
Abstract
Homologous recombination (HR) and non-homologous end joining (NHEJ) are the main pathways ensuring the repair of DNA double-stranded breaks (DSBs) in eukaryotes. It has long been known that cell cycle stage is a major determinant of the type of pathway used to repair DSBs in vivo. However, the mechanistic basis for the cell cycle regulation of the DNA damage response is still unclear. Here we show that a major DSB sensor, the Mre11–Rad50–Xrs2 (MRX) complex, is regulated by cell cycle-dependent phosphorylation specifically in mitosis. This modification depends on the cyclin-dependent kinase Cdc28/Cdk1, and abrogation of Xrs2 and Mre11 phosphorylation results in a marked preference for DSB repair through NHEJ. Importantly, we show that phosphorylation of the MRX complex after DNA damage and during mitosis are regulated independently of each other by Tel1/ATM and Cdc28/Cdk1 kinases. Collectively, our results unravel an intricate network of phosphoregulatory mechanisms that act through the MRX complex to modulate DSB repair efficiency during mitosis.
Introduction
DNA is subject to multiple types of damages resulting from cellular metabolism or environmental hazards. The formation of a DNA double-stranded break (DSB) is one of the most damaging events for a cell, as it can result in gross chromosomal rearrangements or cell death if improperly repaired (reviewed in ref. Citation1). In yeast and humans, this type of DNA damage is repaired by 2 fundamentally different mechanisms: non-homologous end-joining (NHEJ) and homologous recombination (HR). It has been established over the last several years that those 2 processes can compete for the repair of DSBs.Citation2,Citation3 Since this competition can sometimes lead to negative consequences on cell survival,Citation3,Citation4 the regulation of the balance between NHEJ and HR appears to be an issue of major importance to ensure high-efficiency DSB repair in eukaryotes (reviewed in refs. Citation5 and Citation6).
Cell cycle progression from interphase to mitosis has been identified as a major determinant that governs the preference for either NHEJ or HR during DSB repair. Indeed, it is now well established that HR is the preferred DSB repair pathway when sister chromatids are available in late S, G2, and M phases of the cell cycle. In contrast, NHEJ can occur at any time during the cell cycle, but is mostly favored in G1, when no sister-chromatid is present (reviewed in refs. Citation5 and Citation6). The cyclin-dependent kinase Cdk1 is a major factor responsible for the promotion of HR over NHEJ in the late stages of the cell cycle.Citation7,Citation8 A key target in this process is the CtIP/Sae2 protein, whose phosphorylation by Cdk1 has been shown to stimulate DNA end resection and HR.Citation9,Citation10 The cell cycle-dependent expression of CtIP/Sae2 protein also contributes to the mitosis-specific promotion of end resection.Citation11,Citation12 More recently, it was reported that phosphorylation of Dna2 by Cdk1 also plays an important role in determining the extent of DSB end resection.Citation13 Since other DNA repair factors are targets of Cdk1 (e.g., ref. Citation14), it is likely that stimulation of HR during mitosis is coordinated at multiple levels of the DNA damage response (DDR).Citation5
Although recent work has provided important insights into the mechanisms that stimulate HR in the late stages of the cell cycle, it is still not clear whether this preferential use of HR also requires active suppression of the NHEJ pathway. This question is relevant, because NHEJ components such as the Ku70/80 complex can inhibit HR repair at a stage that precedes the end resection step of this pathway.Citation4,Citation15,Citation16 It is thus possible that the rapid recruitment of the Ku70/80 complex to DSBs could suppress the stimulatory effects of Cdk1 on end resection and, ultimately, prevent the establishment of a positive bias for HR in mitosis. Consistent with this view, it was recently demonstrated that the Ku70/80 complex is actively displaced from DNA ends during mitosis.Citation15-Citation17 This removal is mediated in yeast by the Mre11-Rad50-Xrs2 (MRX) complex, a central regulator of DSB repair in eukaryotes.Citation18,Citation19 The MRX complex is unique among DSB repair factors, in that it plays critical roles in both NHEJ and HR repair pathways, and it is one of the first components of the DNA damage response to be recruited to DSBs in vivo. As such, this complex is well placed to modulate the balance between NHEJ and HR in mitosis. This view is supported by the fact that Mre11 acts together with a key target of Cdk1, CtIP/Sae2, to promote the initial resection step of HR in mitosis.Citation9,Citation10,Citation20 Recent studies have also shown that Cdk1/2 modulate DSB repair by direct modification of the Nbs1 protein in human cells.Citation21,Citation22 Whether the Cdk1 regulation of Mre11 complex components promotes HR by also suppressing competition from the NHEJ pathway is currently unknown.
In the present study, we address this important question by showing that the MRX complex promotes DSB repair by NHEJ in a phosphorylation-dependent manner. In particular, we reveal that 2 subunits of the complex, Mre11 and Xrs2, are targeted by Cdk1 phosphorylation as cells progress into mitosis. We found that this phosphorylation specifically inhibits NHEJ, whereas removal of the phosphosites in Xrs2 and Mre11 stimulates DSB repair by NHEJ. We also show that the DNA damage-induced phosphorylation of the MRX complex is functionally distinct from the Cdk1-dependent regulation of this complex. Altogether, our results indicate that the MRX complex promotes DNA repair by acting as a central integrator of various phosphoregulatory signals during the cellular response to DNA damage.
Results
DNA damage-independent phosphorylation of Xrs2 during the cell cycle
Previous studies have demonstrated that members of the MRX complex are regulated by phosphorylation in response to DNA damage.Citation23-Citation25 We noticed that in the absence of DNA damage Xrs2 migrates as a diffuse band during electrophoresis, and considered the possibility that Xrs2 might be a substrate for a cell cycle-regulated kinase. To verify this hypothesis, we arrested an exponential culture of yeast cells in G1 using α-factor and subsequently released this population of cells into a synchronous cell cycle. Samples were taken at regular intervals to evaluate Xrs2 electrophoretic mobility together with the appearance of cell cycle landmarks (i.e., budding, mitotic spindle length, and DNA synthesis). This analysis revealed that Xrs2 migrates in acrylamide gels as either 1 or 2 bands with distinct electrophoretic mobilities depending on cell cycle stage. Specifically, prior to S phase, Xrs2 migrated mostly as a single band (, times 0–30). A second band with reduced electrophoretic mobility accumulated above the main band of Xrs2 as cells progressed through S phase and into mitosis (, times 40–50). The relative intensity of the slow-migrating band became maximal during the period corresponding to the end of DNA replication up to metaphase (, times 50–80). Passage through anaphase, execution of mitotic exit, and return to interphase () were associated with a progressive reduction in the relative importance of the slow-migrating species of Xrs2 (, times 90–120). We conclude from this experiment that Xrs2 is targeted by a cell cycle-specific modification that becomes apparent in late S phase and persists until mid-mitosis.
Figure 1. Phosphorylation of Xrs2 occurs in G2/M. Yeast cells carrying HA-tagged Xrs2 were synchronized in G1 with α-factor and released synchronously in the cell cycle. Samples were taken at various times to examine Xrs2 protein migration by SDS-PAGE and western blot (A). Samples were also taken to monitor spindle formation (B) and DNA content by FACS (C). The graph in (B) also shows the relative ratios of intensity of shifted vs. unshifted bands of Xrs2 during the experiment. (D) Immunoprecipitated Xrs2 was subjected to dephosphorylation with lambda phosphatase (or mock treatment) prior to SDS-PAGE and immunoblotting.
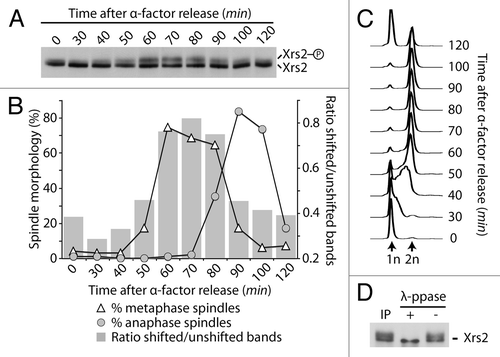
We next sought to determine if the cell cycle modification responsible for Xrs2’s slow-migrating behavior was phosphorylation. To achieve this, Xrs2 was immunoprecipitated from mitotic cells, and the purified material was subjected to lambda phosphatase treatment. This treatment converted the slow-migrating form of Xrs2 into a single species with high electrophoretic mobility (). Taken together with our previous results, this experiment indicates that Xrs2 is regulated by cell cycle-specific phosphorylation in vivo.
