
Abstract
The elegant choreography of metazoan development demands exquisite regulation of cell-division timing, orientation, and asymmetry. In this review, we discuss studies in Drosophila and C. elegans that reveal how the cell cycle machinery, comprised of cyclin-dependent kinase (CDK) and cyclins functions as a master regulator of development. We provide examples of how CDK/cyclins: (1) regulate the asymmetric localization and timely destruction of cell fate determinants; (2) couple signaling to the control of cell division orientation; and (3) maintain mitotic zones for stem cell proliferation. These studies illustrate how the core cell cycle machinery should be viewed not merely as an engine that drives the cell cycle forward, but rather as a dynamic regulator that integrates the cell-division cycle with cellular differentiation, ensuring the coherent and faithful execution of developmental programs.
Introduction
During metazoan development, cell division and cell differentiation must be spatially and temporally coordinated in order to generate the correct quantity and quality of cells in the right place at the right time. Since the timing of cell division is ultimately controlled by the conserved cell cycle machinery, understanding how the cell cycle influences cellular differentiation programs and, conversely, how developmental decisions feed into the regulation of cell cycle machinery is a fundamental challenge of developmental biology.
Cyclin-dependent kinases (CDK) associate with specific regulatory subunits called cyclins to drive cell cycle progression.Citation1 Cyclins, as the name implies, oscillate through the cell cycle and impart to CDK substrate specificity that is appropriate to the particular cell cycle phase. CDK1 and cyclin B form the archetypical CDK/cyclin complex known as mitotic (M)-phase-promoting factor (MPF), which is essential for mitotic and meiotic cell cycle progression.Citation1 CDK2 and cyclin E, on the other hand, mediate the entry of cells into the DNA synthesis (S) phase and the expression of S-phase-specific genes.Citation4 Some animal genomes encode an extensive repertoire of CDKs and cyclins. For example, the human genome encodes more than 20 CDK genes and more than 10 cyclin genes.Citation2,Citation3 With the expansion of the CDK and cyclin families, the roles they play have also expanded to include diverse functions in transcription, signal transduction, epigenetic regulation, metabolism, stem cell self-renewal, neuronal functions, and spermatogenesis.Citation3
Two recurrent themes in development that demand coordination between the cell cycle and differentiation programs are asymmetric (or unequal) cell division and the maintenance of cell-proliferation zones. Asymmetric cell division occurs when a cell divides to generate 2 daughter cells that are committed to different cell fates. Molecular mechanisms governing this process have been studied extensively from yeast to multicellular organisms.Citation5-Citation9 During asymmetric cell division, cells appear to become polarized in response to an intrinsic cue, or in response extrinsic signals from neighboring cells. The generation of daughter cells with distinct developmental fates then requires that the mitotic apparatus orient itself such that the polarized contents of the cell are partitioned unequally to the daughters. We will discuss recent studies that suggest how the core cell cycle machinery may coordinate this process to ensure proper cell division orientation and timing. Another major theme in development is the maintenance of cell-proliferation zones essential for growth, organ homeostasis, and stem-cell renewal. Recently, new insights into how a variety of proliferation signals modulate core cell cycle factors are beginning to shed light on this fascinating process. The studies reviewed here elucidate the developmental roles of CDKs and cyclins and provide insights into how developmental decisions may be coupled with cell cycle regulation.
CDK1 and Cyclin E in Asymmetric Cell Division in Flies
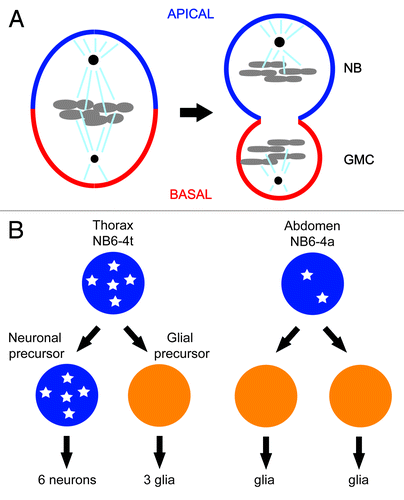
One of the first examples of CDK1’s role in differentiation came from the studies of asymmetric cell division of neuroblasts in the central nervous system of the fruit fly.Citation10 Fly neuroblasts undergo multiple rounds of asymmetric cell division along the apical/basal axis to produce the neurons that populate the central nervous system. Each asymmetric division produces a larger apical cell that retains neuroblast (NB) identity and a smaller basal cell, called the ganglion mother cell (GMC), which divides once giving rise to 2 cells with neuronal or glial identity ().Citation11 A set of conserved apical and basal proteins function to set up a polarity along the apico-basal axis of the neuroblast and cause the basal cell fate determinants Prospero and Numb to asymmetrically localize on the basal cortex as crescents during neuroblast cytokinesis. The asymmetric distribution of cell fate determinants and alignment of the mitotic spindle along the apico–basal axis ensures that the basal cell fate will be induced in each basal daughter cell. Mutations that perturb the asymmetric localization or function of the basal cell fate determinants result in symmetric neuroblast division and fate-specification defects of its daughter cells.Citation5,Citation7,Citation9
Figure 1. (A) Asymmetric segregation of cell fate determinants in asymmetric neuroblast cell division of Drosophila. Apical factors (such as Insecutable and Pins, and the evolutionary conserved Par complex consisting of Par-6, Bazooka, and Cdc42) shown in blue and basal factors (such as Miranda, Prospero, and Numb) shown in red are asymmetrically localized on the cortex along the apico–basal axis of neuroblast (left) and segregated asymmetrically to larger apical cell that retains neuroblast (NB) fate and smaller basal cell called ganglion mother cell (GMC). Mitotic spindle that orients along the apico–basal axis ensures asymmetric inheritance of the cortical cell fate determinants. Cdc2 activity is required to maintain the cortical asymmetry during cell division. (B) Asymmetric cell division of thoracic neuroblast 6-4 (NB6-4). NB6-4t divides asymmetrically to produce a neuroblast (blue) and gliablast (orange), whereas NB6-4a divides symmetrically giving rise to 2 glial cells. The neuroblast identity is maintained by high levels of Cyclin E (white stars), which is significantly reduced in NB6-4a.
