2AX, was significantly reduced in stably Rsf-1-depleted cells (). Furthermore, reintroduction of Rsf-1 WT restored the foci formation of γH2AX and MDC1, whereas reintroduction of Rsf-1 3SA mutant was unable to restore the foci formation of those signaling proteins (; Fig. S3). In addition, Rsf-1 3SA did not significantly affect the foci formation of 53BP1, suggesting that 53BP1 might be regulated by a different pathway in DDR. Taken together, these results demonstrate that Rsf-1 is necessary for DNA damage checkpoint signaling in ATM-dependent manner, and it is also enabled to work as an upstream for modulating DNA damage checkpoint cascades.
Figure 5. Depletion of Rsf-1 attenuated DNA damage checkpoints. (A) Stably Rsf-1-depleted (shRsf-1) or control (shCtrl) cells (HeLa) were treated with phleomycin (50 μg/ml) and harvested at each indicated time points. Whole-cell extracts were directly lysed with sample buffer. The activation of DNA damage checkpoints was examined by western blot with the indicated antibodies. U, untreated. (B) FACS profiles of stably Rsf-1-depleted (shRsf-1) or control (shCtrl) cells during continuous DNA damage with phleomycin. (C) Stably Rsf-1-knockdown or control U2OS cells were immunostained with anti-Rsf-1 and anti-γH2AX, or anti-Rsf-1 and anti-pATM at 1 h after laser micro-irradiation. Scale bar, 10 μm. (D) H2AX wild-type (WT) or H2AX mutant (S139A) was transfected into U2OS cells. Cells were immunostained with anti-Rsf-1 at 10 min after micro-irradiation. (E) Stably Rsf-1-depleted cells (U2OS) were treated with phleomycin for 2 h and immunostained with anti-γH2AX, anti-MDC1, and anti-53BP1. The average number (mean ± S.E.M.) of foci per cell was counted from over 100 cells. **P < 0.01, ***P < 0.005 by Student t test. (F) Stably Rsf-1-knock-downed U2OS cells were transfected with EGFP, Rsf-1 WT, and Rsf-1 3SA mutant and immunostained with anti-γH2AX, anti-MDC1, and anti-53BP1. The number of foci formation per cell was calculated. ***P < 0.005 by Student t test. N.S., not significant.
Rsf-1 facilitates homologous recombination repair by recruiting resection factors
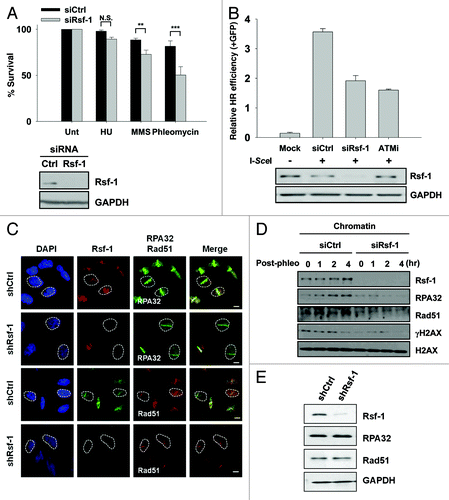
Finally, to evaluate the biological implication of Rsf-1 in DNA damage repair, we performed a survival assay in Rsf-1-depleted or control cells (U2OS). After treatment with different DNA damaging agents, such as HU, MMS, and phleomycin, we found that depletion of Rsf-1 compromised the colony generation, especially in phleomycin and MMS, rather than HU (). This result indicates that Rsf-1-depleted cells are sensitive to DNA double-strand breaks. Next, we focused on whether Rsf-1 is required for HRR. Notably, the relative GFP-positive cells were reduced in Rsf-1-depleted cells as well as in KU55933-treated cells, compared with control cells (). Since it has been shown that chromatin remodelers are required for homologous recombination repair (HRR) by recruiting resection factors, replication protein A (RPA) and Rad51, which are key molecules in searching for homologous template in HRR.Citation10,Citation40 We tested again whether Rsf-1 is critical for recruiting DNA end resection factors RPA32 and Rad51 in HRR. In stably Rsf-1-depleted cells, the recruitment of both resection factors, RPA32 and Rad51, to DSBs were dramatically declined at 1 h after micro-irradiation, which was converse to wild-type cells, even if the total protein level of both factors was unchanged in stably Rsf-1-depleted and control cells (). Again, in post-phleomycin conditions, we confirmed that these resection factors were less accumulated in chromatin in transiently Rsf-1-depleted cells (). Together, our findings suggest that Rsf-1 facilitates HRR through recruiting resection factors RPA32 and Rad51.
