
Abstract
p68 (DDX5) acts both as an ATP-dependent RNA helicase and as a transcriptional co-activator of several cancer-associated transcription factors, including the p53 tumor suppressor. p68 is aberrantly expressed in a high proportion of cancers, but the oncogenic drive for, or the consequences of, these expression changes remain unclear. Here we show that elevated p68 expression in a cohort of human breast cancers is associated significantly with elevated levels of the oncogenic protein kinase, Polo-like kinase-1 (PLK1). Patients expressing detectable levels of both p68 and PLK1 have a poor prognosis, but only if they also have mutation in the TP53 gene (encoding p53), suggesting that p68 can regulate PLK1 levels in a manner that is suppressed by p53. In support of this hypothesis, we show that p68 stimulates expression from the PLK1 promoter, and that silencing of endogenous p68 expression downregulates endogenous PLK1 gene expression. In the absence of functional p53, p68 stimulates the expression of PLK1 both at basal levels and in response to the clinically relevant drug, etoposide. In keeping with a role as a transcriptional activator/co-activator, chromatin immuno-precipitation analysis shows that p68 is associated with the PLK1 promoter, irrespective of the p53 status. However, its recruitment is stimulated by etoposide in cells lacking p53, suggesting that p53 can oppose association of p68 with the PLK1 promoter. These data provide a model in which p68 and p53 interplay regulates PLK1 expression, and which describes the behavior of these molecules, and the outcome of their interaction, in human breast cancer.
Keywords: :
Introduction
p68 (DDX5) is a prototypic member of the DEAD box family of RNA helicases that encompasses multiple functions. First, as an ATP-dependent RNA helicase, p68 is involved in several cellular processes requiring manipulation of RNA structures, including the processing of pre-mRNA,Citation1-Citation3 rRNA,Citation4,Citation5 and microRNA.Citation4 p68 has also an important and apparently RNA helicase-independent role as a transcriptional co-activator of several cancer-associated transcription factors, including the androgen receptor,Citation6 estrogen receptor α,7 and the p53 tumor suppressor.Citation8 p68 is recruited to the promoters of responsive genes under conditions in which these transcription factors are activated,Citation8,Citation9 suggesting that it functions in transcription initiation. Interestingly, p68 itself has been reported to stimulate expression of Cyclin D1 and c-Myc, and in this context p68 ATPase/helicase activity appears to be required.Citation10 From the perspective of human cancer, p68 is aberrantly expressed/modified in a range of cancers, including those of breast,Citation7 prostate,Citation6 and colon origin.Citation11,Citation12 The selective pressure for changes in p68 expression is not understood, but may be associated with its ability to modulate cancer-associated transcription.
p53 plays a critical role in eliminating tumor cells by coordinating changes in gene expression, leading to permanent cell cycle arrest (senescence) or programmed cell death (apoptosis).Citation13-Citation15 p53 regulates the expression of many genes and, accordingly, loss of p53 function during tumor development can have wide-ranging consequences for the pathology of tumor cells.Citation16,Citation17 Most p53 target genes are actually repressed, as opposed to transactivated, by p53,Citation13 with the outcome that loss of p53 function may lead to dysregulated or even unrestricted levels of oncogenic proteins. We previously demonstrated that p68 is important for the p53 response to DNA damage.Citation8 More recently, we have shown that p68 can play a crucial role as a selectivity factor that favors p53-mediated growth arrest and is dispensable for the induction of apoptosis, both in cultured cells and in vivo.Citation18 Additionally, bone marrow cells from p68-knockout mice are more sensitive to irradiation, suggesting that in some contexts p68 may indeed protect against apoptosis.Citation18 Mechanistically, p68 promotes the recruitment of p53 and RNA polymerase II to the CDKN1 (p21) promoter but not to the BAX or PUMA promoters, thereby providing a means by which the selective inhibition of p21 induction could be achieved. These studies, however, did not address whether, in addition to influencing p53-mediated transactivation, p68 can affect repression by p53.
The protein kinase Polo-like kinase-1 (PLK1) plays a pivotal role in the maturation of centrosomes, entry into M phase, spindle formation, and cytokinesis.Citation19-Citation21 Increased PLK1 activity (e.g., through overexpression) stimulates the transcriptional program required for mitosis by phosphorylating FoxM1, overrides the DNA damage checkpoint, contributes to the induction of invasiveness by phosphorylating vimentin, and can interrupt mitotic integrity leading to aneuploidy (ref. Citation20 and references therein). These functions are consistent with a tumor-promoting role. PLK1 levels are elevated in various human cancers,Citation22-Citation32 including breast cancers, where detectable levels of PLK1 protein are associated with aggressive characteristics such as vascular invasion, markers of proliferative activity, and lack of detectable estrogen receptor.Citation29,Citation33 PLK1 is downregulated by p53 as part of the G2/M cell cycle checkpoint,Citation34-Citation41 and we recently established that this occurs mainly through p53-dependent repression of PLK1 mRNA expression.Citation42 In keeping with this observation, we and others have also shown that p53-null cells are unable to downregulate PLK1 levels in response to clinically relevant genotoxic drugs.Citation38,Citation42 Consistent with this model, we also find a close association between mutation of the TP53 gene (encoding p53) and the presence of elevated PLK1 levels (as detected by immunohistochemistry) in human breast cancers.Citation43 Notably, patients lacking wild-type p53 function and expressing detectable PLK1 protein showed a poor clinical outcome.Citation43
PLK1 is widely considered to be a potential therapeutic target, and several PLK1 inhibitors are currently undergoing clinical trials.Citation20 Our data suggest that targeting of cancers lacking p53 with inhibitors of PLK1 could provide an effective tailored therapeutic strategy. However, we do not yet have a full understanding of the factors and pathways that can influence PLK1 levels or the outcome of inhibiting PLK1 activity, nor whether these are, in turn, dependent upon p53 status. In the present study we have investigated the potential impact of p68 on PLK1 expression and its relationship with TP53 status. Our data highlight a strong association between PLK1 and p68 in human breast cancers that is revealed following inactivation of p53. Moreover, we provide a molecular basis for this relationship by demonstrating, in cultured cells, that p68 is an activator of PLK1 expression that can be overridden by the presence of wild-type p53.
Results
p68 expression in human breast cancers is associated with elevated levels of PLK1 and poor clinical outcome
We previously showed that elevated PLK1 levels (i.e., with an immunohistochemistry score of 4–18 as discussed in “Materials and Methods”) were present in a proportion of human breast cancers (23 out of 215: 10.7%Citation43). A similar analysis of p68 levels in the same cohort indicated that 141 out of 215 (65.6%) of the cancers show elevated levels of p68 (). Stratification of the data revealed that there is a striking association between elevated PLK1 and elevated p68, where patients showing p68 expression are greater than 6 times more likely to have detectable PLK1 expression as compared with those lacking measurable p68 expression (). These data suggest a causal link between PLK1 and p68.