A genetic screen for mutants with defects in the asymmetric localization of apical and basal proteins in neuroblasts identified a hypomorphic missense mutation of CDC2 (CDK1), cdc2E51Q, suggesting the involvement of CDC2 activity in the neuroblast asymmetric cell division.Citation10 In the mutant neuroblast, apical factors such as Inscuteable and Bazooka, as well as basal factors such as Prospero, Pon, and Miranda are all mislocalized, and the mitotic spindle fails to orient along the apico–basal axis of the cell. Detailed analyses using a temperature-sensitive allele of cdc2 as well as rescue experiments using wild-type and kinase-dead alleles of cdc2 demonstrated that the kinase activity of CDC2 is required for the asymmetric cell division of neuroblasts, and that attenuation of CDC2 activity results in the loss of the neuroblast asymmetric cell division without hindering mitosis. Temperature-shift experiments using the temperature-sensitive allele of cdc2 suggested that CDC2 activity is required for maintaining the asymmetric localization of apical complex proteins during mitosis but not for the initial establishment of asymmetry during interphase. Furthermore, genetic studies revealed that anaphase-specific cyclins, namely Cyclin B and Cyclin B3, but not metaphase-specific Cyclin A, are involved in maintaining the asymmetry of apical proteins. These results suggest that the kinase activity of CDK1/Cyclin B complex is specifically required for maintaining the apico–basal polarity of neuroblasts during mitosis.
Individual neuroblasts produced from corresponding positions in thoracic and abdominal segments usually acquire similar fates in the developing fruit fly. However, the neuroblast NB6-4 lineage is a case where variations exist.Citation12 In the thoracic segments of the embryonic central nervous system (CNS), NB6-4t divides asymmetrically to generate one neuronal and one glial precursor cell. The neuronal precursor cell then divides several times in stem-cell mode to generate a neuronal sublineage. The glial precursor cell, however, divides twice to generate 3 glial cells. In the abdominal segment of the embryonic CNS, the corresponding neuroblast (NB6-4a) merely divides symmetrically into 2 glial cells (). Mutations in homeotic genes abd-A and abd-B cause abdominal NB6-4 to transform into the more elaborate thoracic lineage. Conversely, overexpression of abd-A results in the opposite NB6-4t to NB6-4a transformation.Citation13 Genetic and biochemical evidence demonstrate that these homeotic transformations are dependent on Cyclin E. In Cyclin E loss-of-function mutants, thoracic NB6-4 adopts the fate of its abdominal counterpart, generating only 2 glial cells. Conversely, ectopic expression of Cyclin E in abdominal NB6-4 transforms its mode of division to one that resembles thoracic NB6-4. In situ hybridization reveals that Cyclin E is expressed in thoracic NB6-4 but not in the abdominal counterpart (). After the first asymmetric division, Cyclin E mRNA is detected in the neuronal precursor but not in the glial precursor daughter cell. In abd-A mutant embryos, which exhibit an abdominal-to-thoracic NB6-4 transformation, Cyclin E mRNA is detected in the transformed NB6-4a lineages. Importantly, overexpression of abd-A, which causes the opposite transformation, reduces Cyclin E mRNA levels in the thoracic segment. Perhaps most critical, the homeotic transformation observed in abd-A mutants is suppressed by mutations in Cyclin E. This epistatic evidence, along with potential Abd-A-binding sites in the Cyclin E promoter, suggest that homeotic genes in the Bithorax complex regulate the differential expression of Cyclin E, which maintains the neuroblast stem cell fate.Citation13 Interestingly, Cyclin E co-precipitates with Prospero, suggesting that it may play a role in the asymmetric localization of a key basal fate determinant.Citation14
An alternative mode by which Cyclin E regulates differentiation has been described in Drosophila female germline stem cells.Citation15 Germline stem cells in mosaic germaria carrying partial loss-of-function alleles of Cyclin E or Cdk2 display normal rates of proliferation as long as they are within the stem cell niche, reflecting that the cell cycle machinery is still intact. However, these CDK2/Cyclin E compromised germ cells fail to maintain their proliferative fate due to a weakened response to niche bone morphogenetic protein (BMP) signals. BMP ligands expressed in the niche are required for germ stem cell maintenance. Reduced levels of pMad (a reporter of BMP signaling) show that Cyclin E mutant GSC do not respond properly. As a result, germ cells are lost from the stem cell niche, arrest in the G1 stage of the cell cycle, and grow excessively without differentiating. This impaired response to BMP induction suggests that CDK2 and Cyclin E maintain germline stem cells by modulating the response to niche-derived signals.
Cell Fate Specification Regulated by C. elegans CDK1
The C. elegans zygote undergoes 5 rounds of successive asymmetric cell divisions, through which 5 somatic founder cells and 1 germline cell are produced. Mutations that perturb these asymmetric cell divisions are easily recognizable due to the absence of specific tissues and/or the presence of extra tissues, reflecting the misspecification of founder cell fates. cdk-1(ne236) and cdk-1(ne2257) are 2 hypomorphic alleles of C. elegans CDK1 that were initially identified in forward genetic screens for embryonic lethal mutations exhibiting defects in cell fate specification.Citation16 These 2 unique alleles of CDK1, one non-conditional (ne236) and one temperature-sensitive (ne2257), exhibited essentially an identical phenotype, in which the founder cell “C” produces extra intestinal tissue instead of giving rise to hypodermis and body wall muscle. cdk-1 is essential for cell division.Citation17,Citation18 However, the phenotypes exhibited by these mutants were not simply caused by defects in cell cycle progression; for both mutants, the striking defects observed in cell fate specification occur in embryos that show an otherwise wild-type pattern and timing of cell division.Citation16 Consistent with these findings, when immunoprecipitated from embryonic lysates CDK-1(ne2257) was shown to phosphorylate Histone H1, a well-known CDK1 substrate during mitosis, at levels similar to that observed for wild-type CDK-1.Citation16 These observations suggest that the function of cdk-1 in cell fate specification can be uncoupled from its role in driving progression through the cell cycle.