Figure 6. Rsf-1 facilitates homologous recombination repair. (A) The number of U2OS cells transfected with siCtrl and siRsf-1 was seeded and treated with different kinds of drugs; HU (100 mM), MMS (0.0002%), and phleomycin (50 μg/ml). The relative percentage of cell survival in control (siCtrl) and Rsf-1-depleted (siRsf-1) cells was calculated. N.S., not significant. **P < 0.01, ***P < 0.005 by Student t test. Expression level of Rsf-1 in whole-cell lysates was confirmed by western blot. (B) DR-GFP cells were transfected with control siRNA (siCtrl) or Rsf-1 siRNA (siRsf-1). At 48 h after siRNA transfection, I-SceI was transfected to induce DSB. ATM inhibitor (KU55933) was treated before transfection of I-SceI. GFP-positive cells were counted using FACS at another 72 h. (C) U2OS Rsf-1-depleted cells were pre-sensitized with BrdU for 30 h, followed by laser micro-irradiation. These cells were immunostained with anti-Rsf-1 and anti-RPA32 or anti-Rsf-1 and anti-Rad51, and imaged using confocal microscope. (D) RPE1 cells were transfected with Rsf-1 siRNA or control siRNA for 72 h and treated with phleomycin (10 μg/ml) for 1 h. The chromatin fractions were isolated at each time points released after treatment with phleomycin. The level of chromatin-bound proteins was analyzed by western blots. (E) The total level of Rsf-1, RPA32, and Rad51 in stably Rsf-1-depleted and control cells (U2OS).
Discussion
Here, we have uncovered the role of Rsf-1 as a guard in DNA damage signals and homologous recombination repair upon DNA damage response. Furthermore, we have established the foci formation of Rsf-1 at DNA double-strand break site(s) in normal and cancer cells, as shown by cellular analyses of DNA damage response (immunoflourescent microscope, micro-irradiation, stably integrated LacO-LacI nuclease system for single focus) and confirmed by chromatin immunoprecipitation (ChIP) at specifically single-break site on chromosome 3 induced by K230-ZFN nuclease. This means, bona fide, that Rsf-1 is recruited at DSB(s) in vivo. Furthermore, Rsf-1 was immediately recruited at DSBs in company with SNF2h. In particular, Rsf-1 signals to DSBs showed more intensive kinetics than SNF2h signals at the initial step after micro-irradiation, and the recruitment of Rsf-1 and SNF2h to DSBs was earlier than that of γH2AX. γH2AX was disappeared around 10 min after micro-irradiation, meanwhile Rsf-1 and SNF2h signals were still remained at DSBs over 30 min after micro-irradiation. Interestingly, Rsf-1 was continuously accumulated to DSBs even until 2 h after micro-irradiation, whereas SNF2h signals were gradually declined from DSBs (data not shown). Although the RSF complex consists of Rsf-1 and SNF2h, they are unlikely to have the same function in DNA damage response, since depletion of either one does not influence their accumulation to DSBs mutually.Citation39 This suggests that chromatin remodeler Rsf-1 has distinct roles from SNF2h in response to DNA damage.
It has been reported that Rsf-1 is the target of ATM/ATR and has the putative pSQ motifs by ATM/ATR.Citation35,Citation36 We identified that the accumulation of Rsf-1 at DSBs occurred in an ATM-dependent manner, and its phosphorylation on the putative motifs pSQ by ATM was required for maintaining its signal to DSBs from Rsf-1 overexpression and depletion experiments. Recently, Beli et al.Citation36 reported many proteins as ATM/ATR/DNA-PK substrates via proteomic investigation, and Rsf-1 was one of the candidates with more than 10 putative motifs in DNA damage conditions. Thus, even if we provided evidence for the significance of the 3 putative pSQ sites (pS524, pS1226, and pS1325) of Rsf-1 by ATM, there is also the possibility that Rsf-1 might have more phosphorylation sites by ATM besides these sites in response to DNA damage. Furthermore, we found that activity/accumulation of ATM-dependent DNA damage signaling cascades (pNBS1, pChk1/2, γH2AX, and p53BP1/γH2AX, MDC1, and 53BP1, respectively) upon DNA damage is decreased with depletion of Rsf-1 from transiently and stably knocked-down cell lines (U2OS and HeLa), strongly suggesting that Rsf-1 is required for the activation of DNA damage checkpoints. On the other hand, SNF2h/SMARCA5 is dispensable for DNA damage checkpoint activity, but it links poly(ADP-ribosyl) polymerase 1 (PARP1) in DDR.Citation23 Thus, it needs to be more evaluated whether Rsf-1 also cross-talks with PARP1 in PARylation-dependent manner upon DNA damage. In addition to regulation of DNA replication of ACF complex (SNF2h and ACF1), depletion or knockout of SNF2h caused defects in S-phase and cell death upon DNA damage in human.Citation41 Interestingly, abrogation of Rsf-1 caused sensitivity to the DNA damaging agents methyl methanesulfonate (MMS), causing collapsed replication fork; and phleomycin (radio-mimetic drug), causing DSBs. These findings suggest that Rsf-1 might have a dual function to sustain genomic integrity by regulating both DNA replication at S-phase and DNA damage checkpoint at G2/M.
