
Abstract
miRNAs are critically implicated in the initiation process of and progression through cancerogenesis. The mechanisms, however, by which miRNAs interfere with the signalosomes of human cancer cells, are still obscure. We utilized the p53-mutated human keratinocyte cell line HACAT to investigate the biological significance and extent to which miRNAs regulate proliferation, cell growth, and apoptosis in transformed phenotypes. Silencing of the miRNA-processing enzyme Dicer1 resulted in cell cycle arrest at the G1/S border, along with restoration of CDK inhibitor p21CIPexpression. Employing a cell cycle-wide phospho-proteomic approach, we detected neglectable changes in abundance and schedule of overall and cell cycle periodic protein expression despite cell cycle arrest of Dicer1-depleted cells. Instead, we found substantially delayed post-translational modifications of some, but not all, signaling nodes. Phospho-site-specific analyses revealed that pro-apoptotic information elicited by Myc, β-catenin, and other mitotic pathways early in G1 are absorbed and balanced by anti-apoptotic signaling from AKT and NFκB in Dicer1-competent cells. The absence of regulatory miRNAs, however, led to a substantial delay of anti-apoptotic signaling, leaving pro-apoptotic stress unbalanced in Dicer1-deprived cells. We here show that this temporal separation of pro- and anti-apoptotic signaling induced by inhibition of Dicer1 is synergistic and synthetic lethal to low-dose 5-FU chemotherapy in p53-mutated HACAT cells. The findings reported here contribute to the understanding of the complex interactions of miRNAs with the signalosom of transformed phenotypes and may help to design novel strategies to fight cancer.
Introduction
p53 is a versatile transcription factor, which integrates information from unbalanced mitotic, oncogenic, or metabolic stress into graded cellular responses such as cell cycle arrest, senescence, DNA repair, block of angiogenesis, or apoptosis. Because of this cell fate determining position within the cell’s signalosome, p53 is lost, mutated, or functionally inactivated in most human tumor cells. miRNAs are a recently discovered class of small, evolutionary highly conserved non-coding RNAs of 18–25 nucleotides length, which are generated by sequential processing of primary and precursor miRNA by the ribonucleases Drosha and Dicer1. They regulate 60% of all known human mRNAs and play key roles in cell physiology, differentiation, and carcinogenesis. The mechanisms, however, by which miRNAs interfere with cell survival and programmed cell death in transformed cells are still obscure.
Results and Discussion
To study the impact and extent to which miRNA contributes to cell cycle progression in transformed phenotypes, we here employed the p53-mutated human keratinocyte cell line (HACAT) and RNA interference (siRNA) techniques. We synchronized HACAT cells in G0 by serum starvation for 48 h and monitored the dynamics of cell cycle transition at various time points after G1 release. In line with previous reports from other cellular systems, we observed a profound G1/S arrest in Dicer1 deficient HACAT cells, as compared with wild-type (wt) cells treated with non-target siRNA (). These observations corroborate and extend published data indicating that miRNAs are required for stem cells to bypass the G1/S checkpoint.Citation1
Figure 1. Dicer1 depletion causes cell cycle arrest and restoration of p21CIP in HACAT cells. (A) HACAT cells were transiently transfected with Dicer1 siRNA (50 nM) or non-target control siRNA, synchronized in serum-free medium for 48 h, and harvested at the indicated time points after release from starvation. Distribution of cells in different phases of the cell cycle was analyzed by propidium iodide staining using flow cytometry techniques. Histogram plots are representative of 3 experiments. (B) Kinetics of induction of p21 in Dicer1-depleted HACAT cells. Effects of Dicer1 knockdown on p21 levels following serum stimulation were analyzed by western blot. Tubulin was used as loading control. The image shows a representative of 3 experiments.