Structural basis of the peculiar phenotype exhibited by cdk-1(ne2257 and ne236) was suggested from the examination of the crystal structure of the activated mouse CDK2/Cyclin A complex.Citation19 The T-loop of CDK2, which harbors the mutations found in ne2257 (I173F) and ne236 (R176H) (), resides at the interface of CDK2 and Cyclin A subunits of the activated kinase complex, suggesting that amino acid substitution therein could have a subtle effect on cyclin binding (). For example, the amino acid affected in cdk-1(ne2257), Ile-173, is positioned in the co-crystal structure in very close proximity to Ile-114 of CYB-3 (Cyclin B3 in C.elegans). Perhaps accommodating the bulkier amino acid, phenylalanine, as found in cdk-1(ne2257), alters the conformation of the active kinase complex sufficiently to perturb substrate-recognition interactions important for the cell fate specification without preventing interactions important for cell cycle progression. Consistent with this idea, a screen for mutants that can suppress the embryonic lethality of cdk-1(ne2257), identified a compensatory mutation in which Ile-114 of CYB-3 is changed to a much smaller amino acid, valine (T.I. and C.M., unpublished results).
Figure 2. (A) Alignment of amino acid sequences surrounding the T-loop region of CDK1 from worm, fly, and mouse, showing the positions of conserved amino acid sequence mutated in the hypomorphic alleles of cdk-1 (ne2257 [I173F] and ne236 [R176H]) isolated in genetic screens using C.elegans, boxed in red or yellow, respectively. Invariant amino acids are indicated with an asterisk. (B) Crystal structure of activated mouse CDK2/CyclinA complex. Amino acids corresponding to those affected in the hypomorphic alleles of cdk-1, Ile173 in the case of ne2257, and Arg176 in the case of ne236 are shown in red and yellow, respectively. The amino acids corresponding to those affected in 2 cyb-3 suppressor alleles of cdk-1(ne2257) are also shown in magenta (ne4276) and light blue (ne4277).
![Figure 2. (A) Alignment of amino acid sequences surrounding the T-loop region of CDK1 from worm, fly, and mouse, showing the positions of conserved amino acid sequence mutated in the hypomorphic alleles of cdk-1 (ne2257 [I173F] and ne236 [R176H]) isolated in genetic screens using C.elegans, boxed in red or yellow, respectively. Invariant amino acids are indicated with an asterisk. (B) Crystal structure of activated mouse CDK2/CyclinA complex. Amino acids corresponding to those affected in the hypomorphic alleles of cdk-1, Ile173 in the case of ne2257, and Arg176 in the case of ne236 are shown in red and yellow, respectively. The amino acids corresponding to those affected in 2 cyb-3 suppressor alleles of cdk-1(ne2257) are also shown in magenta (ne4276) and light blue (ne4277).](/cms/asset/0053db91-873e-400c-b6d1-17ad6f70d404/kccy_a_10928656_f0002.gif)
Detailed genetic and biochemical analysis of the phenotypes caused by these subtle mutations in cdk-1 has uncovered at least 2 biological contexts in which cell cycle-independent functions of CDK-1 contribute to the asymmetric inheritance of cell fate determinants during early embryogenesis. In the first of these, CDK-1 plays a role in the transition from oocyte to embryo by timing the elimination of the OMA-1 protein after the first cell division. In the second, CDK-1 activity couples the orientation of the cell-division axis of the EMS blastomere with a polarizing signal in the 4-cell stage embryo.
CDK-1 regulates the transition from oocyte to embryo in C.elegans
The first evidence linking cdk-1 to the asymmetric localization of cell fate determinants came from the realization that the cell fate transformation observed in cdk-1(ne2257 and ne236) was essentially identical to that seen in a gain-of-function mutation of oma-1(zu405). OMA-1, and its closely related homolog OMA-2, are CCCH-type zinc-finger proteins expressed in oocytes and zygote and required redundantly for oocyte maturation.Citation20,Citation21 A study using GFP-tagged OMA-1 protein revealed that while OMA-1 protein resides uniformly throughout the cytoplasm during oogenesis and in early one-cell stage embryo, it undergoes an abrupt degradation during the first mitotic cell division. OMA-1 protein level is further reduced during the second division, after which it becomes almost non-detectable at the 4-cell stage.Citation22 Interestingly, in the gain-of-function oma-1(zu405) mutant, as well as in cdk-1(ne2257 and ne236), OMA-1 degradation fails to occur, causing OMA-1 protein to persist beyond the 4-cell stage. Reduction of OMA-1 protein level by loss-of-function mutations or by RNAi rescued the embryonic lethal phenotype of oma-1(zu405) and, importantly, also rescued the cell fate transformation and lethality of cdk-1(ne2257). Further genetic studies implicated cyclin B3 but not cyclin A in the regulation of OMA-1 stability.Citation16 Thus, a CDK-1/cyclin B3 complex promotes OMA-1 protein degradation during the first cell cycle to ensure proper cell fate specification in the initial asymmetric cell divisions of the C. elegans embryo.
Recent studies have elegantly demonstrated that the OMA proteins are multi-functional repressors that prevent the premature activation of developmental gene expression in oocytes and early embryos, and that they achieve this regulation through both transcriptional and translational repression.Citation23,Citation24 First, in the zygote as well as in the germline cell in the 2-cell stage embryo, the OMA proteins repress the RNA polymerase II complex (TFIID) by binding to and sequestering TAF-4, an essential component of TFIID.Citation23 Second, in 2-cell and 4-cell embryos the OMA proteins directly bind the 3′ UTR and inhibit the translation of ZIF-1, a substrate-binding subunit of a CUL-2-containing E3-ubiquitin ligase that degrades PIE-1.Citation24 PIE-1 is a CCCH homolog of OMA-1 and like OMA-1 appears to function as a global repressor of transcription in the germline.Citation25,Citation26 Thus, persistence of OMA-1 leads to loss of PIE-1 function, which, in turn, leads to de-repression of SKN-1, the master regulator of intestinal development, leading, among other defects, to the ectopic production of intestine by descendants of the 4-cell stage germline blastomere, P2.