Table 1. Association of PLK1 with p68 expression in a cohort of human breast cancers
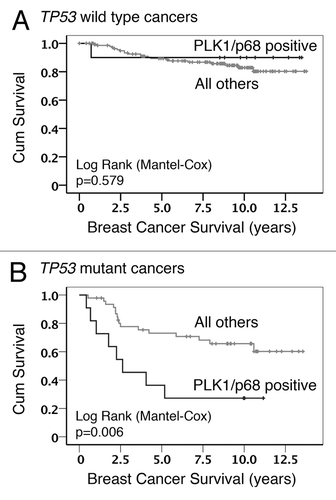
Our previous study also identified a link between PLK1 levels and the status (wild-type vs. mutant) of the TP53 gene (encoding the p53 tumor suppressor protein), such that patients lacking functional p53 were 3 times more likely to show elevated PLK1 levels.Citation43 In order to determine whether the TP53 status had any influence on the association between PLK1 and p68, the data were further examined by Kaplan–Meier analysis following stratification. (As discussed in “Materials and Methods”, a score of >3 was considered as elevated levels for both PLK1 and p68.) This approach revealed that, in tumors harboring a mutation(s) in the TP53 gene, there was a significant association between elevated levels of both PLK1 and p68 and poor survival as compared with tumors that show elevated levels of only one of these proteins or of neither (all other patients). In contrast, there was no such association when the tumors retained wild-type p53 (). These data suggest a cooperative effect of PLK1 and p68 that favors cancer progression that is only evident in the absence of functional p53. Given that p68 can act as a transcriptional co-regulator of p53, and that PLK1 expression is tightly regulated by p53, we hypothesized that p68 can also regulate the expression of PLK1.
Figure 1. Kaplan–Meier survival analysis. Data from the analysis of breast cancer cohort TMA24 were stratified to show the influence of p53 status on the association between PLK1 staining, p68 staining, and overall survival: (A) cancers with wild-type TP53 in which PLK1/p68 double positive were compared with all other combinations; (B) cancers with mutant TP53 in which PLK1/p68 double positive were compared with all other combinations.
p68 activates expression from the PLK1 promoter
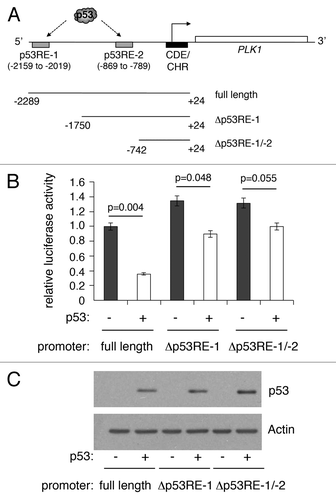
We previously reported that PLK1 expression is repressed by p53 in response to DNA damage and to treatment of cells with the MDM2 inhibitor, Nutlin-3.Citation42 Our study identified 2 p53-responsive elements in the PLK1 promoter (termed p53RE-1 [distal] and p53RE-2 [proximal]: ). To confirm that expression from the PLK1 promoter is repressed by p53, a PLK1 promoter-luciferase plasmid (containing 2.3 kb of the human PLK1 promoter) was transfected into H1299 cells in the absence or presence of a plasmid expressing wild-type human p53. Subsequent luciferase analysis indicated that p53 was able to repress the PLK1 promoter by as much as 60% (). Deletion of either p53RE-1 alone, or both p53RE-1 and p53RE-2, led to a significant reduction in, but not abolition of, repression. (Curiously, deletion of the region encompassing p53RE-1 led to a small but significant increase in the basal level of expression, suggesting that this region contains a p53-independent repressor element.) No luciferase activity was detected when the parent plasmid, pGL3, was used in place of the PLK1-promoter plasmid (data not shown). These data are consistent with our recently published findings that PLK1 repression is mediated through p53 binding at either or both these sites.Citation42
Figure 2. Deletion of p53-responsive elements reduces repression of the PLK1 promoter by p53. (A) Schematic representation of the PLK1 promoter indicating the positions of 2 previously identified p53-responsive elements, p53RE-1 and p53RE-2. The starting positions of 2 deletion mutants lacking, respectively, p53RE-1 alone or both p53RE-1 and p53RE-2, are indicated. (B) Luciferase reporter plasmids containing the full-length PLK1 promoter (A) or deletion mutants lacking p53RE-1 alone or p53RE-1 and p53RE-2 were transfected into H1299 cells without and with a plasmid expressing wild-type human p53 (10 ng), and assayed for luciferase activity 24 h post-transfection. The activity of the PLK1 promoter-Luciferase vector alone was taken as reference (set to 1) and the data converted into fold change. The results presented are mean ± SD of 3 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant. (C) Western blot showing the levels of p53 expressed in H1299 cells, with actin serving as loading control.
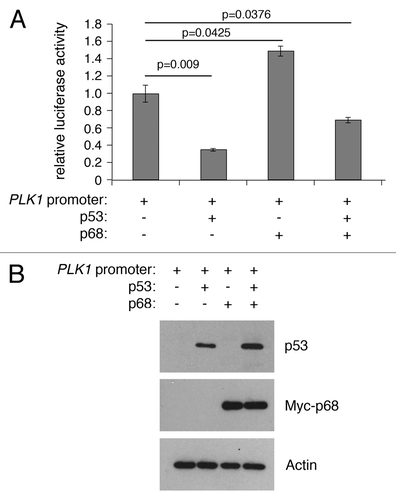
The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell cycle arrest after DNA damage both in cultured human cell lines and in mouse tissues in vivo.Citation8,Citation18 To determine whether p68 could also play a role in p53-mediated repression, luciferase expression levels were measured from the PLK1 promoter-luciferase plasmid in the absence or presence of p53 and p68 (). Curiously, in this case, p68 expression actually stimulated PLK1 promoter activity and partially relieved repression by p53 (). These data suggest that p68 can behave as an activator of PLK1, independently of p53.
Figure 3. p68 and p53 have opposing effects on expression from the PLK1 promoter in a luciferase reporter assay. (A) H1299 cells were transfected with the PLK1 promoter/pGL3 vector with p68 or p53 expression vectors and assayed for luciferase activity after 24 h. p53 represses expression from the PLK1 promoter, whereas p68 activates its expression independently of p53. The results presented are mean ± SD of 3 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant. (B) Western blot showing the levels of p53 and p68 expressed in H1299 cells, with actin serving as loading control.
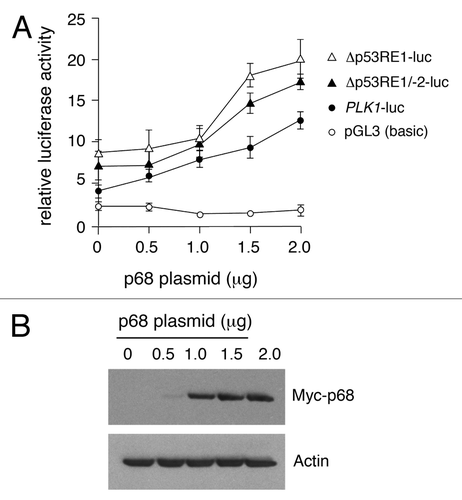
To explore further the stimulation of PLK1 promoter activity by p68, the influence of p68 on the PLK1 promoter-deletion plasmids was analyzed (). In this case, while both deletion derivatives showed an increased basal promoter activity, consistent with the region encompassing p53RE-1 harboring a p53-independent repressor element (as in ), the dose-dependent stimulation by increasing amounts of p68 was unaffected. These data suggest that p68 not only acts independently of p53, but also of the p53 binding sites, p53RE-1 and p53RE-2.
Figure 4. Dose-dependent activation of the PLK1 promoter and 5′ deletion mutants by p68. (A) The full-length PLK1 promoter or its deletion derivatives were transfected individually into H1299 cells that lack functional p53, together with increasing amounts of p68, and assayed for luciferase activity 24 h post-transfection with empty pGL3 vector as control. The results presented are mean ± SD of 3 independent experiments, each performed in triplicate. (B) Western blot showing increasing amounts of Myc-tagged p68 expressed in H1299 cells, with actin serving as loading control. Similar results were obtained whether p68 was Myc-tagged or un-tagged.