Cdk1 is an MRX kinase
We next wanted to identify the kinase responsible for the DNA damage-independent phosphorylation of Xrs2 during the cell cycle. Previous mass spectrometry analyses of the yeast phosphoproteome have identified several phosphorylated residues in Xrs2 that fit the core consensus for phosphorylation by Cdk1 kinase.Citation14,Citation26,Citation27 In fact, examination of Xrs2 sequence revealed that this protein carries a total of 8 serines and threonines that conform to the core consensus for phosphorylation by Cdc28/Cdk1 (i.e., Ser/Thr–Pro, ). Among these, 7 are localized in a specific region of Xrs2 that has a high tendency for structural disorder (), a characteristic feature of surface-accessible residuesCitation28 that associates frequently with Cdk1 phosphosites in vivo.Citation14
Figure 2. Xrs2 is phosphorylated by Cdc28 in vivo and in vitro. (A) Schematic representation of Xrs2 and distribution of consensus Cdc28 Ser/Thr-Pro sites within the protein primary amino acid sequence. The predicted disorder of the protein (calculated by IUPred)Citation65 is shown under the schematic. (B) Alignment of the 7 Cdc28-consensus sites found in the middle of Xrs2. The asterisk symbol indicates residues mutated to alanine. (C) Purified Xrs2 and Xrs2–7A proteins were phosphorylated in vitro using Cdc28-Clb2. Reaction mixtures were subsequently analyzed by SDS-PAGE and Coomassie staining. (D) Wild-type and xrs2–7A cultures were released from a G1 arrest and samples were taken at indicated time points. FACS DNA content profiles show that cell cycle progression was similar in wild-type (left) and mutant xrs2. (E) Cells containing pGAL1–10-Cdc28-Clb2 or an empty plasmid were synchronized in G1 and released in hydroxyurea-containing medium to block cells in early S phase. Cdc28-Clb2 was overexpressed in XRS2 and xrs2–7A cells by addition of galactose. Cells remained in G1 or early S phase for the duration of the experiment, as judged by DNA content (right).
To address the possible role of Cdk1 phosphorylation in the regulation of MRX components, we generated a mutant version of Xrs2 with 7 of its 8 putative Ser/Thr Cdk1 sites mutated to alanine residues (i.e., xrs2–7A, ). The candidate Cdk1 site localized in the FHA domain of Xrs2 () was left unchanged to ensure that the phenotypes of phosphomutations do not reflect FHA domain inactivation. Next, we purified the phospho-mutant and wild-type versions of Xrs2 and tested whether they could act as substrates for Cdc28/Cdk1 in vitro. shows that incubation of wild-type Xrs2 with Cdc28-Clb2 and ATP resulted in quantitative phosphorylation of the substrate, as evidenced by the appearance of a new slow-migrating form of Xrs2 after electrophoresis. In contrast, the Xrs2–7A mutant protein was not modified by Cdc28-Clb2 under identical phosphorylation conditions (). These results indicate that Xrs2 can be a direct substrate for Cdk1 in vitro. To test whether this kinase–substrate relationship also occurs in vivo, we constructed a yeast strain carrying the xrs2–7A allele at its endogenous locus and compared Xrs2 phosphorylation in wild-type and mutant cells progressing synchronously in the cell cycle. Remarkably, elimination of the 7 Cdk1 sites in Xrs2 prevented all detectable phosphorylation of this protein during the cell cycle (). This phenotype is not due to differences in cell cycle progression, since both the XRS2 and xrs2–7A strains initiated S phase and completed cytokinesis with similar kinetics (; compare flow cytometry profiles). Mutation of all Cdk1 sites individually or in pairs reduced the extent of Xrs2 gel retardation upon mitotic entry, but did not fully prevent the appearance of slow-migrating species of the protein (Fig. S1A). This result is consistent with previous mass spectrometry analyses showing that Xrs2 is modified on multiple Cdk1 sites in vivo,Citation14,Citation26,Citation27 and suggests that the mitosis-specific gel retardation of the protein is the consequence of multiple phosphorylation events.
Our previous results suggest that Cdc28/Cdk1 is the actual kinase that phosphorylates Xrs2 in vivo. If this were true, one would predict that forcing Cdc28 activation at a stage of the cell cycle when this kinase is not normally active would induce ectopic phosphorylation of its substrates, including Xrs2. To test this prediction, we co-expressed Cdc28 and the B-type cyclin Clb2 in yeast cells released from a G1 block into hydroxyurea (HU)-containing medium. This procedure induces an arrest in early S phase (see flow cytometry profiles in ), a stage of the cell cycle where Clb2-mediated Cdc28 activity is absentCitation29 and little Xrs2 phosphorylation is detected. Under these conditions, overexpression of Cdc28-Clb2 induced a noticeable shift in the electrophoretic mobility of wild-type Xrs2 (, see 90 min time point). In contrast, no change was observed in the electrophoretic behavior of Xrs2–7A under identical Cdc28-Clb2 overexpression conditions (). Taken together, this experiment reveals that Xrs2 is a target of Cdk1 kinase in live cells.
We next asked whether other members of the MRX complex could also be bona fide targets for Cdc28/Cdk1. Mre11 is of particular interest in this respect, because proteome-wide mass spectrometry analyses have identified a putative Cdc28/Cdk1 phosphorylation site on this proteinCitation14,Citation26-Citation28 (), and mre11 mutants interact genetically with cdc28 and clb2 mutants.Citation30,Citation31 To address this question, we conducted in vitro phosphorylation reactions with purified Mre11 and Cdc28-Clb2. Incubation of Cdc28-Clb2 with Mre11 caused an ATP-dependent shift in Mre11 electrophoretic migration, consistent with the notion that this protein is being quantitatively modified during the kinase reaction (). Moreover, removal of the 6 Ser/Thr-Pro motifs for Cdc28 phosphorylation in the Mre11–6A mutant rendered the protein insensitive to in vitro phosphorylation (). This experiment indicates that the Cdk1 consensus sites in Mre11 are directly modified by Cdc28-Clb2 in vitro and this event induces an electrophoretic mobility shift in gels.
Figure 3. Mre11 is a substrate for phosphorylation by Cdc28. (A) Schematic representation of Mre11 and distribution of consensus Cdc28 Ser/Thr-Pro sites within the protein primary amino acid sequence. The predicted disorder of the protein (calculated by IUPred)Citation65 is shown under the schematic. (B) Alignment of putative Cdc28 phosphorylation sites found in Mre11. The asterisk symbol indicates residues mutated to alanine. (C) Purified Mre11 and Mre11–6A proteins were phosphorylated in vitro using Cdc28-Clb2. Reaction mixtures were subsequently analyzed by SDS-PAGE and Coomassie staining. (D) Samples from MRE11-MYC and mre11–6A-MYC cultures were taken at indicated time points after release from a G1 block and subjected to SDS-PAGE analysis. DNA content determined by FACS shows that cell cycle progression was similar in wild-type and mre11–6A cultures (right). (E) Immunoprecipitated Mre11 was dephosphorylated with lambda phosphatase (or mock treated) prior to SDS-PAGE and immunoblot analysis.
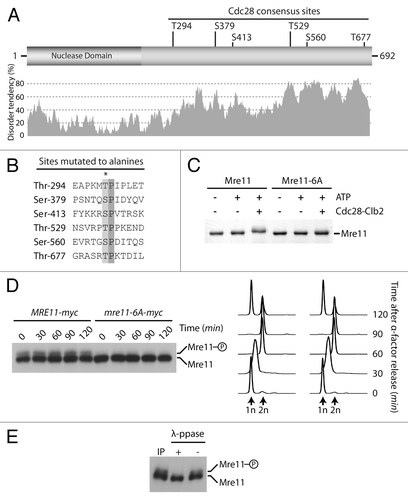
To address whether Mre11 is also phosphorylated in a cell cycle-dependent manner in vivo, samples of a culture of cells progressing synchronously in the cell cycle were taken at regular intervals and processed by SDS-PAGE for immunoblot detection of Myc-tagged Mre11. Analysis of Mre11 in gels containing Phos-tag acrylamideCitation32 revealed that the protein migrates as 2 distinct bands irrespective of cell cycle stage (). As is often the case for phosphorylated proteins, the slower-migrating species of Mre11 was not as abundant as the unmodified form of the protein, and the ratio of slow vs. fast-migrating species did not appear to change during the cell cycle. Lambda phosphatase treatment of immunoprecipitated materials confirmed that protein phosphorylation was responsible for the formation of the slower-migrating species of Mre11 (). Importantly, removal of the 6 Cdk1 core consensus sites in the Mre11–6A mutant prevented the appearance of the slow-migrating species of this protein, while cell cycle progression remained essentially unaffected in the mutant strain (). Taken together with our in vitro kinase experiment, this result indicates that Mre11 is regulated by phosphorylation in vivo, and that Cdk1 may be responsible for this event in cells. However, the cell cycle independence of this process likely indicates that Mre11 is targeted by both G1- and B-type CDKs (as previously shown for Abp1Citation33), or alternatively, that another proline-directed kinase phosphorylates Mre11.
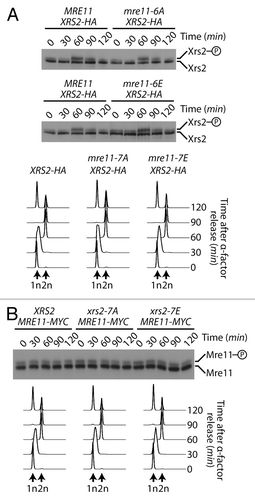
Since MRX complex subunits are genetically epistatic and biochemically interdependent,Citation18 we wondered whether the phosphorylation status of one subunit could affect that of another subunit. To test this notion, we evaluated Xrs2 electrophoretic mobility throughout the cell cycle in an mre11–6A mutant background, as well as in a phospho-mimetic mre11–6E background. While the cell cycle remained unchanged in all backgrounds, we did not detect any difference in the timing and extent of Xrs2 phosphorylation in phospho-deficient and phospho-mimetic mutants of Mre11 (). Likewise, Mre11 phosphorylation level was not altered in xrs2–7A and xrs2–7E backgrounds compared with wild-type cells (). Taken together, our results reveal that Xrs2 and Mre11 are targeted separately of each other for mitosis-specific phosphorylation in live cells.