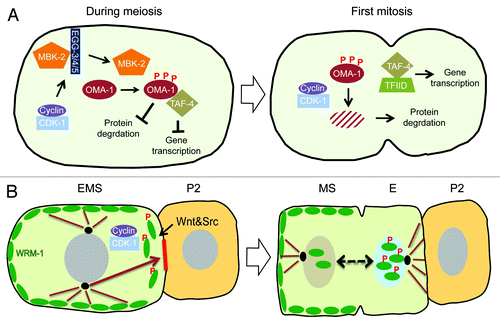
Forward and reverse genetic approaches have revealed that cdk-1 is only one piece of a multi-step mechanism that promotes the timely degradation of OMA-1 at the transition from oogenesis to embryogenesis ().Citation16 A set of biochemical experiments revealed that OMA-1 degradation is promoted by a series of sequential phosphorylations, initiated by a DYRK family kinase MBK-2, a kinase known to phosphorylate various substrates during oocyte-to-embryo transition, marking them for ubiquitin-mediated protein degradation pathway. The phosphorylation of OMA-1 by MBK-2, in turn, serves as the priming phosphorylation for subsequent direct phosphorylations by GSK-3 and Casein Kinase 1, all of which are required for OMA-1 degradation.Citation16,Citation27,Citation28 In addition, MBK-2 phosphorylation promotes the dissociation of OMA-1 from SPN-2 and its interaction with TAF-4,Citation23,Citation24 providing a striking example of how regulations of multiple independent functions and activities of a protein can be coupled. Activation of MBK-2 itself is subject to multi-layered regulation that includes phosphorylation by CDK-1 during oocyte maturation, as well as dynamic regulation of its subcellular localization from the cortex to cytoplasm involving the protein-tyrosine phosphatase-like proteins EGG-3/4/5 that tether MBK-2 to the cortex ().Citation29,Citation30 Thus CDK-1 contributes to OMA-1 degradation in at least 2 ways. First, as noted earlier, CDK-1 activates MBK-2 during the progression though meiosis. Second, since no general defects associated with failure to activate MBK-2 are observed in cdk-1 (ne236 and ne2257) alleles, CDK-1 independently promotes OMA-1 degradation downstream of MBK-2 activation during the first mitosis. However, interestingly, CDK-1 does not directly phosphorylate OMA-1, and target(s) of CDK-1 required for OMA-1 degradation remains mysterious. Despite the fact that the molecular link tying CDK-1 activity to OMA-1 destruction is still missing, it is clear that CDK-1 is embedded in the web of regulatory mechanisms that orchestrate the transition from oogenesis to embryogenesis, for example by providing the timing cue that couples OMA-1 destruction to the first mitosis.
Figure 3. (A) Model for the role of CDK-1 in OMA-1 degradation during oocyte-to-embryo transition. CDK-1 functions together with conserved protein kinases, for example, MBK-2, to promote the timely destruction of zinc-finger protein OMA-1 during the first mitosis. Removal of OMA-1 allows the protein degradation machinery to downregulate the levels of various cell fate determinants for asymmetric inheritance and release of sequestered TAF-4, a component crucial for the assembly of the transcription factor-II D (TFIID) and the RNA polymerase II complex in zygotic gene expression. CDK-1 also functions during the progression through meiosis to phosphorylate and activate MBK-2, which is sequestered on the cortex by EGG-3/4/5, until anaphase-promoting complex degrades EGG-4/5 and releases MBK-2 to cytoplasm. Upon release from the cortex, MBK-2 phosphorylates OMA-1 and starts the sequential phosphorylation events; however, the destruction of OMA-1 is delayed until the first mitosis when CDK-1 is again activated. (B) Model for CDK-1 function in regulating asymmetric cell division of EMS cell. CDK-1 phosphorylation of WRM-1 together with modifications induced by Wnt signaling releases WRM-1, an inhibitor of spindle rotation, from the posterior cortex of EMS, thereby exposing a polarity cue (red solid line) that captures one of the astral microtubules (brown arrow) and rotates the mitotic spindle in alignment with the anterior–posterior axis of embryo. Wnt signal and Src signal reinforce the capturing and/or pulling of astral microtubule. Modified WRM-1 (for example by phosphorylation) released from the posterior cortex of EMS accumulates preferentially in the posterior nascent nucleus and drives the endodermal fate.
Asymmetric localization of WRM-1/β-catenin depends on CDK-1 in C. elegans
In the 4-cell stage C. elegans embryo, the neighboring blastomeres P2 and EMS undergo a complex inductive interaction that polarizes the EMS blastomere, causing it to divide asymmetrically along the anterior–posterior axis of the embryo, producing an anterior daughter cell, MS, committed to the mesodermal fate, and a posterior daughter cell, E, committed to endoderm differentiation. In the absence of this inductive signal the EMS cell divides left-right instead of anterior-posterior, producing 2 MS-like daughter cells and no endoderm. The Wnt- and Src-signaling pathways function together to specify both endoderm and EMS division orientation; however, genetic studies suggest that cell fate specification and division orientation can be uncoupled.Citation31-Citation35 WRM-1, 1 of 4 worm homologs of β-catenin, plays a central role in transducing the P2-EMS signal.Citation36 WRM-1 exhibits a dynamic localization pattern during EMS mitosis, where signaling drives both cortical and nuclear WRM-1 asymmetries in EMS and its daughters. At the beginning of the 4-cell stage WRM-1 is symmetrically localized on the cortex of EMS. At the onset of M-phase, in response to P2-EMS signaling, WRM-1 disappears from the posterior cortex of EMS proximal to P2. After division, WRM-1 accumulates asymmetrically to higher levels in the nucleus of the posterior daughter cell E relative to the anterior daughter MS.Citation37,Citation38 This asymmetric nuclear localization of WRM-1 depends on both Wnt signaling and Src-signaling. On the other hand, the asymmetric cortical disappearance of WRM-1 depends primarily on Wnt signaling, suggesting that these 2 localizations of WRM-1 are regulated independently.Citation38 In genetic backgrounds where WRM-1 remains cortical, such as in Wnt signaling mutants, removal of the parallel Src-signaling pathway results in a highly penetrant defect in aligning the EMS mitotic spindle to the anterior-posterior axis. Significantly, removal of WRM-1 activity from these mutant backgrounds was found to rescue the spindle orientation defect, supporting a negative role for cortical WRM-1 in regulating spindle rotation in EMS. These observations lead to a model in which removal of cortical WRM-1 exposes an intrinsic cue on the posterior cortex that captures astral microtubules and rotates the EMS mitotic spindle onto the anterior–posterior axis of the embryo.Citation38
Interestingly, in early embryos harboring hypomorphic alleles of cdk-1 (ne236 and ne2257), the EMS cell fails to orient its division axis properly and, instead, divides left-right, suggesting a link between cdk-1 and regulation of spindle rotation by the P2-EMS signal.Citation38 Moreover, at non-permissive temperature cdk-1(ne2257) mutant embryos retain cortical WRM-1 throughout the EMS cell cycle just as in Wnt signaling mutants, suggesting that CDK-1 acts together with Wnt signaling to dissociate WRM-1 from the posterior cortex.Citation38 In addition, biochemical studies have shown that CDK-1 directly phosphorylates 2 CDK-1 target sites in the N terminus of WRM-1 in vitro. These phosphorylation sites are conserved in other nematode β-catenin homologs, but not in vertebrate or insect β-catenin. Mutating the CDK-1 consensus sequence of WRM-1 resulted in the persistence of WRM-1 on the posterior EMS cortex.Citation38 Taken together, these findings suggest that CDK-1 directly phosphorylates WRM-1 in vivo, and that this phosphorylation together with Wnt signaling controls the temporal and spatial aspects of WRM-1 dissociation from the EMS cortex. Whether or how CDK-1 targets WRM-1 only on the posterior cortex is not known, but could involve the signal-dependent reorganization of the Wnt signalosome on the posterior cortex that exposes WRM-1 to activated CDK-1/Cyclin B3 at the onset of M-phase.