Stimulation of expression from the PLK1 promoter by p68 requires its ATPase function
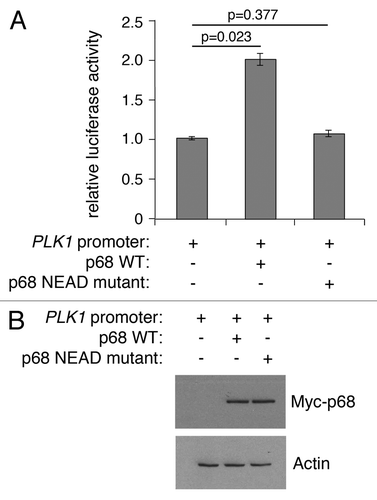
Like other DEAD box proteins, p68 encompasses an RNA helicase activity that is dependent on intrinsic ATPase activity.Citation44 In contrast, its ability to act as a transcriptional co-activator of p53, estrogen receptor, and androgen receptor is independent of its ATPase function,Citation6-Citation8 although, interestingly, p68 ATPase activity is required for β-catenin-mediated transcription of Cyclin D1.Citation10 To determine whether ATPase activity is important for the ability of p68 to stimulate PLK1 promoter activity, the effect of a mutant p68 protein was determined, in which the characteristic DEAD motif (required for ATPase activity) was changed to NEAD (). While wild-type p68 could stimulate PLK1 promoter function by up to 2-fold, the NEAD mutant was completely ineffective. These findings suggest that ATPase/helicase activities are required for stimulation of PLK1 transcription, as shown previously for Cyclin D1.Citation10
Figure 5. Wild-type, but not mutant, p68 activates expression from the PLK1 promoter. (A) H1299 cells were transfected with the PLK1 promoter-luciferase plasmid together with plasmids expressing wild-type p68 or an ATPase/helicase mutant p68 (NEAD-p68) and assayed for luciferase activity. The results represent the mean ± SD of 3 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant. (B) Western blot showing the level of Myc-tagged p68 expressed in H1299 cells, with actin serving as loading control.
Endogenous p68 plays a dual role as an activator of PLK1 expression and a co-repressor of p53
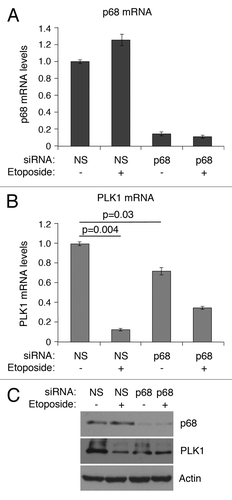
To determine whether p68 can influence endogenous PLK1 expression, p68 expression was silenced by siRNA, and PLK1 mRNA levels were determined in MCF7 cells (human breast cancer-derived expressing wild-type p53) following mock treatment or treatment with 20 μM etoposide for 24 h (). (Etoposide was used as a representative genotoxic drug on the basis that: (1) it is used clinically to treat breast cancers; and (2) it has been used in many studies as a DNA damage-inducing stimulus to activate the p53 pathway.) Treatment of the cells with etoposide in the absence of p68 silencing led to a small stimulation of p68 mRNA levels (panel A), but not protein (panel C) (as observed previouslyCitation18), and a greater than 80% repression of PLK1 mRNA (panel B), consistent with our previously published data.Citation42 In contrast, when p68 expression was silenced (levels reduced by about 90%: ) the basal levels of PLK1 mRNA were reduced by about 25% (). Additionally, under these conditions, the repression of PLK1 expression following etoposide treatment was reduced from 10-fold to 2-fold (). These observations were supported by measurements of the protein levels (). These data, therefore, confirm that p68 is an activator of PLK1 expression. Curiously, however, and in contrast to the luciferase data, they also indicate that repression by p53 is less effective in the absence of p68, suggesting that p68 may have a dual role: as a p53-independent activator of PLK1 expression and, mirroring its role in p53-dependent stimulation of p21 expression, as a co-repressor of p53.
Figure 6. siRNA depletion of p68 expression leads to reduction of expression of PLK1 in MCF7 cells. mRNA levels of p68 (A) and PLK1 (B) were measured, relative to actin, by quantitative real-time PCR. The results presented are mean ± SD of 2 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant. The protein levels of p68 and PLK1 were detected by western blotting, with actin serving as loading control (C). p < 0.05 is significant.
In the absence of p53 function p68 co-repressor activity is lost and its ability to stimulate PLK1 expression becomes dominant
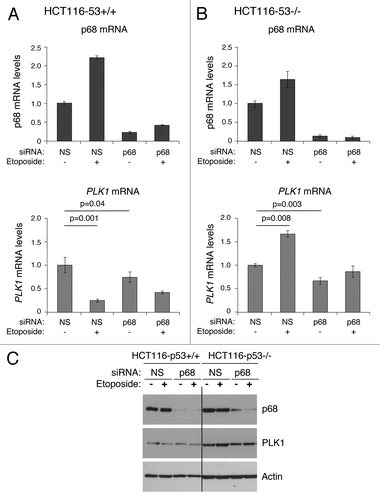
To explore further the p53-dependent and -independent functions of p68, experiments were conducted using the wild-type p53-expressing line, HCT116, and an established derivative “p53-null” line, in which a portion of the p53 gene has been eliminated by homologous recombination-based mutagenesis.Citation45 (While this line retains a portion of the p53 gene, it lacks full-length functional p53 protein. Although of colonic origin, not breast, it allows us to discern the effects of completely eliminating full-length wild-type p53 function, which is not possible using RNAi.) Treatment of both the p53-competent and p53-null cells with etoposide led to a similar increase in p68 mRNA, indicating that this increase is p53-independent, although, as seen previously,Citation18 this did not result in an increase in p68 protein (). Etoposide treatment of HCT116 p53-competent cells (panel A) led to repression of PLK1 expression in a manner similar to that observed in the MCF7 cells (as shown in and ref. Citation42). Also, reflecting the MCF7 data, silencing of p68 reduced PLK1 mRNA levels in the absence of etoposide treatment but reduced the ability of p53 to repress PLK1 expression in response to etoposide (). In contrast, in the cells lacking p53 (panel B), etoposide stimulated PLK1 mRNA levels by >50%, but only when p68 was present (). These data confirm that: (1) stimulation of PLK1 expression by p68 is not simply a feature of a single cell line; (2) the repression of PLK1 by p53 is stimulated by p68; and (3) in the absence of functional p53, the role of p68 becomes predominantly that of an activator of PLK1 expression, and the activation is enhanced by etoposide treatment. Moreover, consistent with the breast cancer data () these findings suggest that the aberrant expression of p68 may lead to changes (increases) in the levels of oncogenic PLK1 protein in cancers lacking functional p53.