Figure 4. Phosphorylation of Mre11 and Xrs2 by Cdk1 are independent events. (A and B) Yeast cells were synchronized in G1 with α-factor and released synchronously into the cell cycle. The electrophoretic mobility of Xrs2 protein in cells expressing MRE11, mre11–6A, and mre11–6E alleles (A), or that of Mre11 protein in cells expressing XRS2, xrs2–7A, and xrs2–7E alleles (B) was evaluated by SDS-PAGE and immunoblotting. DNA content profiles were determined by flow cytometry and show that cell cycle progression was comparable in all yeast strains.
The DNA damage- and cell cycle-specific phosphorylation of MRX are independent events
The identification of Cdc28/Cdk1 as a novel Xrs2/Mre11 kinase uncovers an unexpected mode of regulation of the MRX complex that is independent of the presence of exogenous DNA damage. However, some of the mitotic Cdk1 sites that we identified on MRX components were also identified in large-scale mass spectrometry analyzes of cells treated with DNA damaging agents (namely, Ser349, Ser445, and Ser534 on Xrs2, and Ser560 on Mre11Citation14,Citation26,Citation27). We confirmed this observation using a targeted mass spectrometry analysis of Xrs2 isolated from damaged cells (Fig. S2). These data hint at the possibility that Cdk1 sites on Xrs2 and Mre11 might be phosphorylated under 2 distinct conditions: in response to DNA damage and during mitosis. Alternatively, the phosphorylation of MRX components on mitotic sites during the DNA damage response could be an indirect consequence of checkpoint-induced arrest in G2/M,Citation34 a stage of the cell cycle with elevated Cdk1 activity.Citation35
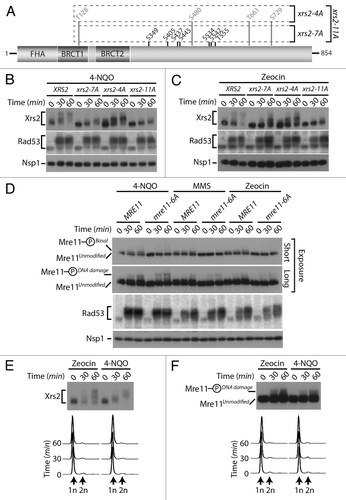
To discriminate between these possibilities, we decided to investigate the impact of losing Cdk1 sites on the DNA damage-induced phosphorylation of Xrs2 (). Cultures of asynchronous cells were exposed to 2 different genotoxic agents, namely 4-nitroquinoline 1-oxide (4-NQO) and zeocin. As control for DNA damage induction, we monitored the activation of Rad53 checkpoint kinase using an in situ activity (ISA) assay.Citation36 In agreement with previous studies,Citation23,Citation25 wild-type Xrs2 experienced a large phosphorylation-induced electrophoretic shift following induction of DNA damage, with a state of maximal phosphorylation being reached 60 min following addition of the genotoxic agents (). Interestingly, the Cdk1 site mutant of Xrs2 also experienced a quantitative retardation in electrophoretic mobility following DNA damage exposure, although one of reduced magnitude compared with that of the wild-type protein (). This result suggests that Xrs2 Cdk1 phospho-sites are not critical targets of the DNA damage response, although they are necessary to achieve maximal phosphorylation of the protein after treatment with DNA-damaging agents.
Figure 5. DNA damage-dependent phosphorylation of Xrs2 and Mre11 mutants (A) Schematic representation of the mutations introduced in the relevant XRS2 phospho-mutants. (B and C) Cultures of asynchronous log-phase cells carrying HA-tagged Xrs2 were treated with either 25 µM 4-NQO or 100 µg/mL zeocin for 60 min. Samples were taken at indicated time points following addition of the genotoxic drugs, and Xrs2 mobility was determined by western blot analysis. Checkpoint-dependent activation of Rad53 was monitored in parallel using an in situ assay (ISA). (D) Cultures of asynchronous log-phase cells carrying MYC-tagged Mre11 were treated with 25 µM 4-NQO, 0.02% MMS or 100 µg/mL zeocin, and processed as in (B) to monitor phosphorylation-induced changes in electrophoretic mobility. (E) Cells expressing HA-tagged Xrs2 were blocked in G1 with α-factor prior to being treated with 100 µg/mL zeocin or 25 µM 4-NQO. DNA content profiles of the arrested cultures are shown under the gel. (F) Cells carrying MYC-tagged Mre11 were treated as in panel (E) to monitor G1-specific DNA damage-induced phosphorylation.
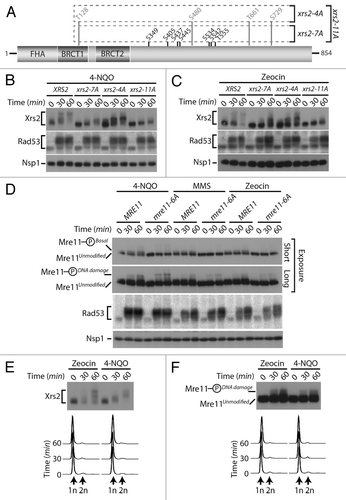
We next wanted to compare the relative contribution of Cdk1 and ATM-family of kinases on the DNA damage-induced phosphorylation of Xrs2. In yeast, Tel1 and Mec1 are the ATM-like kinases responsible for phosphorylation of the MRX complex during the DNA damage response,Citation23,Citation25 and both Mre11 and Xrs2 contain several serine/threonine-glutamine (Ser/Thr-Gln) consensus motifs that are preferred targets for this family of kinases.Citation37 We thus generated a mutant of Xrs2 having lost its 4 serines/threonines highly susceptible to Tel1/Mec1 phosphorylation (i.e., xrs2–4A; ) and evaluated the phosphorylation of the resulting mutant after DNA damage. Compared with wild-type Xrs2, the electrophoretic mobility shift observed with the Xrs2–4A protein following DNA damage was only marginally affected by the loss of Tel1/Mec1 consensus sites (). Remarkably, combining the Cdk1 and Tel1/Mec1 mutations from both xrs2–7A and xrs2–4A alleles (to generate an xrs2–11A allele, ) did reduce Xrs2 phosphorylation to some extent after DNA damage, although not completely (, compare xrs2–7A and xrs2–11A mutants). These results suggest that the optimal Cdk1 and Tel1/Mec1 motifs are not the only sites that are phosphorylated in Xrs2 during the cellular response to DNA damage. Consistent with this hypothesis, removal of additional residues (serines/threonines followed by prolines, aspartates, glycines, and glutamates, Fig. S3A) that are targeted in a number of substrates of ATM-like kinases in vivoCitation38-Citation42 completely abrogated Xrs2 phosphorylation in response to DNA damaging agents (i.e., xrs2–27A, Fig. S3B). Altogether, this analysis reveals that residues that do not fit the Tel1/Mec1 optimal target motif contribute to the DNA damage-induced phosphorylation of Xrs2. Interestingly, we did not detect defects in MRX complex formation or DNA damage sensitivity in xrs2–27A mutants (Figs. S1B and S3C), thereby suggesting that the loss of multiple phosphorylation sites did not inactivate the Xrs2 mutant protein in an unspecific manner.
We next wanted to determine whether the phosphorylation of Mre11 was regulated in a similar fashion to that of Xrs2 following genotoxic stress. As described above, Mre11 migrated as 2 distinct electrophoretic species under normal conditions: an unmodified species and the previously described isoform phosphorylated on Cdk1 consensus sites (hereafter referred to as Mre11-PhosphoBasal). However, following exposure to DNA damage, a novel slower-migrating phospho-species of Mre11 appeared on gel (Mre11-PhosphoDNA damage, ), consistent with previous reports that Mre11 becomes hyperphosphorylated in response to genotoxic stress.Citation23,Citation25 Importantly, elimination of the Cdk1 phosphosites in the Mre11–6A protein did not affect its DNA damage-dependent phosphorylation. Specifically, the Mre11-PhosphoDNA damage isoform persisted at the same position and with the same signal intensity in mre11–6A mutant and wild-type cells treated with DNA-damaging agents, in contrast to the Mre11-PhosphoBasal isoform, which disappeared upon removal of Cdk1 sites ().
The experiments described above with xrs2 and mre11 phosphosite mutants appear to exclude a role for Cdk1 activity in the DNA damage-induced phosphorylation of these proteins. To confirm this conclusion in a context where Xrs2 and Mre11 sequences are not mutated, we investigated whether the DNA damage-specific modification of Xrs2 and Mre11 could still be induced when Cdk1 activity is completely absent. To achieve this, we blocked cells in G1 using α-factor to prevent both G1 (Cln-Cdc28) and mitotic (Clb-Cdc28) Cdk1 activation and exposed them to either zeocin or 4-NQO. Under these conditions, we observed normal DNA damage-induced phosphorylation of both Xrs2 and Mre11 30 and 60 min following treatment (). Flow cytometry analyses confirmed that cells remained arrested in G1 throughout the experimental period (). This result indicates that the DNA damage-induced phosphorylation of Xrs2 and Mre11 does not require cell cycle progression, nor Cdk1 activity. Collectively, the lines of evidence presented herein are consistent with the existence of 2 regulatory systems acting independently of each other to phosphorylate components of the MRX complex in response to DNA damage and during mitosis.