Thus, in the context of P2-EMS signaling in the 4-cell stage embryo, CDK-1 promotes the asymmetric dissociation of cortical WRM-1 from the posterior cortex, to control the orientation of the EMS mitotic spindle (). In this way, the EMS cell couples the spatial cue provided by an external inductive signaling to the temporal cue provided by the core cell cycle machinery, so that the alignment of the cell division axis and the asymmetric nuclear accumulation of WRM-1 are synchronized and executed faithfully.
The Role of Cyclin E in Regulating the Transition from Germ Cell Proliferation to Meiotic Differentiation in C. elegans
The C. elegans germline consists of 2 U-shaped gonad arms, one anterior and one posterior, that are connected at the uterus.Citation39 In each gonad arm, the distal end (relative to the uterus) harbors a stem cell niche of proliferating germ cells known as the “mitotic zone” (). However, unlike other stem cell systems, in which a small number of stem cells divide asymmetrically, the C. elegans mitotic zone harbors more than 200 syncytial cells that divide into equivalent daughters. The fate of these daughters is not determined by asymmetric division, but rather by their position relative to the niche signal. The proliferative mode of the mitotic zone is promoted by Notch signaling from the somatic–gonadal distal tip cell (DTC).Citation40,Citation41 The DTC expresses the transmembrane LAG-2/Delta ligand, which activates the GLP-1/Notch receptor in germ cells, triggering cleavage and nuclear translocation of the Notch intracellular domain (ICD). The GLP-1 ICD interacts with CSL family transcription factors to regulate the expression of genes necessary for the proliferation of germ cells ().Citation42 Thus, under the influence of the DTC, germ cells are maintained in mitosis. However cell division within the distal region gradually causes germ cell nuclei to move away from the DTC and to enter the transition zone, where they exit mitosis and initiate meiosis. GLD-1 is a KH domain RNA binding protein that accumulates at low levels in the distal end of the germline but gradually rises until reaching maximum levels in the transition zone. In the absence of GLP-1/Notch signaling, GLD-1 is ectopically expressed in the distal region of the germline.Citation43 Conversely, overexpression of GLD-1 in the distal end abrogates the proliferative fate and causes germ cells to enter meiosis prematurely. Consistent with these observations, GLD-1 binds the 3′UTR of glp-1 mRNA and represses its translation in the embryo and meiotic region of the germline.Citation44 Collectively, these genetic and biochemical interactions suggest a mutual repressive feedback between Notch-backed proliferation and GLD-1-based differentiation ().
Figure 4. (A) Transition from mitosis into meiosis in C.elegans germline. The Distal Tip Cell (DTC in green) caps the distal end of the germline and induces mitotic proliferation (blue). Beyond the reach of the DTC Notch signaling, germ cells transition into the differentiation zone in a GLD-1 dependent manner, where they undergo meiosis (red). (B) Model for Cyclin E in maintenance of germ stem-cell fate in C. elegans. The boundary between the proliferative and transition zones is maintained by 2 mutual negative regulations: CDK-2/ CYE-1 and GLP-1 inhibit accumulation of GLD-1 in the mitotic proliferation zone, while GLD-1 represses the translation of cye-1 and glp-1 transcripts in the transition zone. It remains to be seen if CYE-1 can maintain proliferation zone by enhancing GLP-1 ICD induction in the similar way it does in vulval development.
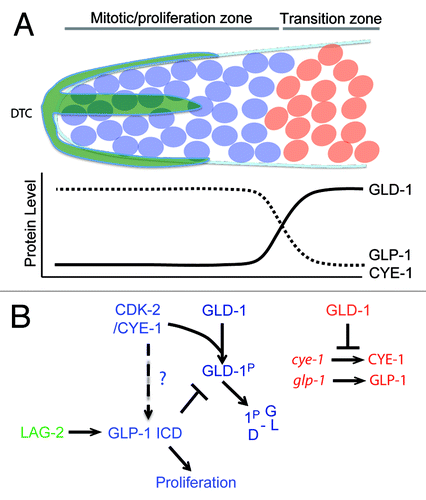
The proliferative fate has also been shown to depend on CDK-2/CYE-1(Cyclin E), since premature meiotic differentiation is enhanced in a weak glp-1 mutant background upon cye-1 or cdk-2 RNAi.Citation45 CDK-2/CYE-1 directly phosphorylates GLD-1 in vitro, and cye-1 RNAi leads to an increase in GLD-1 levels in the distal end of the gonad. Since mutating the phosphorylation site in GLD-1 phenocopies this increase in distal GLD-1 levels, it is likely that that CDK-2/CYE-1 functions in the distal region of the gonad to block the premature switch to meiotic differentiation by targeting GLD-1 directly ().Citation46 Interestingly, GLD-1 binds to the 3′UTR of cye-1 and represses its translation, emphasizing the mutual negative feedbacks that govern proliferation vs. differentiation decisions in the germline ().Citation48 However interestingly, reduction of cdk-2 or cye-1 function leads to premature meiotic entry even in the absence of gld-1. Therefore CDK-2/CYE-1 must also promote proliferation through a GLD-1 independent pathway.