WSTF, a member of WICH complex, was also isolated from RSF complex purified at mononucleosome level.Citation42 In DDR, the WICH complex interacts with H2AX and regulates H2AX phosphorylation (pY142), and its phosphorylation by WSTF is critical for maintaining H2AX phosphorylation (pS139) at the earliest step after DNA damage.Citation43 We also found that Rsf-1 facilitates the propagation of γH2AX at the initial step and is required for homologous recombination repair at a late step in DDR. In regards to SNF2h ATPase, as a major factor in ISWI family, SNF2h ATPase enables to switch the binding partners in DDR, at the earliest step to open the chromatin. It is also valuable to test whether swapping among their partners in ISWI family is necessary for DNA damage response and repair mechanism.
During the preparation of our manuscript, Helfricht et al.Citation39 found that Rsf-1 is required for NHEJ and HR pathway. Specially, Rsf-1 is necessary for deposition of centromeric proteins CENP-A, CENP-S, CENP-X, and NHEJ factor XRCC4 at DSBs, and these centromeric proteins at DSBs promote non-homologous end-joining (NHEJ). However, these proteins are dispensable for homologous recombination repair (HRR), which means Rsf-1 might be involved in HR in a different way (e.g., chromatin structure change or recruitment of homologous recombination factors). At a later step in DDR, chromatin remodeling complex also implicates the relaxation and eviction of histone variants from DSBs to promote HR repair.Citation40 In HR, double-stranded DNA should be resected to make single-stranded filaments for wrapping with replication protein A (RPA), and, in turn, this is replaced by Rad51 to search for homologous template.Citation10 Thus, in this step, RPA and Rad51 are key molecules to bind to single-stranded DNA by resection in homologous recombination repair (HRR), and their activation is important for HRR intermediate. Finally, we demonstrated that Rsf-1 is essential to recruit these recombination factors, RPA32 and Rad51, at DSBs, meaning that Rsf-1 needs to facilitate resection of broken DNA, coordinating with resection factors in HR pathway. One possible scenario is that Rsf-1 does modify chromatin structure (e.g., eviction or relaxation of histones) adjacent to DSBs, and the opened chromatin provides easy to access resection or recombination factors to facilitate HRR. Again, it will be interesting to delineate how Rsf-1 regulates recruitment and activity of these resection factors.
In summary, we have demonstrated the role of Rsf-1 in DDR. These findings provide critical insights linking chromatin remodeler Rsf-1 to DNA damage checkpoint and repair. Further investigation is necessary to identify the function of Rsf-1 in DDR, whether the function of Rsf-1 might be important to relax the chromatin structure at the sites of DNA damage, or whether Rsf-1 has a yet-to-be indentified function distinct from chromatin remodeler responding to DNA damage. Furthermore, finding the way to control Rsf-1 function might have clinical implications along with DNA damage reagents for malignancy.
Materials and Methods
Cell culture, reagents, and treatment
Human U2OS osteosarcoma and 293T embryonic kidney cells were grown in DMEM containing 10% fetal bovine serum (Gibco). RPE1 retinal pigment epithelial cells were cultured in DMEM/F12 (Gibco) containing 10% FBS and 10 µg/ml hygromycin. yz5 (human fibroblast) and S7 (human A-T fibroblast) cells were grown in DMEM with 15% FBS and 100 µg/ml hygromycin. To make the stable cell lines, U2OS and HeLa cells were infected with Rsf-1-shRNA plasmid using a lentiviral system and selected under puromycin (2 µg/ml) for 4 wk. Hydroxyurea, methyl methanesufonate, and phleomycin were purchased from Sigma-Aldrich. KU55933 was from Selleckchem.