DSB resection and HR are not defective in phospho-deficient mutants of the MRX complex
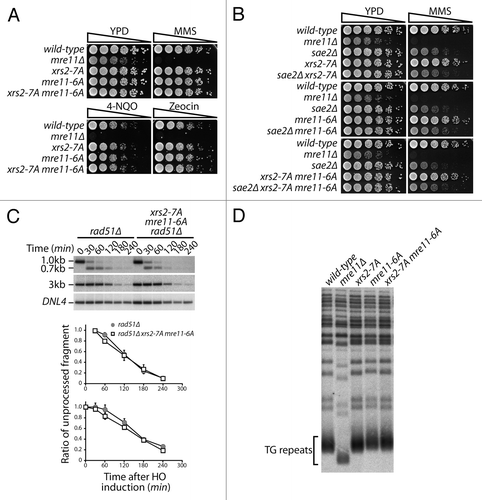
We next wanted to determine the biological significance of the cell cycle-dependent phosphorylation of the MRX complex. Previous studies have shown that Cdk1 activity can promote repair of DSBs through the HR pathway, with one of the key substrates in this process being the Mre11-interacting protein Sae2/CtIP.Citation9 Taking this into consideration, it seemed likely that phosphorylation of Mre11 and Xrs2 by Cdk1 would influence HR repair of DSBs. However, we performed several independent experiments that argue against a role for Xrs2 and Mre11 phosphorylation in HR repair in budding yeast. For instance, processes that are heavily reliant on effective HR, such as resistance to chronic doses of DNA damage or execution of meiosis, were not detectably affected by phospho-deficient alleles of XRS2 and/or MRE11 (; Fig. S4A and B). The absence of defects in those assays was not the result of functional redundancy in the Cdk1 phosphorylation of Sae2/CtIP and MRX components, because combination of xrs2–7A and mre11–6A alleles with a SAE2 deletion did not give rise to enhanced DNA damage sensitivity in the double mutants (relative to the sae2Δ single mutant, ). Moreover, analysis of DSB resection at the HO locusCitation17 indicated that Cdk1 phosphorylation of Xrs2 and Mre11 does not detectably affect the formation of single-strand DNA at damaged sites (). Finally, epistatic analysis revealed that xrs2–7A and mre11–6A mutations did not exacerbate the resection phenotype of strains lacking Exo1 or Sgs1, 2 key regulators of DSB end processing (Fig. S4C and D).Citation13,Citation17,Citation43 Taken together with the normal telomere length of xrs2–7A and mre11–6A mutants (), these results indicate that phosphorylation of the MRX complex by Cdk1 is not essential for HR repair of DSBs in mitosis and meiosis.
Figure 6. Cdc28 phosphorylation of the MRX complex is not required for HR. (A) Five-fold serial dilutions of wild-type and phospho-defective yeast mutant grown on solid YPD medium containing no drug, 0.005% MMS, 0.250 µM 4-NQO, or 5 µg/mL zeocin. The relevant genotypes of the yeast strains are described on the left of the panels. (B) Genetic analysis of the DNA damage sensitivity of sae2Δ and Cdk1-mutants of Mre11 and Xrs2. (C) DNA end resection of an HO-induced DSB at the MAT locus was monitored by Southern blot analysis, as previously described.Citation17 Resection of the DSB was measured by following the disappearance of the 0.7-kb and 3-kb bands by densitometry. Graphics represent the signal ratio of the resected band over that of the loading control band. Top graph corresponds to the signal ratio of the 0.7-kb fragment using the 30 min time point as control, while the bottom graph corresponds to the signal ratio of the 3-kb fragment using the initial time point as control. Data are presented as the mean of 3 independent experiments ± SEM. (D) Telomere length was determined in various yeast mutants using Southern blotting with a probe against the TG repeats of telomeres.
Xrs2 and Mre11 phosphorylation inhibits DSB repair by NHEJ
The Mre11 complex is a known regulator of DSB repair by NHEJ in several eukaryotes.Citation18 This DNA repair pathway is under cell cycle regulation, since its relative importance for DSB repair is reduced (relative to HR) in the late stages of the cell cycle.Citation6,Citation44 Importantly, this reduction correlates with the period of maximal Cdk1 phosphorylation of Xrs2, which suggests that there might be a functional connection between these 2 events.
To test this hypothesis, we measured the ability of mutant cells to rejoin linearized plasmids carrying 5′ or 3′ overhang in vivo. Specifically, rad51-defective yeast cells were transformed with linear DNA substrates carrying the URA3 gene, and their ability to reconnect the ends of these plasmids by NHEJ was measured on solid medium lacking uracil.Citation45 Remarkably, this assay revealed that NHEJ activity was increased by 30 to 40% over control in xrs2–7A mre11–6A phospho-mutants (). This observation was true irrespective of the nature of the overhangs used in repair templates (compare 5′ and 3′ overhang substrates in ). Moreover, xrs2–7E mre11–6E phospho-mimetic mutants displayed the exact opposite phenotype in end-joining assays. Indeed, we observed that NHEJ was reduced by 40% relative to control in the xrs2–7E mre11–6E mutant (). The reciprocal nature of the results observed in NHEJ assays with the phospho-mimetic and phospho-mutant alleles of XRS2 and MRE11, combined with the normal behavior of these alleles in other assays () and normal MRX complex formation (Fig. S1B), argue in favor of a specific inhibitory role of Cdk1 phosphorylation in the NHEJ process. Interestingly, the increase in the NHEJ activity of xrs2–7A mre11–6A phospho-mutants was not at the expense of the precision of the end-joining process, since both wild-type yeast and phospho-mutant strains were capable of religating DNA ends with similar precision (). Moreover, the NHEJ efficiency of individual xrs2–7A or mre11–6A phospho-mutants was near wild-type levels (data not shown), which indicates that these phosphorylation events must act in a concerted manner to effectively regulate the end-joining activity of the MRX complex. Taken together, our results show that the phosphorylation of MRX complex components regulates the efficiency of DSB repair by NHEJ in vivo.
Figure 7. Phospho-dependent regulation of the MRX complex affects NHEJ in vivo. (A) NHEJ activity in mre11 and xrs2 phosphomutants. The relative end-joining activity of mrx phosphomutants and control cells was determined using SphI (3′overhang) or XmaI (5′ overhang)-digested plasmid templates. Experiments were performed in cell defective in Rad51 activity to avoid HR-based repair of plasmid templates.Citation45 The NHEJ efficiency of cells carrying wild-type XRS2 and MRE11 was set arbitrarily to 100%. The mean of 3 or more independent experiments is shown ± SEM (B) Effects of Cdk1 mutations in MRE11 and XRS2 on the precision of NHEJ in yeast cells. The experiment was performed as in (A), except that the NHEJ template plasmid carried both URA3 and LEU2 genes, and was cut in the LEU2 coding sequence with XcmI. The overall efficiency and accuracy of plasmid end-joining at the XcmI site was monitored by selection of yeasts on growth media lacking the appropriate supplements (see “Materials and Methods” for details). The mean of 3 independent experiments is shown ± SEM (C) Survival to an acute dose of DNA damage at the rDNA locus. I-PpoI expression was induced for 2 h by addition of 2% galactose to exponentially growing cell. Cellular viability is expressed as the ratio of the number of colony-forming units obtained prior to and after I-PpoI induction, multiplied by 100. The values shown represent the average of 3 independent experiments performed with 2 separate clones for each genotype. The error bars reflect SEM. P value was calculated using a 2-tailed unpaired Student t test, and the asterisk symbol represents a P value of less than 0.05.
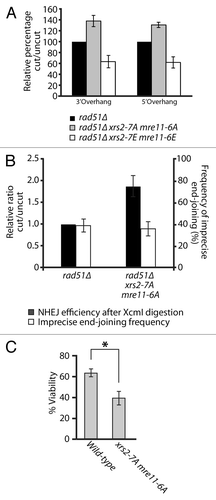
The results described above may explain why MRX phospho-mutants did not show DNA damage sensitivity in previous assays (). Indeed, under normal circumstances in yeast, the physiological contribution of NHEJ to DSB repair is modest relative to HR and can be revealed only with very high or acute doses of DNA damaging agents.Citation45-Citation47 Consistent with this, our previous DNA damage sensitivity assays were performed under conditions of chronic or constant exposure to genotoxic drugs, a setting that necessitates the use of mild/moderate doses of DNA damaging agents to allow cellular growth (). Therefore, we asked whether the NHEJ defect of MRX phospho-mutants diminished the ability of cells to resist to an acute dose of DNA damage. To achieve this, we overexpressed the I-PpoI enzyme in yeast, a meganuclease that recognizes a 15-bp site repeated ~100 times in the ribosomal DNA (rDNA) array on yeast chromosome 12.Citation48 This level of damage represents a heavy burden on DSB repair pathways, and it is expected that any defect in DSB repair pathways would result in loss of viability under such severe stress. As expected, even a short pulse of expression (2 h) of I-PpoI using the inducible GAL10-1 promoter reduced the viability of wild-type cells to 65% of that of non-induced controls (). Under identical conditions, xrs2–7A mre11-6A mutants experiencing I-PpoI expression had a much more severe reduction in viability (). Specifically, loss of phosphorylation ability reduced the viability of xrs2 mre11 mutants to approximately 43% of non-induced controls, a reduction significantly greater than that of wild-type cells (P < 0.05, ). Taken together, these results indicate that phosphorylation of the MRX complex plays an important role in the regulation of NHEJ efficiency and in the cellular resistance to acute DNA damage.