Figure 7. Effect of silencing p68 expression on PLK1 levels. Isogenic HCT116 cell lines (p53+/+ and p53−/−) were transfected with non-silencing siRNA (NS) as control, or p68 siRNA. The cells were subsequently exposed to the DNA damaging agent etoposide (20 μM) for 24 h, following which total RNA and protein were extracted. (A and B) p68 and PLK1 mRNA levels before and after etoposide treatment were measured relative to actin (control) by quantitative real-time PCR (qRT-PCR) in HCT116-p53+/+ (A) and HCT116-p53−/− (B) cells. The results presented are mean ± SD of 2 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant. (C) The extracted proteins were western blotted onto a nitrocellulose membrane and probed for p68 and PLK1 protein using p68- and PLK1-specific antibodies. Actin served as a control.
p68 is recruited to the PLK1 promoter in response to etoposide treatment
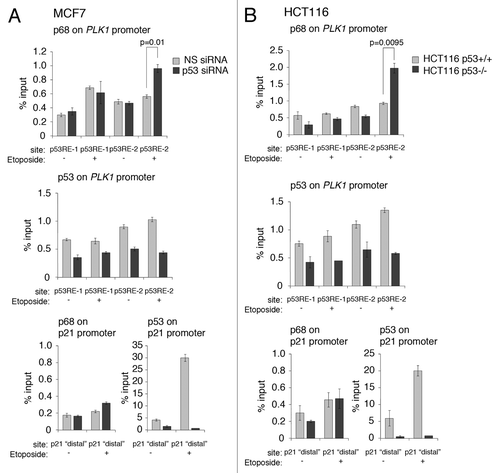
To explore in greater depth the ability of p68 to modulate PLK1 expression, the recruitment of p68 to the PLK1 promoter was examined by chromatin immuno-precipitation (). These experiments were performed in 2 independent cells lines, MCF7 and HCT116, and performed in the presence and absence of wild-type p53; (endogenous p53 expression was silenced by RNAi in the MCF7 cells; for the HCT116 cells, the p53knockout derivative discussed above was usedCitation45). The data indicate that p68 was present in the vicinity of both the p53RE-1- and p53RE-2-responsive elements, independently of the presence of p53 (, top panels). Substitution of the p68 antibody in these experiments with a non-specific IgG, or elimination of p68 by RNAi, gave background levels for the MCF7 and HCT116 cells of 0.1% and 0.2% of input, respectively (data not shown). The data also show that p68 was recruited to the vicinity of the p53RE-1 in response to etoposide treatment, independently of the p53 status, in the MCF7 cells. Likewise, p68 was recruited at or near p53RE-1 in HCT116 cells, although there was a higher basal recruitment in p53-competent cells that was not increased upon etoposide treatment. There was a modest increase in p68 recruitment in the p53-null derivative. Strikingly, while there was only a modest recruitment of p68 to the vicinity of p53RE-2 in both cell backgrounds, when p53 was present, the level of occupancy of p68 in this region was stimulated significantly in both lines in the absence of p53. These data suggest that p53 may act to block/occlude, either directly or indirectly, the association of p68 with the region encompassing p53RE-2. The data are also consistent with the findings (, above) that etoposide treatment stimulates PLK1 expression only when p53 is absent. The data also confirmed that p53 is present within the vicinities of the p53-responsive elements, p53RE-1 and p53RE-2Citation42 (i.e., within 500 bp, given that the resolution of the ChIP is about 500 bp). (We note that the background levels of p53 recruitment in the ChIP experiments with the PLK1 promoter were consistently about 0.5% of input under conditions of p53 depletion by siRNA in the MCF7 cells or in the HCT116 cells lacking p53. It is likely that this can be attributed to the high amount of C/G nucleotides in this promoter. In particular, we were unable to generate reproducible data with low background for the core promoter region, where there is an unusually high proportion of C/G nucleotides, in spite of testing a number of different primers corresponding to this region.) Additionally, etoposide treatment of the cells did not lead to any significant increase in p53 levels at p53RE-1, but did give rise to a small but significant increase in recruitment to the p53RE-2 region, thereby confirming previous observations.Citation42 The effectiveness of the knockdown/lack of p53 expression and the etoposide treatment were confirmed by ChIP analysis of the p21 promoter, which showed that (1) p53 knockdown/absence gave rise to only a background signal, and (2) significant recruitment of p53 occurred only when the cells had been treated with etoposide.
Figure 8. p68 is recruited to the vicinity of the p53-responsive elements, p53RE-1 and p53RE-2, on the PLK1 promoter. (A) p53 expression was silenced or mock-silenced in MCF7 cells. The cells were subsequently treated with 20 μM etoposide for 6 hours (+) or left untreated (−) as control. (B) HCT116 cells expressing wild-type p53 (HCT116 p53+/+) or lacking p53 expression (HCT116 p53−/−) were treated with 20 μM etoposide for 6 h (+) or left untreated (−) as control. In both panels, the presence of p68 and p53 on the p53RE-1 and p53RE-2 regions of the PLK1 promoter were determined by ChIP/qRT-PCR. The effectiveness of the silencing/absence of p53 expression and the etoposide treatments were tested on the p21 promoter, which is a well-established target for p53 and p68. The results presented are mean ± SD of 2 independent experiments, each performed in triplicate. P values were calculated using Student paired t test, where P < 0.05 is considered to be significant.
Discussion
In the present study we find a strong association between the presence of the DEAD box protein, p68, and the oncogenic protein kinase, PLK1, in an exploratory cohort of human breast cancers, as measured by immunohistochemistry. While this cohort is small as judged by present day approaches, the association between these 2 cancer-associated proteins is underpinned both by the statistical analysis of the data and by the high odds ratio, whereby there is a greater than 6-fold likelihood that cancers expressing elevated p68 will also express PLK1 (). Additionally, the added value of studying this particular cohort lies in the fact that, in addition to a full and informative set of clinical data, the status of a range of different cancer-associated genes/proteins has previously been established for this set of cancers, and can thus be interrogated for possible interactions/associations between the current protein of interest (p68) and those that were previously measured.Citation43,Citation46 Importantly, the cohort has also provided us with a hypothesis on which we have explored regulatory interactions that may underpin the development of breast cancer.
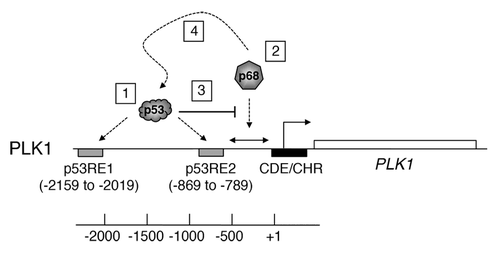
The data from the molecular analyses suggest that p68 plays a dual function at the PLK1 promoter. In this model, p53 binds to 2 previously established p53-responsive elements, p53RE-1 and p53RE-2, respectively, both under normal un-stimulated conditions and following induction of p53 by DNA damage stimuli or the MDM2 inhibitor, Nutlin-3Citation42: (see ). The data from the promoter–reporter assays in indicate that the ability of p68 to stimulate PLK1 promoter function is independent of these p53-responsive elements and involves a sequence(s) between −742 and +24 of the PLK1 promoter region. Taken together with the ChIP experiments (), where the resolution is limited to the order of about 500 bp, the data suggest that p68 is likely to stimulate PLK1 expression through an element(s) within 500 bp downstream of −742 bp. The presence of p68 on the promoter, most likely within this region, is stimulated by etoposide and, strikingly, significantly enhanced in the absence of p53. This is consistent with our finding that PLK1 mRNA levels are stimulated by etoposide treatment in the HCT116−/− cells (lacking p53), and that this stimulation is lost upon depletion of p68 by siRNA. These data suggest that p53 occupancy of p53RE-2 either directly or, more likely, indirectly (as suggested by the data in ) inhibits the recruitment of p68 to this region. The data () also indicate that p68 is present at a site(s) within the vicinity of p53RE-1 (see ). The presence of p68 here may contribute either to stimulating PLK1 expression or could have a bearing on repression by p53 (or perhaps both). There is no evidence that p68 itself binds DNA directly; therefore, it is presumably recruited to chromatin via its interactions with partner proteins. The different contributions to regulating PLK1 expression (p53-dependent and -independent) may reflect an ability of p53 to govern which interactions are available for p68 at this promoter.