Plasmids and RNA interference
pcDNA6-Rsf-1-V5, kindly gifted from Dr Ie-Ming Shin (Johns Hopkins Medical Institutions), was subcloned into pEGFP-N1 (GenBank Accession) to generate Rsf-1-GFP. The siRNA for Rsf-1 purchased from Ambion was transfected with neon electroporation (Invitrogen) or Lipofectamine 2000 (Invitrogen) and incubated for 72 h. The sequence for siRNA was 5′-GGAAAAUGUC AAACCCAUU-3′. H2AX-GFP, SNF2h-GFP, FokI wild-type, and mutant D450A, Mre11-YFP, shATM/shATR, or K230-ZFN was supported from Jongbum Kwon, Akira Yasui, Brendan D Price, Ho Chul Kang, Young-Joo Jang, or Jin-Soo Kim, respectively.
Mutagenesis
3SA-GFP (S524, S1226A, and S1325A) and H2AX-GFP (S139A) were constructed by using QuickChange Multi Site-Directed Mutagenesis (Stratagene). The following primers were used for mutagenesis:
5′-GTTCAATATC AGTCTTAGAG ATCCATGCTC AAAAAGCACA AATAGAGG-3′ for S524A, 5′-AGAAGACTCA GAAAGTGACG GTGCCCAGAA GAGTT-3′ for S1226A, and 5′-GCCTGCCCGT GACGCCCAGC CTAGGGTC-3′ for S1325A, 5′-CCAGGCGTCC CAGGAGGCCA AGCTTCGAAT TCTG-3′ for H2AX-S139A.
Laser micro-irradiation
For laser micro-irradiation, cells were seeded onto 35-mm round glass bottom dishes (SPL) and 5-bromo-2-deoxyuridine (BrdU, final 10 μM) was added to the medium for 30 h prior to micro-irradiation. For confocal imaging, we used an A1 confocal microscope (Nikon) with a 60× oil objective and a temperature-controlled chamber (37 °C, 5% CO2). Damage sites were induced by micro-irradiation with a 405 nm laser and 1.2 s irradiation time (32 lines/sec). Images were acquired every 1 s for 10 min, 30 min, or 2 h. At least 10 cells were irradiated in every experiment, and representative data are shown.
Immunofluorescence microscopy
Cells were grown on coverslips or 35-mm round glass bottom dishes, and then treated with phleomycin, or we performed micro-irradiation experiments as indicated, respectively. Coverslips or glass-bottom dishes were washed once with PBS, fixed with 4% paraformaldehyde and 0.1% TritonX-100 in PBS, and blocked for 30 min at room temperature in blocking buffer (3% BSA in PBS). Primary antibodies were incubated for 2 h at room temperature or overnight at 4 °C in the same buffer. Cells were then washed off 3 times in PBS before incubation with Alexa 488, 594, or cy3 in blocking buffer for 40 min at room temperature. Cells were again washed 3 times in PBS and mounted onto glass slides with DAPI-containing mounting solution. Images were analyzed using an Eclipse Ti (Nikon) or an A1 confocal microscope (Nikon) with a Nikon PlanApo 60× oil objective lens.
Survival assay
At 48 h after siRNA transfection into U2OS cells, cells were re-seeded at each density on 6-well plates and were treated with phleomycin, MMS, and HU. Cells were incubated in DMEM containing drugs for 1 h, washed out, and fresh media was added. Cells were allowed to form colonies for 10 d at 37 °C and stained with a mixture of 6.0% glutaraldehyde and 0.5% crystal violet, and the surviving colonies were counted.
Antibodies
We used the following antibodies: Rsf-1 (Abnova or abcam), pATM (pS1981; abcam), MDC1 (abcam), RPA32 (abcam), γH2AX (Millipore), SNF2h (Millipore), pATR (pS428; Cell signaling), pSQ/pTQ (Cell signaling), p53BP1 (Cell signaling), 53BP1 (Cell signaling), pChk1 (pS296; Cell signaling), pChk2 (pT68; Cell signaling), pNBS1(pS343; Cell signaling), H2AX (Cell signaling), Rad51 (Santa cruz), GAPDH (Santa cruz), GFP (Santa cruz). The HRP-conjugated anti-mouse IgG and anti-rabbit IgG secondary antibodies were purchased from Invitrogen. The following antibodies were used as secondary antibodies in immunofluorescence microscopy: Alexa Fluor 488, Alexa Fluor 594 (Molecular Probes), Cy3 (Jackson).