Discussion
The MRX complex is a critical component of the DNA damage response, having essential roles in checkpoint signaling as well as in repair of DSBs through HR and NHEJ.Citation18,Citation19 The multifaceted involvement of the MRX complex in the DNA damage response is a likely consequence of its ability to act as an early sensor for the presence of DSBs within the genome. As such, the MRX complex sits at a unique position to influence the efficiency and type of repair pathway cells use to respond to genotoxic stress. Consistent with this view, our findings reveal the existence of a novel phosphorylation mechanism that acts on Xrs2 and Mre11 to regulate its function in NHEJ repair of DSBs. We show here that components of the MRX complex are subject to Cdk1-mediated cell cycle phosphorylation as well as ATM/Tel1-mediated phosphorylation in response to DNA damage. These 2 regulatory events do not appear to be connected to each other functionally, since removal of Cdk1 phosphorylation does not prevent the DNA damage-induced modification of MRX components. Taken together, our results implicate Cdk1/Cdc28-mediated phosphorylation as a key determinant of the efficiency of DSB repair by NHEJ.
It has been firmly established over the last several years that NHEJ and HR—2 pathways competing for the repair of DSBs—are preferentially used in different stages of the cell cycle (reviewed in refs. Citation5 and Citation6). Furthermore, it has been argued that the choice for using a specific pathway over the other is primarily made at the level of DSB resection, a process promoted by Cdk1 phosphorylation.Citation5,Citation6 The rationale supporting this view is compelling, because the increase in Cdk1 activity that accompanies cell cycle progression is temporally connected with the appearance of sister chromatids, the optimal template for effective HR. Furthermore, Cdk1 has the ability to specifically phosphorylate several HR factors, most notably CtIP/Sae2, thus promoting 5′-to-3′ resection of DSBs and HR.Citation9,Citation10 The cell cycle-regulated abundance of CtIP/Ctp1—which peaks simultaneously with B-type CDK activity—may also play an important role in the promotion of DNA end resection.Citation11,Citation12 Ultimately, resection of DSB is known to inhibit NHEJ, and it is generally thought that activation of the resection process acts as a switch that favors HR-based repair of DSBs at the expense of NHEJ in late S, G2, and M phases of the cell cycle. A key question with respect to this model is whether the preferential choice for HR-based repair of DSB in S/G2/M can be fully explained by an increase in end-resection activity, or whether a direct negative regulation of the NHEJ process is also required to promote HR repair. Indeed, previous studies have shown that the NHEJ pathway is active throughout the cell cycle, acts prior to the initiation of HR,Citation49 and can inhibit HR in a dominant manner via Ku-mediated end-binding.Citation2,Citation3,Citation50,Citation51 These facts, together with additional evidencesCitation52,Citation53 argue that increased HR activity alone may not be sufficient to explain pathway preference for DSB repair in S/G2/M phases. Our data provide key insight into this issue by indicating that the choice of DSB repair pathway may also be governed by direct inhibition of NHEJ. Indeed, we show that Cdk1-mediated phosphorylation of the MRX complex reduces the ability of cells to perform NHEJ in vivo. Removal of Cdk1 consensus sites in MRX complex components increases significantly the efficiency of plasmid-based NHEJ, whereas introducing phospho-mimetic mutations causes a corresponding decrease in end joining. This reciprocal relationship, together with the absence of detectable defects when assessing phospho-mutants for other MRX-specific phenotypes, strongly suggests that the effects of Cdk1 phosphorylation on the MRX complex are specific. Our data fit into a model whereby Cdk1 regulation of the Mre11 complex facilitates HR repair of DSBs by limiting competition from the NHEJ machinery. Taken together, our findings unravel a novel mechanism that underpins pathway preference for DNA repair during the cell cycle.
The fact that the MRX complex is a key target for this mechanism is consistent with the observations that it plays a very early role in the DNA damage response,Citation54 and that it regulates both NHEJ and HR repair pathways.Citation18 Interestingly, multiple lines of evidence suggest that the mechanisms unraveled herein are conserved evolutionarily. Indeed, it was recently reported that human Mre11 interacts directly with Cdk2 (a homolog of yeast Cdc28Citation55), thus making the complex a likely target for Cdk2 phosphorylation in mammals. Moreover, 2 studies recently reported that human Nbs1, the functional homolog of Xrs2, is phosphorylated directly by Cdk1/2 on Ser432.Citation21,Citation22 Both studies showed that removal of this phosphosite affected the ability of cells to survive DNA damage, but the reported effects of the Nbs1 phospho-mutation on DNA-end resection and HR efficiency differed in the 2 studies. Although we did not detect any defect in DSB resection or HR efficiency in our mutants, we note that removal of Ser432 in human Nbs1 resulted in an apparent increase in NHEJ efficiency relative to control cells (see Fig. 3D in ref. Citation21). This observation is reminiscent of the NHEJ phenotype seen in xrs2 phospho-mutants described herein, although the magnitude of the increase in the human system is not as strong as in yeast. It is conceivable that a stronger NHEJ stimulation would be observed if all putative Cdk1 consensus sites would be removed from the human protein in the experiments described above. It is also important to note that Wohlbold et al.Citation22 did not detect any sensitivity to chronic doses of DNA damaging agents in human cells expressing the Nbs1 S432A phosphomutant, whereas clear hypersensitivity was detected in the same cells after acute exposure to genotoxic stress. This behavior is highly similar to that of our phosphomutants in yeast and suggests that the impact of Cdk1 phosphorylation on MRX complex components will be more relevant in contexts where cells experience severe DNA damage. Collectively, these observations highlight the conserved features of the phosphoregulation of MRX complex components throughout evolution.
We have previously shown that Xrs2 and Mre11 are phosphorylated in response to genotoxic stress.Citation23 Interestingly, we and others have determined that this phosphorylation event was dependent solely on the ATM family of kinases, since inactivation of checkpoint kinases downstream of Tel1/Mec1 had no detectable effect on MRX phosphorylation.Citation23,Citation25 However, a subsequent study revealed that removal of the preferred ATM phospho-consensus sites in Xrs2 caused no detectable consequence on DNA damage sensitivity.Citation37 Our analysis of Xrs2/Mre11 phosphorylation provides some insight on this surprising result. Indeed, we show here that removal of the canonical ATM consensus sites on Xrs2 is insufficient to abrogate DNA damage-dependent phosphorylation of this substrate. Complete elimination of this regulatory event necessitates the removal of phosphorylation sites that do not fit the canonical consensus for ATM kinase. This result is consistent with the observation that members of the ATM/ATR family of kinases—like many other kinases—are sometimes capable of phosphorylating substrates on residues that do not fit their canonical consensus motif.Citation39-Citation42 It is also important to note that Xrs2 has been predicted to be a target of Rad53 kinase, a central effector of the Tel1/Mec1-dependent checkpoint response.Citation56 Although this prediction remains to be verified, it is possible that a small, but nevertheless significant, fraction of Xrs2 DNA damage-dependent phosphorylation may be catalyzed by Rad53, thereby explaining the involvement of non-consensus Tel1 sites in this phosphoregulatory process.
In conclusion, we show in this study that the MRX complex is regulated by distinct phospho-dependent regulatory pathways in cells. These pathways respond to different signals—cell cycle status and DNA damage—and result in differing consequences for the regulation of NHEJ. Our data indicate that the preference for using an HR-based mechanism to repair DSBs in G2/M phase of the cell cycle is not only regulated at the level of the HR machinery itself, as previously thought, but also at the level of the NHEJ machinery. The regulatory mechanisms proposed here are likely to be relevant in higher eukaryotes, since both ATM and the MRN complex positively regulate DNA repair through NHEJ.Citation57-Citation59 In this regard, it will be exciting to determine whether the mechanisms described herein contribute to the DNA damage sensitivity and poor prognostic associated with human diseases such as the Nijmegen breakage syndrome and ataxia telangiectasia.
Materials and Methods
Yeast strains, mutant construction, and growth conditions
Yeast genetics and molecular biology manipulations were performed according to St-Pierre et al.Citation60 All yeast strains used in this study are listed in Table S1. For mutant generation, XRS2 and MRE11 genes were cloned in a 2 μ URA3 leu2-d-containing shuttle plasmid and pFA6A-derived plasmid, respectively, and mutations were introduced by site-directed mutagenesis (Quickchange, Stratagene). The xrs2–27A mutant was partially synthesized as a xrs2–23A fragment (BioBasic), which was cloned in a YIplac204-XRS2 plasmid replacing its wild-type counterpart. Further mutations were introduced by site-directed mutagenesis of this construct. Modified genes were integrated in the genome by recombination, replacing the wild-type versions. After transformation, the genomic loci of all phospho-mutant alleles of XRS2 and MRE11 used in this study were sequenced to confirm the presence of the relevant mutations and the absence of secondary mutation. For DNA damage sensitivity assays, cells were spotted in 5-fold dilution series on YPD medium containing different concentrations of DNA damaging agents. Cells were grown for 2–3 d at 30 °C until individual colonies became visible.