Figure 9. Model describing the role of p68 at the PLK1 promoter. The PLK1 promoter/gene is shown as a schematic and is not to scale. (1) p53 binds to 2 previously established p53-responsive elements: p53RE-1 and p53RE-2, respectively, both under normal un-stimulated conditions and following induction of p53 by DNA damage stimuli or the MDM2 inhibitor, Nutlin-3.Citation42 (2) Based on the data in and , p68 is likely to be recruited to a site(s) located approximately between −742 and +24, independently of the presence of p53. (3) The data also suggest that p53 occupancy of p53RE-2 occludes, either directly or indirectly, the recruitment of p68 to this region. (4) In the presence of wild-type p53, p68 recruitment may actually assist repression of PLK1 expression by p53.
The model is also consistent with the following observations:
1) It has previously been established that p68 can interact with, and may help recruit, RNA polymerase II to core promoters.Citation18,Citation47-Citation49 It is also highly possible that it carries out a helicase-dependent role through its association with RNA polymerase II, possibly for release or processing of nascent transcripts.Citation2,Citation50,Citation51 If so, this would fit with the data in , which indicate that the stimulation of expression from the PLK1 promoter is dependent, at least in part, upon the ATPase activity of p68. Although in many cases, transcriptional co-regulator function of p68 within promoter regions appears to be independent of its ATPase/helicase functions,Citation6-Citation8 in other cases ATPase activity is required.Citation10 Additionally, p68 has been shown to be recruited to promoter regions containing E2F-1, SP1, and JunD binding sites;Citation48,Citation52 interestingly, these factors have also been identified as binding to the PLK1 promoter as defined by ChIP-seq (ENCODE project: UCSC genome Browser). It is therefore possible that p68 recruitment may be mediated or assisted through interaction with one or more of these transcription factors.
2) Strikingly, the binding of p68 downstream of p53RE-2 (beyond −742) is inhibited by the presence of p53. This offers a plausible explanation as to why repression of PLK1 promoter activity by p53 is dominant over stimulation of expression by p68. As suggested above, p53 may prevent p68 associating with a chromatin-bound factor(s), either by interacting with that factor itself, or with a nearby factor, thus sterically blocking association of the relevant factor with p68. Alternatively, direct binding of p53 to p68 (which, it has been established, does occurCitation8) may prevent recruitment of p68 to the appropriate stimulatory site(s) in the chromatin.
3) Importantly, the model also offers a plausible explanation for the association between elevated p68 and elevated PLK1 in human breast cancers (; ). Thus, while other mechanisms may additionally operate, our data support the idea that elevated p68 levels in the cancers lead to, or at least contribute to, increased levels of PLK1 expression. The model also fits with the very striking observations that: (1) the presence of wild-type p53 in the tumor cells is dominant over any possible effect of, or collaboration between, elevated p68, and PLK1 (), thereby favoring a good prognosis, and (2) there is evidence of tumor-promoting function and collaboration between p68 and PLK1 following loss of wild-type p53 in the cancers.
It is well established that p68 contributes to p53-mediated transcriptional activation for at least some promoters, as demonstrated for the CDKN1A (p21) promoter.Citation8,Citation18 The data in the present study also raise the possibility that p68 can contribute to p53-mediated repression of PLK1 on the basis that repression of PLK1 expression in response to etoposide is proportionately reduced (from approximately 10-fold to 2-fold) upon silencing of p68 expression (). These data argue for a dual role for p68 in controlling events at the PLK1 promoter. Our data show p68 to be present in the vicinity (±500 bp) of both the p53RE-1 and p53RE-2 sites in the absence of etoposide stimulation in 2 independent cell lines (MCF7 and HCT116; ). We speculate that recruitment of p68 to different regions of the promoter may differentially mediate contributions to repression or transactivation. While not within the scope of the present study, future analyses should address this issue.
In conclusion, our study provides a plausible model to explain the observed association between p68 and PLK1 levels in human breast cancers and its relationship to the TP53 status of the cancers. The data also suggest that treatment with genotoxic drugs may actually elevate levels of an oncogenic protein kinase in cancer cells lacking p53. To counter this, the finding that p68 and PLK1 can collaborate in cancer cells raises the possibility that novel PLK1 inhibitors could be used to target specifically those cancers in which the TP53 gene is mutant. Consideration of such an approach would be attractive given the poor prognosis of patients whose cancers lack p53 but show elevated p68 and PLK1, as indicated by the stratified survival curves ().
Materials and Methods
Patients
The cohort used in this study (TMA24) has been reported previouslyCitation43,Citation46 and comprises 215 unselected pre- and post-menopausal women (aged 28–89; median 62 y) with primary, previously untreated breast cancer. Samples were obtained at the time of surgery with informed consent from each patient prior to surgery. Tissue microarrays (TMAs) were prepared as described previously;Citation46 all TMAs included 6 cores per tumor from across the tumor. The values stated are the mean across all 6 cores. Ethical approval was given by the Tayside Research and Ethics committee (ref. 07/S1402/90). The PLK1 and TP53 analysis of this cohort was reported recently.Citation43 p68 analysis was previously performed for a separate study.Citation53
Immunohistochemistry (IHC)
Immunohistochemistry was performed as described previously.Citation6,Citation43 Antibodies specific for other proteins used in the study have been reported elsewhere.Citation43,Citation46 The mutation status of the TP53 gene was also reported previously.Citation43,Citation54
Scoring and data analysis
Scoring was conducted as described previously.Citation43 Scoring was semi-quantitative, using the Quickscore methodCitation55 for the intensity (0–3) and proportion (1–6) of cells stained for the p68 antibody. Multiplication of intensity and proportion scores obtained an overall result of 0–18; in line with our previous study,Citation43 overall scores of 0–3 were taken as low and 4–18 as high/elevated. Data were analyzed using an internally developed automated data analysis system, INSPIRECitation56 as described previously.Citation43
Cell culture
H1299 (p53-null lung carcinoma-derived cells) and MCF7 (breast carcinoma-derived cells expressing wild-type p53) were cultured in Dulbecco modified Eagle medium (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (Life Technologies), 2 mM L-glutamine (Life Technologies), and 50 units/ml penicillin plus 50 μg/ml streptomycin (Life Technologies). HCT116 p53+/+ and p53−/− (isogenic colon carcinoma derived cells), described previously,Citation45 were cultured in McCoy 5A medium (Life Technologies) supplemented with 10% (v/v) fetal bovine serum (Life Technologies) and 2 mM L-glutamine (Life Technologies). All cells were maintained in a humidified 5% CO2 atmosphere at 37 °C.