Immunoblotting
Cells were washed with PBS 3 times and directly lysed in sample buffer (0.3M Tris, pH 6.8, 12% SDS, 30% β-mercaptoethanol, 0.6% Bromophenol blue, 40% glycerol) with protease inhibitor cocktail (Roche). Proteins were separated by the gradient gel (4–20% acrylamide gel) and transferred onto nitrocellulose membrane (Whatman) or PVDF (Millipore). The membranes were blocked for 1 h with 3% BSA in TBST (Tris-buffered saline with 0.1% triton X-100, pH 7.6) and blotted with the appropriate primary antibodies 4 °C for overnight. After washing the membranes with TBST for 1 h, the membranes were incubated with the appropriated secondary antibodies (Invitrogen). Proteins were detected with ECL reagents (GE Healthcare).
Immunoprecipitation and chromatin fractionation
Cells were harvested and lysed in NETN buffer (50 mM TRIS-HCl, pH 8.0, 150 mM NaCl, 0.5% NP-40, and 5 mM EDTA) with protease and phosphatase inhibitors. The lysate was sonicated and centrifuged at 13 000rpm for 15 min at 4 °C. The supernatant was measured by Bradford assay and the equal amount of protein lysate was incubated with primary antibody for overnight at 4 °C. The immunoprecipitates were captured by Protein A sepharose Fast-Flow (GE Healthcare). The beads were washed 4 times with NETN buffer and boiled in 2× sample buffer. For chromatin fractionation, cells were processed as described previously.Citation32
Homologous recombination repair assay
For HR repair analysis, cells were processed as described previously.Citation44
Chromatin immunoprecipitation (ChIP)
For chromatin immunoprecipitation, cells were processed as described previously.Citation34
FokI assays
For the recruitment of Rsf-1-GFP and H2AX-GFP, or Mre11-YFP, cells were processed as described previously.Citation33
| Abbreviations: | ||
| Rsf-1 | = | remodeling and spacing factor-1 |
| DDR | = | DNA damage response |
| DSBs | = | DNA double-strand breaks |
| NHEJ | = | non-homologous end-joining |
| HR | = | homologous recombination |
| ChIP | = | chromatin immunoprecipitation |
Additional material
Download Zip (7.2 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank the members of Hyeseong Cho’s laboratory and Youngsoo Lee’s laboratory for critical discussions and Ie-Ming Shin, Brendan D Price, Jeremy Stark, Akira Yasui, Takashi Kohno, Ho Chul Kang, Jin-Soo Kim, Jongbum Kwon, and Yong Won Choi for kindly providing valuable reagents. The DSB reporter cell line (U2OS) was provided by S Janicki and D Spector (Wistar Institute and Cold Spring Harbor Laboratories, respectively). This work was supported by National Research Foundation of Korea grants funded by the Korea government (MSIP) (No. 2011-0030043:SRC) and (No. 2011-0017635:Mid-career Researcher Program).
Author Contributions
SM, SJ, JHJ, and HC designed experiments. SM, SJ, and JHJ performed most of the experiments and participated in data analysis. HSL and JSL supported the stable cell lines (HeLa). SC contributed to set up the micro-irradiation experiments. SM, SJ, JHJ, and HC wrote the paper.