The role of Cyclin E in C. elegans vulval development may offer a clue to this additional mode of action. During vulva development in C. elegans, the anchor cell (AC) signals the induction of a primary vulval precursor cell (VPC) P6.p, via the EGFR signaling pathway. Upon induction, P6.p then signals its neighboring VPCs P5.p and P7.p, via the LIN-12/Notch signaling pathway, to adopt secondary fates. By utilizing a NICD::GFP reporter, it was demonstrated that CYE-1 exerts a stabilizing effect on the intercellular pool of NICD in VPCs.Citation47 It is enticing to surmise that CYE-1 may exert a similar stabilizing effect on GLP-1 ICD in the germline mitotic zone. If this indeed is the case, then Cyclin E would maintain the proliferative mode of mitotic germ cells by both positively stabilizing the GLP-1/Notch proliferative cue and negatively targeting GLD-1 to prevent the transition to meiosis ().
Conclusion
In addition to executing DNA synthesis and mitosis, dividing cells must compute crucial developmental decisions such as division orientation and identity. Because of their central role in regulating cell division, CDK cyclin complexes are ideal factors for mediating the multifaceted decisions required to ensure the overall coherence of developmental programs. The examples reviewed here identify common themes that cells use to couple cell division and differentiation. External cues provide spatial orientation to dividing cells within a larger developmental context (e.g., P2, DTC, and AC). Internal cellular components, such as centrosomes and microtubules, provide early reference points for future daughter cells. Cyclins provide temporal orientation, defining the phase through which a dividing cell is passing. CDKs provide a molecular switch, modifying targets to regulate their activity or stability. Using these tools, cells are able to segregate cell fate determinants to specific daughter cells, activate proteins at a certain stage of division and within a certain spatial context, or destroy them after fulfilling their role. The coordination of these events ensures the accurate progression of development. It seems likely that the molecular connections between the cell cycle machinery and developmental regulation will become more prominent in the future, as additional CDK substrates important for specific developmental processes are identified. For example, Xenopus CDK14 (a CDK1 variant) bound to membrane-tethered Cyclin Y promotes Wnt signal transduction by phosphorylating the low-density lipoprotein receptor-related protein 5 and 6 (LRP5/6) co-receptor during the G2/M phase of the cell cycle.Citation49 The identification of CDK1 substrates important for developmental regulation will improve our understanding of the role of CDK1/cyclin in regulating development.
A recurrent challenge facing the analysis of a major cell cycle regulator such as CDK1 in development has been its obligatory role in cell cycle progression. The studies summarized in this review provided unique opportunities to identify additional roles of CDK1 by genetically uncoupling developmental decisions from cell cycle regulation. These studies also underscore the value of forward genetics in model organisms to identify rare and useful alleles of essential genes, for which the strong loss-of-function (e.g., null alleles or RNAi) cause pleiotropy that masks more subtle functions. Given the conservation and fundamental importance of CDKs and cyclins, their roles in regulating differentiation as identified in flies and worms are very likely to be conserved in mammals.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We thank Dr Darryl Conte for critical reading of the manuscript and helpful comments, Dr David Lambright for help with Figure 2, and NIH and HHMI for funding. We also apologize to authors whose work could not be cited.
Related Research Data
References
- Morgan DO. Cyclin-dependent kinases: engines, clocks, and microprocessors. Annu Rev Cell Dev Biol 1997; 13:261 - 91; http://dx.doi.org/10.1146/annurev.cellbio.13.1.261; PMID: 9442875
- Malumbres M, Harlow E, Hunt T, Hunter T, Lahti JM, Manning G, Morgan DO, Tsai LH, Wolgemuth DJ. Cyclin-dependent kinases: a family portrait. Nat Cell Biol 2009; 11:1275 - 6; http://dx.doi.org/10.1038/ncb1109-1275; PMID: 19884882
- Lim S, Kaldis P. Cdks, cyclins and CKIs: roles beyond cell cycle regulation. Development 2013; 140:3079 - 93; http://dx.doi.org/10.1242/dev.091744; PMID: 23861057
- Möröy T, Geisen C, Cyclin E. Cyclin E. Int J Biochem Cell Biol 2004; 36:1424 - 39; http://dx.doi.org/10.1016/j.biocel.2003.12.005; PMID: 15147722
- Gönczy P. Mechanisms of asymmetric cell division: flies and worms pave the way. Nat Rev Mol Cell Biol 2008; 9:355 - 66; http://dx.doi.org/10.1038/nrm2388; PMID: 18431399
- Li R. The art of choreographing asymmetric cell division. Dev Cell 2013; 25:439 - 50; http://dx.doi.org/10.1016/j.devcel.2013.05.003; PMID: 23763946
- Knoblich JA. Mechanisms of asymmetric stem cell division. Cell 2008; 132:583 - 97; http://dx.doi.org/10.1016/j.cell.2008.02.007; PMID: 18295577