Plasmids
Plasmids expressing wild-type p68, mutant p68 (NEAD) and wild-type p53 have been described previously.Citation6,Citation8 The human PLK1 promoter (2.3 kb) and the two 5′-deletion mutants were amplified by PCR using, as template, bacterial artificial chromosome (BAC) clone RP11–141E3 (MRC Geneservice: GenBank accession number X90725) with primer pairs flanking NheI and BamHI restriction enzyme sites, respectively, and cloned into the luciferase reporter vector pGL3-Basic (Promega). The two 5′-deletion mutants retained the transcriptional start site but lacked, respectively, previously identified p53-responsive elements: either p53RE-1 alone or both p53RE-1 and p53RE-2. The primer pairs used are as follows:
PLK1 promoter forward primer: 5′-CCGCTAGCAA CAAAAAGGAA TTAAGGAGGA-3′
PLK1 promoter reverse primer: 5′-ATGGATCCGC TCCTCCCCGA ATTCA-3′
p53RE-1 deletion mutant forward primer: 5′-ATGCTAGCGA CTGTGGGAGG CTTACACC-3′
p53RE-1 deletion mutant reverse primer: 5′- ATGGATCCGC TCCTCCCCGA ATTCA-3′
p53RE-1/p53RE-2 deletion mutant forward primer: 5′- ATGCTAAGCG TTGCAGTGAG CTGAGATCG-3′
p53RE-1/p53RE-2 deletion mutant reverse primer: 5′- ATGGATCCGC TCCTCCCCGA ATTCA -3′
Luciferase reporter assay
p53-null H1299 cells (3 × 104) were seeded onto 24-well plates and transfected with the relevant plasmids 24 h post-seeding using FuGENE 6 reagent (Promega) as directed by the manufacturer. Transfections were done in triplicate and repeated at least 3 times. Cells were harvested 24 h post-transfection and assayed for luciferase activity using Dual-Luciferase Reporter Assay System (Promega) according to manufacturer’s instructions. Values from each triplicate were averaged and plotted on graph with standard deviations as error bars. Statistical analysis was done using Student paired t test.
Western blotting
SDS-PAGE and western blotting were performed using standard conditions. The following antibodies were used: β-actin (mouse monoclonal ab6276, Abcam), p68 (mouse monoclonal PAb204, Millipore), p53 (mouse monoclonal DO1, Santa Cruz Biotechnology), PLK1 (rabbit monoclonal 208G4, Cell Signaling Technologies), Myc-epitope (mouse monoclonal 9E10, Sigma-Aldrich). Anti-mouse and anti-rabbit secondary antibodies were from DAKO.
siRNA transfection, RNA extraction, and quantitative RT-PCR
These were performed as described previously.Citation18 In brief, 30 pmol of p68-specific (AACUCUAAUG UGGAGGCGAC), p53-specific (GACUCCAGUG GUAAUCUAC), or non-specific siRNA (CAGUCGCGUU UGCGACUGG) as control (Dharmacon) were transfected into 1.5 × 106 MCF7 or HCT116 cells in 10-cm plates using Lipofectamine RNAiMax. Total cell RNA was extracted using RNeasy (Qiagen) and cDNA was synthesized as described previously.Citation18 Quantitative PCR for p68 (Hs00189323_m1) and PLK1 mRNA (Hs00983229_m1) expression, measured relative to β-actin (Hs99999903_m1) as control, was performed using TaqMan probes (Applied Biosystems; the catalog numbers for probes are given in the parentheses). Fold changes were calculated using ∆∆Ct method in MS Excel.
Chromatin immuno-precipitation
Chromatin was crosslinked by treating cells with 1.5% formaldehyde and sonicated to yield fragments of appropximately 500 base pairs in length. Approximately 500 μg of protein-bound chromatin was pre-cleared with Protein A sepharose beads (Sigma) with sheared salmon sperm DNA and then immuno-precipitated using 3 μg of DO1 or PAb204 antibody (see “Western blotting”), respectively, as described previously.Citation8,Citation18 Quantitative PCR was performed using SYBR Green QuantiTect Mastermix (Qiagen) according to manufacturer’s instructions. Promoter occupancy was calculated as percentage of input DNA using the ΔΔCt method in Microsoft Excel. Primer sequences for PLK1 p53RE-1, p53RE-2, and p21 promoter (positive control) have been described previously.Citation18,Citation42
| Abbreviations: | ||
| PLK1 | = | Polo-like kinase-1 |
| p53RE-1 and -2 | = | p53-responsive elements-1 and -2 |
| TMA | = | tissue microarray |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This study was funded through contributions from Cancer Research UK, the Breast Cancer Campaign, and the University of Dundee.
Related Research Data
References
- Dardenne E, Pierredon S, Driouch K, Gratadou L, Lacroix-Triki M, Espinoza MP, Zonta E, Germann S, Mortada H, Villemin JP, et al. Splicing switch of an epigenetic regulator by RNA helicases promotes tumor-cell invasiveness. Nat Struct Mol Biol 2012; 19:1139 - 46; http://dx.doi.org/10.1038/nsmb.2390; PMID: 23022728
- Guil S, Gattoni R, Carrascal M, Abián J, Stévenin J, Bach-Elias M. Roles of hnRNP A1, SR proteins, and p68 helicase in c-H-ras alternative splicing regulation. Mol Cell Biol 2003; 23:2927 - 41; http://dx.doi.org/10.1128/MCB.23.8.2927-2941.2003; PMID: 12665590
- Liu ZR. p68 RNA helicase is an essential human splicing factor that acts at the U1 snRNA-5′ splice site duplex. Mol Cell Biol 2002; 22:5443 - 50; http://dx.doi.org/10.1128/MCB.22.15.5443-5450.2002; PMID: 12101238
- Fukuda T, Yamagata K, Fujiyama S, Matsumoto T, Koshida I, Yoshimura K, Mihara M, Naitou M, Endoh H, Nakamura T, et al. DEAD-box RNA helicase subunits of the Drosha complex are required for processing of rRNA and a subset of microRNAs. Nat Cell Biol 2007; 9:604 - 11; http://dx.doi.org/10.1038/ncb1577; PMID: 17435748
- Jalal C, Uhlmann-Schiffler H, Stahl H. Redundant role of DEAD box proteins p68 (Ddx5) and p72/p82 (Ddx17) in ribosome biogenesis and cell proliferation. Nucleic Acids Res 2007; 35:3590 - 601; http://dx.doi.org/10.1093/nar/gkm058; PMID: 17485482
- Clark EL, Coulson A, Dalgliesh C, Rajan P, Nicol SM, Fleming S, Heer R, Gaughan L, Leung HY, Elliott DJ, et al. The RNA helicase p68 is a novel androgen receptor coactivator involved in splicing and is overexpressed in prostate cancer. Cancer Res 2008; 68:7938 - 46; http://dx.doi.org/10.1158/0008-5472.CAN-08-0932; PMID: 18829551