Protein purification
Xrs2 and Mre11 were overexpressed in yeast strains D1374 and D2540, respectively. These strains express either Xrs2-His-HA or Mre11-His-StrepTagII under the control of the GAL1 promoter on 2 μ URA3 leu2-d-derived plasmids. Conditions for culture and preparation of cell lysate are as previously described.Citation61 Xrs2 and Mre11 were purified by metal-chelate chromatography using standard procedures (Ni-NTA; Qiagen). Mutant Xrs2–7A and Mre11–6A were purified by the same methods in strains D2446 and D2541, respectively. Active Cdc28-Clb2 kinase was purified according to published procedures.Citation62
In vitro phosphorylation and dephosphorylation of Xrs2 and Mre11
Purified wild-type and mutant Xrs2 or Mre11 were incubated in kinase bufferCitation64 supplemented with 100 µM ATP and purified Cdc28-Clb2. Reactions were performed at 30 °C for 15 min and stopped by addition of SDS-PAGE sample buffer. For dephosphorylation experiments, asynchronous cells were lysedCitation61 in lysis buffer (50 mM TRIS-HCl pH 7.5, 100 mM KCl, 100 mM NaF, 10% glycerol, 0.1% tween 20, 1 mM tungstate, 1 mM DTT, 10 µM AEBSF, 10 μM pepstatin A, 10 μM E-64). Xrs2-HA was immunoprecipitated using anti-HA 16B12 antibody bound to Gammabind Plus Sepharose beads (GE Healthcare). Beads were resuspended in lambda-ppase buffer (NEB) and were incubated for 30 min at 30 °C with or without purified lambda phosphatase.Citation62
Protein samples and SDS-PAGE
Protein samples were prepared from yeast cultures using the TCA-glass bead method described in Foiani et al.Citation63 For Xrs2 cell cycle/Cdk1 phosphorylation analysis, protein samples were resolved on 8% SDS–polyacrylamide gels in a BioRad Mini-Protean gel system. The modification status of Mre11 after in vitro phosphorylation was determined by migration on a 10% SDS–polyacrylamide gel using the same conditions. For the DNA damage-dependent phosphorylation analysis of Xrs2, samples were resolved on 7.5% NextGel acrylamide gels. In vivo Mre11 phosphorylation events were analyzed by migration on 6% SDS–polyacrylamide gels supplemented with 20 µM Phos-Tag and 40 µM MnCl2. Xrs2 and Mre11 protein samples from DNA damage and cell cycle experiments, respectively, were run in Hoefer SE 600 Chroma electrophoresis apparatus overnight. For western blotting, proteins were transferred on nitrocellulose membranes by semi-dry transfer. Xrs2-HA or Mre11-MYC were detected with a mouse 12CA5 antibody (1:1000) or a mouse 9E10 antibody (1:1000), respectively.Citation64 Primary antibody–antigen complexes were revealed with sheep anti-mouse IgGs conjugated to HRP.
DSB resection assays
Analysis of ssDNA formation at DSBs was performed according to previously published procedures.Citation17 Briefly, cells were grown in YEP–Raffinose medium to an OD600 of 0.5 before HO endonuclease expression was induced by addition of 2% galactose. Yeast genomic DNA was purified from samples taken at 30 min intervals following HO induction. DNA was digested using StyI and XbaI enzymes and separated by electrophoresis on 1% agarose gels. DNA was transferred on Hybond-N+ membrane (GE Healthcare) using a vacuum blotter system (BioRad). ssDNA formation was measured by disappearance of 0.7- and 3-kb signal from probes. A probe annealing in the DNL4 gene was used as loading control.
NHEJ efficiency and accuracy assays
For NHEJ efficiency experiments, YCplac33 plasmid was digested with either SphI (3′overhang) or XmaI (5′overhang) and purified by gel extraction. Equal quantities of DNA were transformed in exponentially growing cells using the standard lithium acetate method. Circular YCplac33 was transformed as a control for plasmid transformation efficiency. Cells were plated on SC-URA medium, and NHEJ efficiency was measured by determination of the number of colonies growing after transformation with a digested plasmid relative to that obtained with an undigested circular plasmid.Citation45 The procedure to determine the accuracy of NHEJ in mutant strains was similar to that described above, with few modifications. Specifically, an YCplac111-derivative plasmid containing 2 selection markers, URA3 and LEU2, was cut within the LEU2 coding sequence with XcmI enzyme. The linearized plasmid was transformed in yeast cells and the overall NHEJ efficiency was determined by counting yeast colonies growing on selective medium lacking uracil (SC-URA), as described above. To determine NHEJ accuracy, colonies growing on SC-URA medium were subsequently tested for growth on selective medium lacking leucine (SC-LEU). Whereas yeast growth on SC-URA medium reflects overall plasmid repair/re-circularization, growth on SC-LEU medium specifically indicates that the XcmI site in the LEU2 gene of the plasmid was precisely repaired by end-joining (i.e., thus reconstituting the wild-type sequence of the LEU2 gene). The imprecise NHEJ frequency was calculated as the fraction of URA3-positive yeast colonies unable to grow on SC-LEU medium.
Telomere length analysis
DNA from yeast cells in stationary phase was extracted and digested by XhoI overnight. Digested DNA was separated by migration on 1% agarose gel and transferred on Hybond-N+ membrane (GE healthcare) using a vacuum blotter system (BioRad). Telomeres were detected by Southern blotting using a radiolabeled 280-bp probe consisting of TG repeats.
I-Ppo viability
For I-Ppo viability experiments, cells were grown overnight in SC-URA supplemented with 0.5% glucose to prevent leaky expression from the GAL1 promoter. Cells were diluted to O.D.600 0.4 in SC-URA supplemented with 2% raffinose and were allowed to grow for 4 h at room temperature. I-PpoI expression was induced by addition of 2% galactose and blocked after 2 h by addition of 2% glucose. To control for potential differences in plating efficiency of yeast strains (i.e., differences unrelated to I-PpoI expression), the ability of mutant and wild-type cells to repair I-PpoI damage is expressed as the ratio in the number of viable colony-forming units (CFU) immediately before and after 2 h of induction of I-PpoI with galactose. Three independent experiments were performed, in which the average viability of 2 independent clones was used.
| Abbreviations: | ||
| DDR | = | DNA damage response |
| DSBs | = | DNA double-stranded breaks |
| HR | = | homologous recombination |
| HU | = | hydroxyurea |
| ISA | = | in situ activity |
| MRX | = | Mre11-Rad50-Xrs2 |
| MMS | = | methyl methanesulfonate |
| 4-NQO | = | 4-nitroquinoline 1-oxide |
| NHEJ | = | non-homologous end joining |
| rDNA | = | ribosomal DNA |
Additional material
Download Zip (375.1 KB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We would like to thank Drs Julie St-Pierre and Lea Harrington for their comments on the manuscript. We would also like to thank Dr Robert Driscoll for discussions on xrs2 phenotypes. Research in D.D. laboratory is supported by grants from the Canadian Institutes of Health Research (CIHR; MOP 82912), from the Cancer Research Society (CRS), and from the Canadian Cancer Society Research Institute (CCSRI; #020304). D.D. is a recipient of a Tier II Canada Research Chair in Cell Cycle Regulation and Genomic Integrity. X.R. is supported by a post-doctoral fellowship from the Cole Foundation, whereas A.S. was supported by graduate scholarships from CIHR and FRSQ.