- Horvitz HR, Herskowitz I. Mechanisms of asymmetric cell division: two Bs or not two Bs, that is the question. Cell 1992; 68:237 - 55; http://dx.doi.org/10.1016/0092-8674(92)90468-R; PMID: 1733500
- Betschinger J, Knoblich JA. Dare to be different: asymmetric cell division in Drosophila, C. elegans and vertebrates. Curr Biol 2004; 14:R674 - 85; http://dx.doi.org/10.1016/j.cub.2004.08.017; PMID: 15324689
- Tio M, Udolph G, Yang X, Chia W. cdc2 links the Drosophila cell cycle and asymmetric division machineries. Nature 2001; 409:1063 - 7; http://dx.doi.org/10.1038/35059124; PMID: 11234018
- Chia W, Somers WG, Wang H. Drosophila neuroblast asymmetric divisions: cell cycle regulators, asymmetric protein localization, and tumorigenesis. J Cell Biol 2008; 180:267 - 72; http://dx.doi.org/10.1083/jcb.200708159; PMID: 18209103
- Akiyama-Oda Y, Hosoya T, Hotta Y. Asymmetric cell division of thoracic neuroblast 6-4 to bifurcate glial and neuronal lineage in Drosophila. Development 1999; 126:1967 - 74; PMID: 10101130
- Berger C, Pallavi SK, Prasad M, Shashidhara LS, Technau GM. A critical role for cyclin E in cell fate determination in the central nervous system of Drosophila melanogaster. Nat Cell Biol 2005; 7:56 - 62; http://dx.doi.org/10.1038/ncb1203; PMID: 15580266
- Berger C, Kannan R, Myneni S, Renner S, Shashidhara LS, Technau GM. Cell cycle independent role of Cyclin E during neural cell fate specification in Drosophila is mediated by its regulation of Prospero function. Dev Biol 2010; 337:415 - 24; http://dx.doi.org/10.1016/j.ydbio.2009.11.012; PMID: 19914234
- Ables ET, Drummond-Barbosa D. Cyclin E controls Drosophila female germline stem cell maintenance independently of its role in proliferation by modulating responsiveness to niche signals. Development 2013; 140:530 - 40; http://dx.doi.org/10.1242/dev.088583; PMID: 23293285
- Shirayama M, Soto MC, Ishidate T, Kim S, Nakamura K, Bei Y, van den Heuvel S, Mello CC. The Conserved Kinases CDK-1, GSK-3, KIN-19, and MBK-2 Promote OMA-1 Destruction to Regulate the Oocyte-to-Embryo Transition in C. elegans. Curr Biol 2006; 16:47 - 55; http://dx.doi.org/10.1016/j.cub.2005.11.070; PMID: 16343905
- Boxem M, Srinivasan DG, van den Heuvel S. The Caenorhabditis elegans gene ncc-1 encodes a cdc2-related kinase required for M phase in meiotic and mitotic cell divisions, but not for S phase. Development 1999; 126:2227 - 39; PMID: 10207147
- Polinko ES, Strome S. Depletion of a Cks homolog in C. elegans embryos uncovers a post-metaphase role in both meiosis and mitosis. Curr Biol 2000; 10:1471 - 4; http://dx.doi.org/10.1016/S0960-9822(00)00808-3; PMID: 11102813
- Jeffrey PD, Russo AA, Polyak K, Gibbs E, Hurwitz J, Massagué J, Pavletich NP. Mechanism of CDK activation revealed by the structure of a cyclinA-CDK2 complex. Nature 1995; 376:313 - 20; http://dx.doi.org/10.1038/376313a0; PMID: 7630397
- Detwiler MR, Reuben M, Li X, Rogers E, Lin R. Two zinc finger proteins, OMA-1 and OMA-2, are redundantly required for oocyte maturation in C. elegans. Dev Cell 2001; 1:187 - 99; http://dx.doi.org/10.1016/S1534-5807(01)00026-0; PMID: 11702779
- Shimada M, Kawahara H, Doi H. Novel family of CCCH-type zinc-finger proteins, MOE-1, -2 and -3, participates in C. elegans oocyte maturation. Genes Cells 2002; 7:933 - 47; http://dx.doi.org/10.1046/j.1365-2443.2002.00570.x; PMID: 12296824
- Lin R. A gain-of-function mutation in oma-1, a C. elegans gene required for oocyte maturation, results in delayed degradation of maternal proteins and embryonic lethality. Dev Biol 2003; 258:226 - 39; http://dx.doi.org/10.1016/S0012-1606(03)00119-2; PMID: 12781695
- Guven-Ozkan T, Nishi Y, Robertson SM, Lin R. Global transcriptional repression in C. elegans germline precursors by regulated sequestration of TAF-4. Cell 2008; 135:149 - 60; http://dx.doi.org/10.1016/j.cell.2008.07.040; PMID: 18854162
- Guven-Ozkan T, Robertson SM, Nishi Y, Lin R. zif-1 translational repression defines a second, mutually exclusive OMA function in germline transcriptional repression. Development 2010; 137:3373 - 82; http://dx.doi.org/10.1242/dev.055327; PMID: 20826530
- Mello CC, Schubert C, Draper B, Zhang W, Lobel R, Priess JR. The PIE-1 protein and germline specification in C. elegans embryos. Nature 1996; 382:710 - 2; http://dx.doi.org/10.1038/382710a0; PMID: 8751440
- Seydoux G, Mello CC, Pettitt J, Wood WB, Priess JR, Fire A. Repression of gene expression in the embryonic germ lineage of C. elegans. Nature 1996; 382:713 - 6; http://dx.doi.org/10.1038/382713a0; PMID: 8751441
- Nishi Y, Lin R. DYRK2 and GSK-3 phosphorylate and promote the timely degradation of OMA-1, a key regulator of the oocyte-to-embryo transition in C. elegans. Dev Biol 2005; 288:139 - 49; http://dx.doi.org/10.1016/j.ydbio.2005.09.053; PMID: 16289132
- Stitzel ML, Pellettieri J, Seydoux G. The C. elegans DYRK Kinase MBK-2 Marks Oocyte Proteins for Degradation in Response to Meiotic Maturation. Curr Biol 2006; 16:56 - 62; http://dx.doi.org/10.1016/j.cub.2005.11.063; PMID: 16338136