- Wortham NC, Ahamed E, Nicol SM, Thomas RS, Periyasamy M, Jiang J, Ochocka AM, Shousha S, Huson L, Bray SE, et al. The DEAD-box protein p72 regulates ERalpha-/oestrogen-dependent transcription and cell growth, and is associated with improved survival in ERalpha-positive breast cancer. Oncogene 2009; 28:4053 - 64; http://dx.doi.org/10.1038/onc.2009.261; PMID: 19718048
- Bates GJ, Nicol SM, Wilson BJ, Jacobs AM, Bourdon JC, Wardrop J, Gregory DJ, Lane DP, Perkins ND, Fuller-Pace FV. The DEAD box protein p68: a novel transcriptional coactivator of the p53 tumour suppressor. EMBO J 2005; 24:543 - 53; http://dx.doi.org/10.1038/sj.emboj.7600550; PMID: 15660129
- Métivier R, Penot G, Hübner MR, Reid G, Brand H, Kos M, Gannon F. Estrogen receptor-alpha directs ordered, cyclical, and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003; 115:751 - 63; http://dx.doi.org/10.1016/S0092-8674(03)00934-6; PMID: 14675539
- Yang L, Lin C, Zhao S, Wang H, Liu ZR. Phosphorylation of p68 RNA helicase plays a role in platelet-derived growth factor-induced cell proliferation by up-regulating cyclin D1 and c-Myc expression. J Biol Chem 2007; 282:16811 - 9; http://dx.doi.org/10.1074/jbc.M610488200; PMID: 17412694
- Causevic M, Hislop RG, Kernohan NM, Carey FA, Kay RA, Steele RJ, Fuller-Pace FV. Overexpression and poly-ubiquitylation of the DEAD-box RNA helicase p68 in colorectal tumours. Oncogene 2001; 20:7734 - 43; http://dx.doi.org/10.1038/sj.onc.1204976; PMID: 11753651
- Shin S, Rossow KL, Grande JP, Janknecht R. Involvement of RNA helicases p68 and p72 in colon cancer. Cancer Res 2007; 67:7572 - 8; http://dx.doi.org/10.1158/0008-5472.CAN-06-4652; PMID: 17699760
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 12; http://dx.doi.org/10.1038/nrm2395; PMID: 18431400
- Vousden KH, Lane DP. p53 in health and disease. Nat Rev Mol Cell Biol 2007; 8:275 - 83; http://dx.doi.org/10.1038/nrm2147; PMID: 17380161
- Vousden KH, Prives C. Blinded by the Light: The Growing Complexity of p53. Cell 2009; 137:413 - 31; http://dx.doi.org/10.1016/j.cell.2009.04.037; PMID: 19410540
- Goh AM, Coffill CR, Lane DP. The role of mutant p53 in human cancer. J Pathol 2011; 223:116 - 26; http://dx.doi.org/10.1002/path.2784; PMID: 21125670
- Muller PA, Vousden KH. p53 mutations in cancer. Nat Cell Biol 2013; 15:2 - 8; http://dx.doi.org/10.1038/ncb2641; PMID: 23263379
- Nicol SM, Bray SE, Black HD, Lorimore SA, Wright EG, Lane DP, Meek DW, Coates PJ, Fuller-Pace FV. The RNA helicase p68 (DDX5) is selectively required for the induction of p53-dependent p21 expression and cell-cycle arrest after DNA damage. Oncogene 2013; 32:3461 - 9; http://dx.doi.org/10.1038/onc.2012.426; PMID: 22986526
- Archambault V, Glover DM. Polo-like kinases: conservation and divergence in their functions and regulation. Nat Rev Mol Cell Biol 2009; 10:265 - 75; http://dx.doi.org/10.1038/nrm2653; PMID: 19305416
- Strebhardt K. Multifaceted polo-like kinases: drug targets and antitargets for cancer therapy. Nat Rev Drug Discov 2010; 9:643 - 60; http://dx.doi.org/10.1038/nrd3184; PMID: 20671765
- Strebhardt K, Ullrich A. Targeting polo-like kinase 1 for cancer therapy. Nat Rev Cancer 2006; 6:321 - 30; http://dx.doi.org/10.1038/nrc1841; PMID: 16557283
- Dietzmann K, Kirches E, von Bossanyi, Jachau K, Mawrin C. Increased human polo-like kinase-1 expression in gliomas. J Neurooncol 2001; 53:1 - 11; http://dx.doi.org/10.1023/A:1011808200978; PMID: 11678424
- Ito Y, Miyoshi E, Sasaki N, Kakudo K, Yoshida H, Tomoda C, Uruno T, Takamura Y, Miya A, Kobayashi K, et al. Polo-like kinase 1 overexpression is an early event in the progression of papillary carcinoma. Br J Cancer 2004; 90:414 - 8; http://dx.doi.org/10.1038/sj.bjc.6601540; PMID: 14735186
- Knecht R, Elez R, Oechler M, Solbach C, von Ilberg C, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in squamous cell carcinomas of the head and neck. Cancer Res 1999; 59:2794 - 7; PMID: 10383133
- Kneisel L, Strebhardt K, Bernd A, Wolter M, Binder A, Kaufmann R. Expression of polo-like kinase (PLK1) in thin melanomas: a novel marker of metastatic disease. J Cutan Pathol 2002; 29:354 - 8; http://dx.doi.org/10.1034/j.1600-0560.2002.290605.x; PMID: 12135466
- Takahashi T, Sano B, Nagata T, Kato H, Sugiyama Y, Kunieda K, Kimura M, Okano Y, Saji S. Polo-like kinase 1 (PLK1) is overexpressed in primary colorectal cancers. Cancer Sci 2003; 94:148 - 52; http://dx.doi.org/10.1111/j.1349-7006.2003.tb01411.x; PMID: 12708489
- Tokumitsu Y, Mori M, Tanaka S, Akazawa K, Nakano S, Niho Y. Prognostic significance of polo-like kinase expression in esophageal carcinoma. Int J Oncol 1999; 15:687 - 92; PMID: 10493949
- Weichert W, Denkert C, Schmidt M, Gekeler V, Wolf G, Köbel M, Dietel M, Hauptmann S. Polo-like kinase isoform expression is a prognostic factor in ovarian carcinoma. Br J Cancer 2004; 90:815 - 21; http://dx.doi.org/10.1038/sj.bjc.6601610; PMID: 14970859
- Weichert W, Kristiansen G, Winzer KJ, Schmidt M, Gekeler V, Noske A, Müller BM, Niesporek S, Dietel M, Denkert C. Polo-like kinase isoforms in breast cancer: expression patterns and prognostic implications. Virchows Arch 2005; 446:442 - 50; http://dx.doi.org/10.1007/s00428-005-1212-8; PMID: 15785925
- Weichert W, Schmidt M, Gekeler V, Denkert C, Stephan C, Jung K, Loening S, Dietel M, Kristiansen G. Polo-like kinase 1 is overexpressed in prostate cancer and linked to higher tumor grades. Prostate 2004; 60:240 - 5; http://dx.doi.org/10.1002/pros.20050; PMID: 15176053
- Wolf G, Elez R, Doermer A, Holtrich U, Ackermann H, Stutte HJ, Altmannsberger HM, Rübsamen-Waigmann H, Strebhardt K. Prognostic significance of polo-like kinase (PLK) expression in non-small cell lung cancer. Oncogene 1997; 14:543 - 9; http://dx.doi.org/10.1038/sj.onc.1200862; PMID: 9053852
- Yamada S, Ohira M, Horie H, Ando K, Takayasu H, Suzuki Y, Sugano S, Hirata T, Goto T, Matsunaga T, et al. Expression profiling and differential screening between hepatoblastomas and the corresponding normal livers: identification of high expression of the PLK1 oncogene as a poor-prognostic indicator of hepatoblastomas. Oncogene 2004; 23:5901 - 11; http://dx.doi.org/10.1038/sj.onc.1207782; PMID: 15221005