Related Research Data
References
- Chapman JR, Taylor MR, Boulton SJ. Playing the end game: DNA double-strand break repair pathway choice. Mol Cell 2012; 47:497 - 510; http://dx.doi.org/10.1016/j.molcel.2012.07.029; PMID: 22920291
- Lee SE, Moore JK, Holmes A, Umezu K, Kolodner RD, Haber JE. Saccharomyces Ku70, mre11/rad50 and RPA proteins regulate adaptation to G2/M arrest after DNA damage. Cell 1998; 94:399 - 409; http://dx.doi.org/10.1016/S0092-8674(00)81482-8; PMID: 9708741
- Takata M, Sasaki MS, Sonoda E, Morrison C, Hashimoto M, Utsumi H, Yamaguchi-Iwai Y, Shinohara A, Takeda S. Homologous recombination and non-homologous end-joining pathways of DNA double-strand break repair have overlapping roles in the maintenance of chromosomal integrity in vertebrate cells. EMBO J 1998; 17:5497 - 508; http://dx.doi.org/10.1093/emboj/17.18.5497; PMID: 9736627
- Zhang Y, Hefferin ML, Chen L, Shim EY, Tseng HM, Kwon Y, Sung P, Lee SE, Tomkinson AE. Role of Dnl4-Lif1 in nonhomologous end-joining repair complex assembly and suppression of homologous recombination. Nat Struct Mol Biol 2007; 14:639 - 46; http://dx.doi.org/10.1038/nsmb1261; PMID: 17589524
- Pardo B, Gómez-González B, Aguilera A. DNA repair in mammalian cells: DNA double-strand break repair: how to fix a broken relationship. Cell Mol Life Sci 2009; 66:1039 - 56; http://dx.doi.org/10.1007/s00018-009-8740-3; PMID: 19153654
- Langerak P, Russell P. Regulatory networks integrating cell cycle control with DNA damage checkpoints and double-strand break repair. Philos Trans R Soc Lond B Biol Sci 2011; 366:3562 - 71; http://dx.doi.org/10.1098/rstb.2011.0070; PMID: 22084383
- Ira G, Pellicioli A, Balijja A, Wang X, Fiorani S, Carotenuto W, Liberi G, Bressan D, Wan L, Hollingsworth NM, et al. DNA end resection, homologous recombination and DNA damage checkpoint activation require CDK1. Nature 2004; 431:1011 - 7; http://dx.doi.org/10.1038/nature02964; PMID: 15496928
- Aylon Y, Liefshitz B, Kupiec M. The CDK regulates repair of double-strand breaks by homologous recombination during the cell cycle. EMBO J 2004; 23:4868 - 75; http://dx.doi.org/10.1038/sj.emboj.7600469; PMID: 15549137
- Huertas P, Cortés-Ledesma F, Sartori AA, Aguilera A, Jackson SP. CDK targets Sae2 to control DNA-end resection and homologous recombination. Nature 2008; 455:689 - 92; http://dx.doi.org/10.1038/nature07215; PMID: 18716619
- Huertas P, Jackson SP. Human CtIP mediates cell cycle control of DNA end resection and double strand break repair. J Biol Chem 2009; 284:9558 - 65; http://dx.doi.org/10.1074/jbc.M808906200; PMID: 19202191
- Limbo O, Chahwan C, Yamada Y, de Bruin RA, Wittenberg C, Russell P. Ctp1 is a cell-cycle-regulated protein that functions with Mre11 complex to control double-strand break repair by homologous recombination. Mol Cell 2007; 28:134 - 46; http://dx.doi.org/10.1016/j.molcel.2007.09.009; PMID: 17936710
- Yu X, Baer R. Nuclear localization and cell cycle-specific expression of CtIP, a protein that associates with the BRCA1 tumor suppressor. J Biol Chem 2000; 275:18541 - 9; http://dx.doi.org/10.1074/jbc.M909494199; PMID: 10764811
- Chen X, Niu H, Chung WH, Zhu Z, Papusha A, Shim EY, Lee SE, Sung P, Ira G. Cell cycle regulation of DNA double-strand break end resection by Cdk1-dependent Dna2 phosphorylation. Nat Struct Mol Biol 2011; 18:1015 - 9; http://dx.doi.org/10.1038/nsmb.2105; PMID: 21841787
- Holt LJ, Tuch BB, Villén J, Johnson AD, Gygi SP, Morgan DO. Global analysis of Cdk1 substrate phosphorylation sites provides insights into evolution. Science 2009; 325:1682 - 6; http://dx.doi.org/10.1126/science.1172867; PMID: 19779198
- Clerici M, Mantiero D, Guerini I, Lucchini G, Longhese MP. The Yku70-Yku80 complex contributes to regulate double-strand break processing and checkpoint activation during the cell cycle. EMBO Rep 2008; 9:810 - 8; http://dx.doi.org/10.1038/embor.2008.121; PMID: 18600234
- Foster SS, Balestrini A, Petrini JH. Functional interplay of the Mre11 nuclease and Ku in the response to replication-associated DNA damage. Mol Cell Biol 2011; 31:4379 - 89; http://dx.doi.org/10.1128/MCB.05854-11; PMID: 21876003
- Mimitou EP, Symington LS. Ku prevents Exo1 and Sgs1-dependent resection of DNA ends in the absence of a functional MRX complex or Sae2. EMBO J 2010; 29:3358 - 69; http://dx.doi.org/10.1038/emboj.2010.193; PMID: 20729809
- D’Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317 - 27; http://dx.doi.org/10.1038/nrm805; PMID: 11988766
- Stracker TH, Petrini JH. The MRE11 complex: starting from the ends. Nat Rev Mol Cell Biol 2011; 12:90 - 103; http://dx.doi.org/10.1038/nrm3047; PMID: 21252998
- Nicolette ML, Lee K, Guo Z, Rani M, Chow JM, Lee SE, Paull TT. Mre11-Rad50-Xrs2 and Sae2 promote 5′ strand resection of DNA double-strand breaks. Nat Struct Mol Biol 2010; 17:1478 - 85; http://dx.doi.org/10.1038/nsmb.1957; PMID: 21102445
- Falck J, Forment JV, Coates J, Mistrik M, Lukas J, Bartek J, Jackson SP. CDK targeting of NBS1 promotes DNA-end resection, replication restart and homologous recombination. EMBO Rep 2012; 13:561 - 8; http://dx.doi.org/10.1038/embor.2012.58; PMID: 22565321
- Wohlbold L, Merrick KA, De S, Amat R, Kim JH, Larochelle S, Allen JJ, Zhang C, Shokat KM, Petrini JH, et al. Chemical genetics reveals a specific requirement for Cdk2 activity in the DNA damage response and identifies Nbs1 as a Cdk2 substrate in human cells. PLoS Genet 2012; 8:e1002935; http://dx.doi.org/10.1371/journal.pgen.1002935; PMID: 22927831
- D’Amours D, Jackson SP. The yeast Xrs2 complex functions in S phase checkpoint regulation. Genes Dev 2001; 15:2238 - 49; http://dx.doi.org/10.1101/gad.208701; PMID: 11544181
- Grenon M, Gilbert C, Lowndes NF. Checkpoint activation in response to double-strand breaks requires the Mre11/Rad50/Xrs2 complex. Nat Cell Biol 2001; 3:844 - 7; http://dx.doi.org/10.1038/ncb0901-844; PMID: 11533665
- Usui T, Ogawa H, Petrini JH. A DNA damage response pathway controlled by Tel1 and the Mre11 complex. Mol Cell 2001; 7:1255 - 66; http://dx.doi.org/10.1016/S1097-2765(01)00270-2; PMID: 11430828
- Albuquerque CP, Smolka MB, Payne SH, Bafna V, Eng J, Zhou H. A multidimensional chromatography technology for in-depth phosphoproteome analysis. Mol Cell Proteomics 2008; 7:1389 - 96; http://dx.doi.org/10.1074/mcp.M700468-MCP200; PMID: 18407956
- Smolka MB, Albuquerque CP, Chen SH, Zhou H. Proteome-wide identification of in vivo targets of DNA damage checkpoint kinases. Proc Natl Acad Sci U S A 2007; 104:10364 - 9; http://dx.doi.org/10.1073/pnas.0701622104; PMID: 17563356
- Li X, Gerber SA, Rudner AD, Beausoleil SA, Haas W, Villén J, Elias JE, Gygi SP. Large-scale phosphorylation analysis of alpha-factor-arrested Saccharomyces cerevisiae. J Proteome Res 2007; 6:1190 - 7; http://dx.doi.org/10.1021/pr060559j; PMID: 17330950
- Amon A, Irniger S, Nasmyth K. Closing the cell cycle circle in yeast: G2 cyclin proteolysis initiated at mitosis persists until the activation of G1 cyclins in the next cycle. Cell 1994; 77:1037 - 50; http://dx.doi.org/10.1016/0092-8674(94)90443-X; PMID: 8020094
- Grandin N, Charbonneau M. Mitotic cyclins regulate telomeric recombination in telomerase-deficient yeast cells. Mol Cell Biol 2003; 23:9162 - 77; http://dx.doi.org/10.1128/MCB.23.24.9162-9177.2003; PMID: 14645528
- Enserink JM, Hombauer H, Huang ME, Kolodner RD. Cdc28/Cdk1 positively and negatively affects genome stability in S. cerevisiae. J Cell Biol 2009; 185:423 - 37; http://dx.doi.org/10.1083/jcb.200811083; PMID: 19398760
- Kinoshita E, Kinoshita-Kikuta E, Takiyama K, Koike T. Phosphate-binding tag, a new tool to visualize phosphorylated proteins. Mol Cell Proteomics 2006; 5:749 - 57; http://dx.doi.org/10.1074/mcp.T500024-MCP200; PMID: 16340016
- Garcia B, Stollar EJ, Davidson AR. The importance of conserved features of yeast actin-binding protein 1 (Abp1p): the conditional nature of essentiality. Genetics 2012; 191:1199 - 211; http://dx.doi.org/10.1534/genetics.112.141739; PMID: 22661326