- Cheng KC, Klancer R, Singson A, Seydoux G. Regulation of MBK-2/DYRK by CDK-1 and the pseudophosphatases EGG-4 and EGG-5 during the oocyte-to-embryo transition. Cell 2009; 139:560 - 72; http://dx.doi.org/10.1016/j.cell.2009.08.047; PMID: 19879842
- Parry JM, Velarde NV, Lefkovith AJ, Zegarek MH, Hang JS, Ohm J, Klancer R, Maruyama R, Druzhinina MK, Grant BD, et al. EGG-4 and EGG-5 Link Events of the Oocyte-to-Embryo Transition with Meiotic Progression in C. elegans. Curr Biol 2009; 19:1752 - 7; http://dx.doi.org/10.1016/j.cub.2009.09.015; PMID: 19879147
- Thorpe CJ, Schlesinger A, Carter JC, Bowerman B. Wnt signaling polarizes an early C. elegans blastomere to distinguish endoderm from mesoderm. Cell 1997; 90:695 - 705; http://dx.doi.org/10.1016/S0092-8674(00)80530-9; PMID: 9288749
- Rocheleau CE, Downs WD, Lin R, Wittmann C, Bei Y, Cha YH, Ali M, Priess JR, Mello CC. Wnt signaling and an APC-related gene specify endoderm in early C. elegans embryos. Cell 1997; 90:707 - 16; http://dx.doi.org/10.1016/S0092-8674(00)80531-0; PMID: 9288750
- Schlesinger A, Shelton CA, Maloof JN, Meneghini M, Bowerman B. Wnt pathway components orient a mitotic spindle in the early Caenorhabditis elegans embryo without requiring gene transcription in the responding cell. Genes Dev 1999; 13:2028 - 38; http://dx.doi.org/10.1101/gad.13.15.2028; PMID: 10444600
- Bei Y, Hogan J, Berkowitz LA, Soto M, Rocheleau CE, Pang KM, Collins J, Mello CC. SRC-1 and Wnt signaling act together to specify endoderm and to control cleavage orientation in early C. elegans embryos. Dev Cell 2002; 3:113 - 25; http://dx.doi.org/10.1016/S1534-5807(02)00185-5; PMID: 12110172
- Walston T, Tuskey C, Edgar L, Hawkins N, Ellis G, Bowerman B, Wood W, Hardin J. Multiple Wnt signaling pathways converge to orient the mitotic spindle in early C. elegans embryos. Dev Cell 2004; 7:831 - 41; http://dx.doi.org/10.1016/j.devcel.2004.10.008; PMID: 15572126
- Ishidate T, Kim S, Mello C, Shirayama M. Inductive asymmetric cell division: The WRM leads the way. Worm 2013; 2:e26276; http://dx.doi.org/10.4161/worm.26276; PMID: 24524013
- Nakamura K, Kim S, Ishidate T, Bei Y, Pang K, Shirayama M, Trzepacz C, Brownell DR, Mello CC. Wnt signaling drives WRM-1/beta-catenin asymmetries in early C. elegans embryos. Genes Dev 2005; 19:1749 - 54; http://dx.doi.org/10.1101/gad.1323705; PMID: 16077004
- Kim S, Ishidate T, Sharma R, Soto MC, Conte D Jr., Mello CC, Shirayama M. Wnt and CDK-1 regulate cortical release of WRM-1/β-catenin to control cell division orientation in early Caenorhabditis elegans embryos. Proc Natl Acad Sci U S A 2013; 110:E918 - 27; http://dx.doi.org/10.1073/pnas.1300769110; PMID: 23431196
- Kimble J, Crittenden SL. Germline proliferation and its control. In: Community TCeR, ed. Worm Book, 2005.
- Kimble J, Simpson P. The LIN-12/Notch signaling pathway and its regulation. Annu Rev Cell Dev Biol 1997; 13:333 - 61; http://dx.doi.org/10.1146/annurev.cellbio.13.1.333; PMID: 9442877
- Crittenden SL, Eckmann CR, Wang L, Bernstein DS, Wickens M, Kimble J. Regulation of the mitosis/meiosis decision in the Caenorhabditis elegans germline. Philos Trans R Soc Lond B Biol Sci 2003; 358:1359 - 62; http://dx.doi.org/10.1098/rstb.2003.1333; PMID: 14511482
- Christensen S, Kodoyianni V, Bosenberg M, Friedman L, Kimble J. lag-1, a gene required for lin-12 and glp-1 signaling in Caenorhabditis elegans, is homologous to human CBF1 and Drosophila Su(H). Development 1996; 122:1373 - 83; PMID: 8625826
- Hansen D, Wilson-Berry L, Dang T, Schedl T. Control of the proliferation versus meiotic development decision in the C. elegans germline through regulation of GLD-1 protein accumulation. Development 2004; 131:93 - 104; http://dx.doi.org/10.1242/dev.00916; PMID: 14660440
- Marin VA, Evans TC. Translational repression of a C. elegans Notch mRNA by the STAR/KH domain protein GLD-1. Development 2003; 130:2623 - 32; http://dx.doi.org/10.1242/dev.00486; PMID: 12736207
- Fox PM, Vought VE, Hanazawa M, Lee MH, Maine EM, Schedl T. Cyclin E and CDK-2 regulate proliferative cell fate and cell cycle progression in the C. elegans germline. Development 2011; 138:2223 - 34; http://dx.doi.org/10.1242/dev.059535; PMID: 21558371
- Jeong J, Verheyden JM, Kimble J. Cyclin E and Cdk2 control GLD-1, the mitosis/meiosis decision, and germline stem cells in Caenorhabditis elegans. PLoS Genet 2011; 7:e1001348; http://dx.doi.org/10.1371/journal.pgen.1001348; PMID: 21455289
- Nusser-Stein S, Beyer A, Rimann I, Adamczyk M, Piterman N, Hajnal A, Fisher J. Cell-cycle regulation of NOTCH signaling during C. elegans vulval development. Mol Syst Biol 2012; 8:618; http://dx.doi.org/10.1038/msb.2012.51; PMID: 23047528
- Biedermann B, Wright J, Senften M, Kalchhauser I, Sarathy G, Lee MH, Ciosk R. Translational repression of cyclin E prevents precocious mitosis and embryonic gene activation during C. elegans meiosis. Dev Cell 2009; 17:355 - 64; http://dx.doi.org/10.1016/j.devcel.2009.08.003; PMID: 19758560
- Davidson G, Shen J, Huang YL, Su Y, Karaulanov E, Bartscherer K, Hassler C, Stannek P, Boutros M, Niehrs C. Cell cycle control of wnt receptor activation. Dev Cell 2009; 17:788 - 99; http://dx.doi.org/10.1016/j.devcel.2009.11.006; PMID: 20059949