Related Research Data
References
- Bonner WM, Redon CE, Dickey JS, Nakamura AJ, Sedelnikova OA, Solier S, Pommier Y. GammaH2AX and cancer. Nat Rev Cancer 2008; 8:957 - 67; http://dx.doi.org/10.1038/nrc2523; PMID: 19005492
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 16; http://dx.doi.org/10.1083/jcb.146.5.905; PMID: 10477747
- Lukas C, Melander F, Stucki M, Falck J, Bekker-Jensen S, Goldberg M, Lerenthal Y, Jackson SP, Bartek J, Lukas J. Mdc1 couples DNA double-strand break recognition by Nbs1 with its H2AX-dependent chromatin retention. EMBO J 2004; 23:2674 - 83; http://dx.doi.org/10.1038/sj.emboj.7600269; PMID: 15201865
- Stucki M, Clapperton JA, Mohammad D, Yaffe MB, Smerdon SJ, Jackson SP. MDC1 directly binds phosphorylated histone H2AX to regulate cellular responses to DNA double-strand breaks. Cell 2005; 123:1213 - 26; http://dx.doi.org/10.1016/j.cell.2005.09.038; PMID: 16377563
- Panier S, Durocher D. Regulatory ubiquitylation in response to DNA double-strand breaks. DNA Repair (Amst) 2009; 8:436 - 43; http://dx.doi.org/10.1016/j.dnarep.2009.01.013; PMID: 19230794
- Huertas P. DNA resection in eukaryotes: deciding how to fix the break. Nat Struct Mol Biol 2010; 17:11 - 6; http://dx.doi.org/10.1038/nsmb.1710; PMID: 20051983
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 8; http://dx.doi.org/10.1038/nature08467; PMID: 19847258
- Price BD, D’Andrea AD. Chromatin remodeling at DNA double-strand breaks. Cell 2013; 152:1344 - 54; http://dx.doi.org/10.1016/j.cell.2013.02.011; PMID: 23498941
- Osley MA, Tsukuda T, Nickoloff JA. ATP-dependent chromatin remodeling factors and DNA damage repair. Mutat Res 2007; 618:65 - 80; http://dx.doi.org/10.1016/j.mrfmmm.2006.07.011; PMID: 17291544
- West SC. Molecular views of recombination proteins and their control. Nat Rev Mol Cell Biol 2003; 4:435 - 45; http://dx.doi.org/10.1038/nrm1127; PMID: 12778123
- Lans H, Marteijn JA, Vermeulen W. ATP-dependent chromatin remodeling in the DNA-damage response. Epigenetics Chromatin 2012; 5:4; http://dx.doi.org/10.1186/1756-8935-5-4; PMID: 22289628
- Clapier CR, Cairns BR. The biology of chromatin remodeling complexes. Annu Rev Biochem 2009; 78:273 - 304; http://dx.doi.org/10.1146/annurev.biochem.77.062706.153223; PMID: 19355820
- Morrison AJ, Highland J, Krogan NJ, Arbel-Eden A, Greenblatt JF, Haber JE, Shen X. INO80 and gamma-H2AX interaction links ATP-dependent chromatin remodeling to DNA damage repair. Cell 2004; 119:767 - 75; http://dx.doi.org/10.1016/j.cell.2004.11.037; PMID: 15607974
- van Attikum H, Fritsch O, Hohn B, Gasser SM. Recruitment of the INO80 complex by H2A phosphorylation links ATP-dependent chromatin remodeling with DNA double-strand break repair. Cell 2004; 119:777 - 88; http://dx.doi.org/10.1016/j.cell.2004.11.033; PMID: 15607975
- van Attikum H, Fritsch O, Gasser SM. Distinct roles for SWR1 and INO80 chromatin remodeling complexes at chromosomal double-strand breaks. EMBO J 2007; 26:4113 - 25; http://dx.doi.org/10.1038/sj.emboj.7601835; PMID: 17762868
- Gospodinov A, Vaissiere T, Krastev DB, Legube G, Anachkova B, Herceg Z. Mammalian Ino80 mediates double-strand break repair through its role in DNA end strand resection. Mol Cell Biol 2011; 31:4735 - 45; http://dx.doi.org/10.1128/MCB.06182-11; PMID: 21947284
- Lee HS, Park JH, Kim SJ, Kwon SJ, Kwon J. A cooperative activation loop among SWI/SNF, gamma-H2AX and H3 acetylation for DNA double-strand break repair. EMBO J 2010; 29:1434 - 45; http://dx.doi.org/10.1038/emboj.2010.27; PMID: 20224553