- Wolf G, Hildenbrand R, Schwar C, Grobholz R, Kaufmann M, Stutte HJ, Strebhardt K, Bleyl U. Polo-like kinase: a novel marker of proliferation: correlation with estrogen-receptor expression in human breast cancer. Pathol Res Pract 2000; 196:753 - 9; PMID: 11186170
- Ando K, Ozaki T, Yamamoto H, Furuya K, Hosoda M, Hayashi S, Fukuzawa M, Nakagawara A. Polo-like kinase 1 (Plk1) inhibits p53 function by physical interaction and phosphorylation. J Biol Chem 2004; 279:25549 - 61; http://dx.doi.org/10.1074/jbc.M314182200; PMID: 15024021
- Incassati A, Patel D, McCance DJ. Induction of tetraploidy through loss of p53 and upregulation of Plk1 by human papillomavirus type-16 E6. Oncogene 2006; 25:2444 - 51; http://dx.doi.org/10.1038/sj.onc.1209276; PMID: 16369493
- Kho PS, Wang Z, Zhuang L, Li Y, Chew JL, Ng HH, Liu ET, Yu Q. p53-regulated transcriptional program associated with genotoxic stress-induced apoptosis. J Biol Chem 2004; 279:21183 - 92; http://dx.doi.org/10.1074/jbc.M311912200; PMID: 15016801
- Smits VA, Klompmaker R, Arnaud L, Rijksen G, Nigg EA, Medema RH. Polo-like kinase-1 is a target of the DNA damage checkpoint. Nat Cell Biol 2000; 2:672 - 6; http://dx.doi.org/10.1038/35023629; PMID: 10980711
- Sur S, Pagliarini R, Bunz F, Rago C, Diaz LA Jr., Kinzler KW, Vogelstein B, Papadopoulos N. A panel of isogenic human cancer cells suggests a therapeutic approach for cancers with inactivated p53. Proc Natl Acad Sci U S A 2009; 106:3964 - 9; http://dx.doi.org/10.1073/pnas.0813333106; PMID: 19225112
- van Vugt MA, Medema RH. Getting in and out of mitosis with Polo-like kinase-1. Oncogene 2005; 24:2844 - 59; http://dx.doi.org/10.1038/sj.onc.1208617; PMID: 15838519
- van Vugt MA, Smits VA, Klompmaker R, Medema RH. Inhibition of Polo-like kinase-1 by DNA damage occurs in an ATM- or ATR-dependent fashion. J Biol Chem 2001; 276:41656 - 60; http://dx.doi.org/10.1074/jbc.M101831200; PMID: 11514540
- Zhu H, Chang BD, Uchiumi T, Roninson IB. Identification of promoter elements responsible for transcriptional inhibition of polo-like kinase 1 and topoisomerase IIalpha genes by p21(WAF1/CIP1/SDI1). Cell Cycle 2002; 1:59 - 66; http://dx.doi.org/10.4161/cc.1.1.101; PMID: 12429910
- McKenzie L, King S, Marcar L, Nicol S, Dias SS, Schumm K, Robertson P, Bourdon JC, Perkins N, Fuller-Pace F, et al. p53-dependent repression of polo-like kinase-1 (PLK1). Cell Cycle 2010; 9:4200 - 12; http://dx.doi.org/10.4161/cc.9.20.13532; PMID: 20962589
- King SI, Purdie CA, Bray SE, Quinlan PR, Jordan LB, Thompson AM, Meek DW. Immunohistochemical detection of Polo-like kinase-1 (PLK1) in primary breast cancer is associated with TP53 mutation and poor clinical outcom. Breast Cancer Res 2012; 14:R40; http://dx.doi.org/10.1186/bcr3136; PMID: 22405092
- Hirling H, Scheffner M, Restle T, Stahl H. RNA helicase activity associated with the human p68 protein. Nature 1989; 339:562 - 4; http://dx.doi.org/10.1038/339562a0; PMID: 2471939
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, Sedivy JM, Kinzler KW, Vogelstein B. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 501; http://dx.doi.org/10.1126/science.282.5393.1497; PMID: 9822382
- Hadad SM, Baker L, Quinlan PR, Robertson KE, Bray SE, Thomson G, Kellock D, Jordan LB, Purdie CA, Hardie DG, et al. Histological evaluation of AMPK signalling in primary breast cancer. BMC Cancer 2009; 9:307; http://dx.doi.org/10.1186/1471-2407-9-307; PMID: 19723334
- Caretti G, Schiltz RL, Dilworth FJ, Di Padova M, Zhao P, Ogryzko V, Fuller-Pace FV, Hoffman EP, Tapscott SJ, Sartorelli V. The RNA helicases p68/p72 and the noncoding RNA SRA are coregulators of MyoD and skeletal muscle differentiation. Dev Cell 2006; 11:547 - 60; http://dx.doi.org/10.1016/j.devcel.2006.08.003; PMID: 17011493
- Mazurek A, Luo W, Krasnitz A, Hicks J, Powers RS, Stillman B. DDX5 regulates DNA replication and is required for cell proliferation in a subset of breast cancer cells. Cancer Discov 2012; 2:812 - 25; http://dx.doi.org/10.1158/2159-8290.CD-12-0116; PMID: 22750847
- Rossow KL, Janknecht R. Synergism between p68 RNA helicase and the transcriptional coactivators CBP and p300. Oncogene 2003; 22:151 - 6; http://dx.doi.org/10.1038/sj.onc.1206067; PMID: 12527917
- Buszczak M, Spradling AC. The Drosophila P68 RNA helicase regulates transcriptional deactivation by promoting RNA release from chromatin. Genes Dev 2006; 20:977 - 89; http://dx.doi.org/10.1101/gad.1396306; PMID: 16598038
- Lin C, Yang L, Yang JJ, Huang Y, Liu ZR. ATPase/helicase activities of p68 RNA helicase are required for pre-mRNA splicing but not for assembly of the spliceosome. Mol Cell Biol 2005; 25:7484 - 93; http://dx.doi.org/10.1128/MCB.25.17.7484-7493.2005; PMID: 16107697
- Guo J, Hong F, Loke J, Yea S, Lim CL, Lee U, Mann DA, Walsh MJ, Sninsky JJ, Friedman SLA. A DDX5 S480A polymorphism is associated with increased transcription of fibrogenic genes in hepatic stellate cells. J Biol Chem 2010; 285:5428 - 37; http://dx.doi.org/10.1074/jbc.M109.035295; PMID: 20022962
- Moore HC, Jordan LB, Bray SE, Baker L, Quinlan PR, Purdie CA, Thompson AM, Bourdon JC, Fuller-Pace FV. The RNA helicase p68 modulates expression and function of the Δ133 isoform(s) of p53, and is inversely associated with Δ133p53 expression in breast cancer. Oncogene 2010; 29:6475 - 84; http://dx.doi.org/10.1038/onc.2010.381; PMID: 20818423
- Baker L, Quinlan PR, Patten N, Ashfield A, Birse-Stewart-Bell LJ, McCowan C, Bourdon JC, Purdie CA, Jordan LB, Dewar JA, et al. p53 mutation, deprivation and poor prognosis in primary breast cancer. Br J Cancer 2010; 102:719 - 26; http://dx.doi.org/10.1038/sj.bjc.6605540; PMID: 20104224
- Detre S, Saclani Jotti G, Dowsett M. A “quickscore” method for immunohistochemical semiquantitation: validation for oestrogen receptor in breast carcinomas. J Clin Pathol 1995; 48:876 - 8; http://dx.doi.org/10.1136/jcp.48.9.876; PMID: 7490328
- Quinlan PR, Reed C, Thompson A. INSPIRE: an integrated agent based system for hypothesis generation within cancer datasets. WI-IAT 2008; 3:587-90, 2008 IEEE/WIC/ACM International Conference on Web Intellegence and Intellegent Agent Technology.