- Weinert TA, Hartwell LH. The RAD9 gene controls the cell cycle response to DNA damage in Saccharomyces cerevisiae. Science 1988; 241:317 - 22; http://dx.doi.org/10.1126/science.3291120; PMID: 3291120
- Surana U, Robitsch H, Price C, Schuster T, Fitch I, Futcher AB, Nasmyth K. The role of CDC28 and cyclins during mitosis in the budding yeast S. cerevisiae. Cell 1991; 65:145 - 61; http://dx.doi.org/10.1016/0092-8674(91)90416-V; PMID: 1849457
- Pellicioli A, Lucca C, Liberi G, Marini F, Lopes M, Plevani P, Romano A, Di Fiore PP, Foiani M. Activation of Rad53 kinase in response to DNA damage and its effect in modulating phosphorylation of the lagging strand DNA polymerase. EMBO J 1999; 18:6561 - 72; http://dx.doi.org/10.1093/emboj/18.22.6561; PMID: 10562568
- Mallory JC, Bashkirov VI, Trujillo KM, Solinger JA, Dominska M, Sung P, Heyer WD, Petes TD. Amino acid changes in Xrs2p, Dun1p, and Rfa2p that remove the preferred targets of the ATM family of protein kinases do not affect DNA repair or telomere length in Saccharomyces cerevisiae. DNA Repair (Amst) 2003; 2:1041 - 64; http://dx.doi.org/10.1016/S1568-7864(03)00115-0; PMID: 12967660
- Abraham RT. Cell cycle checkpoint signaling through the ATM and ATR kinases. Genes Dev 2001; 15:2177 - 96; http://dx.doi.org/10.1101/gad.914401; PMID: 11544175
- Cortez D, Wang Y, Qin J, Elledge SJ. Requirement of ATM-dependent phosphorylation of brca1 in the DNA damage response to double-strand breaks. Science 1999; 286:1162 - 6; http://dx.doi.org/10.1126/science.286.5442.1162; PMID: 10550055
- O’Neill T, Dwyer AJ, Ziv Y, Chan DW, Lees-Miller SP, Abraham RH, Lai JH, Hill D, Shiloh Y, Cantley LC, et al. Utilization of oriented peptide libraries to identify substrate motifs selected by ATM. J Biol Chem 2000; 275:22719 - 27; http://dx.doi.org/10.1074/jbc.M001002200; PMID: 10801797
- Kozlov SV, Graham ME, Peng C, Chen P, Robinson PJ, Lavin MF. Involvement of novel autophosphorylation sites in ATM activation. EMBO J 2006; 25:3504 - 14; http://dx.doi.org/10.1038/sj.emboj.7601231; PMID: 16858402
- Yu Y, Mahaney BL, Yano K, Ye R, Fang S, Douglas P, Chen DJ, Lees-Miller SP. DNA-PK and ATM phosphorylation sites in XLF/Cernunnos are not required for repair of DNA double strand breaks. DNA Repair (Amst) 2008; 7:1680 - 92; http://dx.doi.org/10.1016/j.dnarep.2008.06.015; PMID: 18644470
- Shim EY, Chung WH, Nicolette ML, Zhang Y, Davis M, Zhu Z, Paull TT, Ira G, Lee SE. Saccharomyces cerevisiae Mre11/Rad50/Xrs2 and Ku proteins regulate association of Exo1 and Dna2 with DNA breaks. EMBO J 2010; 29:3370 - 80; http://dx.doi.org/10.1038/emboj.2010.219; PMID: 20834227
- Wohlbold L, Fisher RP. Behind the wheel and under the hood: functions of cyclin-dependent kinases in response to DNA damage. DNA Repair (Amst) 2009; 8:1018 - 24; http://dx.doi.org/10.1016/j.dnarep.2009.04.009; PMID: 19464967
- Boulton SJ, Jackson SP. Identification of a Saccharomyces cerevisiae Ku80 homologue: roles in DNA double strand break rejoining and in telomeric maintenance. Nucleic Acids Res 1996; 24:4639 - 48; http://dx.doi.org/10.1093/nar/24.23.4639; PMID: 8972848
- Siede W, Friedl AA, Dianova I, Eckardt-Schupp F, Friedberg EC. The Saccharomyces cerevisiae Ku autoantigen homologue affects radiosensitivity only in the absence of homologous recombination. Genetics 1996; 142:91 - 102; PMID: 8770587
- Milne GT, Jin S, Shannon KB, Weaver DT. Mutations in two Ku homologs define a DNA end-joining repair pathway in Saccharomyces cerevisiae. Mol Cell Biol 1996; 16:4189 - 98; PMID: 8754818
- Berkovich E, Monnat RJ Jr., Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc 2008; 3:915 - 22; http://dx.doi.org/10.1038/nprot.2008.54; PMID: 18451799
- Frank-Vaillant M, Marcand S. Transient stability of DNA ends allows nonhomologous end joining to precede homologous recombination. Mol Cell 2002; 10:1189 - 99; http://dx.doi.org/10.1016/S1097-2765(02)00705-0; PMID: 12453425
- Fukushima T, Takata M, Morrison C, Araki R, Fujimori A, Abe M, Tatsumi K, Jasin M, Dhar PK, Sonoda E, et al. Genetic analysis of the DNA-dependent protein kinase reveals an inhibitory role of Ku in late S-G2 phase DNA double-strand break repair. J Biol Chem 2001; 276:44413 - 8; http://dx.doi.org/10.1074/jbc.M106295200; PMID: 11577093
- Pierce AJ, Hu P, Han M, Ellis N, Jasin M. Ku DNA end-binding protein modulates homologous repair of double-strand breaks in mammalian cells. Genes Dev 2001; 15:3237 - 42; http://dx.doi.org/10.1101/gad.946401; PMID: 11751629
- Karathanasis E, Wilson TE. Enhancement of Saccharomyces cerevisiae end-joining efficiency by cell growth stage but not by impairment of recombination. Genetics 2002; 161:1015 - 27; PMID: 12136007
- Zhang Y, Shim EY, Davis M, Lee SE. Regulation of repair choice: Cdk1 suppresses recruitment of end joining factors at DNA breaks. DNA Repair (Amst) 2009; 8:1235 - 41; http://dx.doi.org/10.1016/j.dnarep.2009.07.007; PMID: 19699692
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004; 118:699 - 713; http://dx.doi.org/10.1016/j.cell.2004.08.015; PMID: 15369670
- Buis J, Stoneham T, Spehalski E, Ferguson DO. Mre11 regulates CtIP-dependent double-strand break repair by interaction with CDK2. Nat Struct Mol Biol 2012; 19:246 - 52; http://dx.doi.org/10.1038/nsmb.2212; PMID: 22231403
- Brinkworth RI, Breinl RA, Kobe B. Structural basis and prediction of substrate specificity in protein serine/threonine kinases. Proc Natl Acad Sci U S A 2003; 100:74 - 9; http://dx.doi.org/10.1073/pnas.0134224100; PMID: 12502784
- Rass E, Grabarz A, Plo I, Gautier J, Bertrand P, Lopez BS. Role of Mre11 in chromosomal nonhomologous end joining in mammalian cells. Nat Struct Mol Biol 2009; 16:819 - 24; http://dx.doi.org/10.1038/nsmb.1641; PMID: 19633668
- Deriano L, Stracker TH, Baker A, Petrini JH, Roth DB. Roles for NBS1 in alternative nonhomologous end-joining of V(D)J recombination intermediates. Mol Cell 2009; 34:13 - 25; http://dx.doi.org/10.1016/j.molcel.2009.03.009; PMID: 19362533
- Taylor EM, Cecillon SM, Bonis A, Chapman JR, Povirk LF, Lindsay HD. The Mre11/Rad50/Nbs1 complex functions in resection-based DNA end joining in Xenopus laevis. Nucleic Acids Res 2010; 38:441 - 54; http://dx.doi.org/10.1093/nar/gkp905; PMID: 19892829
- St-Pierre J, Douziech M, Bazile F, Pascariu M, Bonneil E, Sauvé V, Ratsima H, D’Amours D. Polo kinase regulates mitotic chromosome condensation by hyperactivation of condensin DNA supercoiling activity. Mol Cell 2009; 34:416 - 26; http://dx.doi.org/10.1016/j.molcel.2009.04.013; PMID: 19481522
- Roy MA, Siddiqui N, D’Amours D. Dynamic and selective DNA-binding activity of Smc5, a core component of the Smc5-Smc6 complex. Cell Cycle 2011; 10:690 - 700; http://dx.doi.org/10.4161/cc.10.4.14860; PMID: 21293191
- Ratsima H, Ladouceur AM, Pascariu M, Sauvé V, Salloum Z, Maddox PS, D’Amours D. Independent modulation of the kinase and polo-box activities of Cdc5 protein unravels unique roles in the maintenance of genome stability. Proc Natl Acad Sci U S A 2011; 108:E914 - 23; http://dx.doi.org/10.1073/pnas.1106448108; PMID: 21987786
- Foiani M, Marini F, Gamba D, Lucchini G, Plevani P. The B subunit of the DNA polymerase alpha-primase complex in Saccharomyces cerevisiae executes an essential function at the initial stage of DNA replication. Mol Cell Biol 1994; 14:923 - 33; PMID: 8289832
- Field J, Nikawa J, Broek D, MacDonald B, Rodgers L, Wilson IA, Lerner RA, Wigler M. Purification of a RAS-responsive adenylyl cyclase complex from Saccharomyces cerevisiae by use of an epitope addition method. Mol Cell Biol 1988; 8:2159 - 65; PMID: 2455217
- Dosztányi Z, Csizmok V, Tompa P, Simon I. IUPred: web server for the prediction of intrinsically unstructured regions of proteins based on estimated energy content. Bioinformatics 2005; 21:3433 - 4; http://dx.doi.org/10.1093/bioinformatics/bti541; PMID: 15955779