- Park JH, Park EJ, Lee HS, Kim SJ, Hur SK, Imbalzano AN, Kwon J. Mammalian SWI/SNF complexes facilitate DNA double-strand break repair by promoting gamma-H2AX induction. EMBO J 2006; 25:3986 - 97; http://dx.doi.org/10.1038/sj.emboj.7601291; PMID: 16932743
- Smeenk G, Wiegant WW, Vrolijk H, Solari AP, Pastink A, van Attikum H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J Cell Biol 2010; 190:741 - 9; http://dx.doi.org/10.1083/jcb.201001048; PMID: 20805320
- Larsen DH, Poinsignon C, Gudjonsson T, Dinant C, Payne MR, Hari FJ, Rendtlew Danielsen JM, Menard P, Sand JC, Stucki M, et al. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J Cell Biol 2010; 190:731 - 40; http://dx.doi.org/10.1083/jcb.200912135; PMID: 20805324
- Polo SE, Kaidi A, Baskcomb L, Galanty Y, Jackson SP. Regulation of DNA-damage responses and cell-cycle progression by the chromatin remodelling factor CHD4. EMBO J 2010; 29:3130 - 9; http://dx.doi.org/10.1038/emboj.2010.188; PMID: 20693977
- Lan L, Ui A, Nakajima S, Hatakeyama K, Hoshi M, Watanabe R, Janicki SM, Ogiwara H, Kohno T, Kanno S, et al. The ACF1 complex is required for DNA double-strand break repair in human cells. Mol Cell 2010; 40:976 - 87; http://dx.doi.org/10.1016/j.molcel.2010.12.003; PMID: 21172662
- Smeenk G, Wiegant WW, Marteijn JA, Luijsterburg MS, Sroczynski N, Costelloe T, Romeijn RJ, Pastink A, Mailand N, Vermeulen W, et al. Poly(ADP-ribosyl)ation links the chromatin remodeler SMARCA5/SNF2H to RNF168-dependent DNA damage signaling. J Cell Sci 2013; 126:889 - 903; http://dx.doi.org/10.1242/jcs.109413; PMID: 23264744
- Loyola A, Huang JY, LeRoy G, Hu S, Wang YH, Donnelly RJ, Lane WS, Lee SC, Reinberg D. Functional analysis of the subunits of the chromatin assembly factor RSF. Mol Cell Biol 2003; 23:6759 - 68; http://dx.doi.org/10.1128/MCB.23.19.6759-6768.2003; PMID: 12972596
- Hu BS, Yu HF, Zhao G, Zha TZ. High RSF-1 expression correlates with poor prognosis in patients with gastric adenocarcinoma. Int J Clin Exp Pathol 2012; 5:668 - 73; PMID: 22977663
- Liu S, Dong Q, Wang E. Rsf-1 overexpression correlates with poor prognosis and cell proliferation in colon cancer. Tumour Biol 2012; 33:1485 - 91; http://dx.doi.org/10.1007/s13277-012-0399-y; PMID: 22528946
- Mao TL, Hsu CY, Yen MJ, Gilks B, Sheu JJ, Gabrielson E, Vang R, Cope L, Kurman RJ, Wang TL, et al. Expression of Rsf-1, a chromatin-remodeling gene, in ovarian and breast carcinoma. Hum Pathol 2006; 37:1169 - 75; http://dx.doi.org/10.1016/j.humpath.2006.04.008; PMID: 16938522
- Shih IeM, Sheu JJ, Santillan A, Nakayama K, Yen MJ, Bristow RE, Vang R, Parmigiani G, Kurman RJ, Trope CG, et al. Amplification of a chromatin remodeling gene, Rsf-1/HBXAP, in ovarian carcinoma. Proc Natl Acad Sci U S A 2005; 102:14004 - 9; http://dx.doi.org/10.1073/pnas.0504195102; PMID: 16172393
- Fang FM, Li CF, Huang HY, Lai MT, Chen CM, Chiu IW, Wang TL, Tsai FJ, Shih IeM, Sheu JJ. Overexpression of a chromatin remodeling factor, RSF-1/HBXAP, correlates with aggressive oral squamous cell carcinoma. Am J Pathol 2011; 178:2407 - 15; http://dx.doi.org/10.1016/j.ajpath.2011.01.043; PMID: 21514451
- Sheu JJ, Guan B, Choi JH, Lin A, Lee CH, Hsiao YT, Wang TL, Tsai FJ, Shih IeM. Rsf-1, a chromatin remodeling protein, induces DNA damage and promotes genomic instability. J Biol Chem 2010; 285:38260 - 9; http://dx.doi.org/10.1074/jbc.M110.138735; PMID: 20923775
- Kshirsagar M, Jiang W, Shih IeM. DNA Damage Response is Prominent in Ovarian High-Grade Serous Carcinomas, Especially Those with Rsf-1 (HBXAP) Overexpression. J Oncol 2012; 2012:621685; http://dx.doi.org/10.1155/2012/621685; PMID: 22028712
- Chae S, Ji JH, Kwon SH, Lee HS, Lim JM, Kang D, Lee CW, Cho H. HBxAPα/Rsf-1-mediated HBx-hBubR1 interactions regulate the mitotic spindle checkpoint and chromosome instability. Carcinogenesis 2013; 34:1680 - 8; http://dx.doi.org/10.1093/carcin/bgt105; PMID: 23536579
- Shanbhag NM, Rafalska-Metcalf IU, Balane-Bolivar C, Janicki SM, Greenberg RA. ATM-dependent chromatin changes silence transcription in cis to DNA double-strand breaks. Cell 2010; 141:970 - 81; http://dx.doi.org/10.1016/j.cell.2010.04.038; PMID: 20550933
- Xu Y, Ayrapetov MK, Xu C, Gursoy-Yuzugullu O, Hu Y, Price BD. Histone H2A.Z controls a critical chromatin remodeling step required for DNA double-strand break repair. Mol Cell 2012; 48:723 - 33; http://dx.doi.org/10.1016/j.molcel.2012.09.026; PMID: 23122415
- Matsuoka S, Ballif BA, Smogorzewska A, McDonald ER 3rd, Hurov KE, Luo J, Bakalarski CE, Zhao Z, Solimini N, Lerenthal Y, et al. ATM and ATR substrate analysis reveals extensive protein networks responsive to DNA damage. Science 2007; 316:1160 - 6; http://dx.doi.org/10.1126/science.1140321; PMID: 17525332
- Beli P, Lukashchuk N, Wagner SA, Weinert BT, Olsen JV, Baskcomb L, Mann M, Jackson SP, Choudhary C. Proteomic investigations reveal a role for RNA processing factor THRAP3 in the DNA damage response. Mol Cell 2012; 46:212 - 25; http://dx.doi.org/10.1016/j.molcel.2012.01.026; PMID: 22424773
- Wang H, Wang M, Wang H, Böcker W, Iliakis G. Complex H2AX phosphorylation patterns by multiple kinases including ATM and DNA-PK in human cells exposed to ionizing radiation and treated with kinase inhibitors. J Cell Physiol 2005; 202:492 - 502; http://dx.doi.org/10.1002/jcp.20141; PMID: 15389585
- Ciccia A, Elledge SJ. The DNA damage response: making it safe to play with knives. Mol Cell 2010; 40:179 - 204; http://dx.doi.org/10.1016/j.molcel.2010.09.019; PMID: 20965415
- Helfricht A, Wiegant WW, Thijssen PE, Vertegaal AC, Luijsterburg MS, van Attikum H. Remodeling and spacing factor 1 (RSF1) deposits centromere proteins at DNA double-strand breaks to promote non-homologous end-joining. Cell Cycle 2013; 12:3070 - 82; http://dx.doi.org/10.4161/cc.26033; PMID: 23974106
- Nakamura K, Kato A, Kobayashi J, Yanagihara H, Sakamoto S, Oliveira DV, Shimada M, Tauchi H, Suzuki H, Tashiro S, et al. Regulation of homologous recombination by RNF20-dependent H2B ubiquitination. Mol Cell 2011; 41:515 - 28; http://dx.doi.org/10.1016/j.molcel.2011.02.002; PMID: 21362548
- Collins N, Poot RA, Kukimoto I, García-Jiménez C, Dellaire G, Varga-Weisz PD. An ACF1-ISWI chromatin-remodeling complex is required for DNA replication through heterochromatin. Nat Genet 2002; 32:627 - 32; http://dx.doi.org/10.1038/ng1046; PMID: 12434153
- Perpelescu M, Nozaki N, Obuse C, Yang H, Yoda K. Active establishment of centromeric CENP-A chromatin by RSF complex. J Cell Biol 2009; 185:397 - 407; http://dx.doi.org/10.1083/jcb.200903088; PMID: 19398759
- Xiao A, Li H, Shechter D, Ahn SH, Fabrizio LA, Erdjument-Bromage H, Ishibe-Murakami S, Wang B, Tempst P, Hofmann K, et al. WSTF regulates the H2A.X DNA damage response via a novel tyrosine kinase activity. Nature 2009; 457:57 - 62; http://dx.doi.org/10.1038/nature07668; PMID: 19092802
- Gunn A, Bennardo N, Cheng A, Stark JM. Correct end use during end joining of multiple chromosomal double strand breaks is influenced by repair protein RAD50, DNA-dependent protein kinase DNA-PKcs, and transcription context. J Biol Chem 2011; 286:42470 - 82; http://dx.doi.org/10.1074/jbc.M111.309252; PMID: 22027841