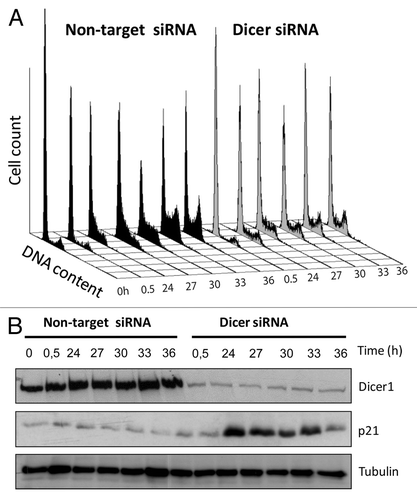
Both, p53 and miRNAs feed into the cyclin/CDK-driven cell cycle clock. p53 arrests cells during G1 progression and G1/S transit by inducing the CDK-inhibitory protein p21CIP1 and microRNA cluster miR-34a. miR-34a inhibits CDK2, CDK4, and CDK6 as well as E2F, a transcription factor that, after release from the retinoblastoma tumor suppressor Rb, initiates S-phase entry. In addition, miRNA clusters miR-17–92, miR-106b, miR-181, and miR-221/222 promote the induction of the CDK inhibitors p21CIP1, p27KIP1, and p57KIP2, thus enhancing the p53-mediated anti-proliferative effect. Consequently, we screened for expression of CDK inhibitors and found substantial upregulation of p21CIP1 in Dicer1-disrupted cells but not in Dicer1-competent (non-target siRNA) controls (). This finding is of utmost importance, since p53 is known to be key for p21 expression.Citation2 The results reported here suggest that depletion of Dicer1 restores p21 expression in p53-mutated cells, and that miRNA critically contributes to cell cycle regulation at least in part through intervening with the cyclin/CDK system and its inhibitors.
To investigate whether the delayed cell cycle progression observed in Dicer1-depleted cells can be attributed to restored p21CIP1 function, we employed a phospho-protein array (Signaling Phospho Antibody Array; Fullmoon Biosystems). Since miRNAs inhibit translation or tag mRNA for degradation, we first analyzed the overall abundance of 248 proteins and phospho-proteins but found no significant difference between Dicer1-deficient and -competent HACAT cells at 6 time points ranging from 0 to 36 h after G1 release. Next, we identified 88 periodic total and phospho-proteins whose expression levels are dynamically regulated within the cell division cycle (). Irrespective of the delayed cell cycle progression observed, 61 out of these 88 signaling molecules occurred on schedule in Dicer1 knockout cells, a finding that was not expected in light of high p21CIP1 expression as reported earlier. These observations, however, are in line with previous reports suggesting that a periodic transcription factor network constitutes a cell cycle oscillator independent of, and in tandem with, the CDK system.Citation3 The most intriguing observation, however, was a profound delay of post-translational regulatory modifications by phosphorylation in some, but not all, interrogated signaling nodules, which dissociate proliferative signals and mitotic stress from balancing anti-apoptotic responses. Precisely, we detected mitotic signals communicated by Myc proteins and others on schedule, but found activation of the AKT and NFkB pathways substantially delayed in Dicer1-disrupted cells. Both, AKT and NFκB confer cell survival and are critically engaged in balancing apoptotic signals ().
Figure 2. Dynamics of periodic proteins in Dicer1 competent (non-target siRNA) and Dicer1 depleted (Dicer1 siRNA) HACAT cells. (A) Heat maps depicting total protein (left) or phospho-protein levels (right) of 88 periodic genes. Each row represents data for the same protein. (B) Cluster of proteins that exhibited altered expression pattern in Dicer1-deficient cells. Expression levels are depicted as log2-fold change vs. mean expression. Low intensities are colored in green; high intensities are colored in red. Expression levels at each point in the time series were mapped onto a cell cycle timeline. The S and G2/M phases of the Dicer1 knockout timeline are given in green, indicating that Dicer1-depleted cells arrest at the G1/S-phase transition. (C) Network display: nodes are either colored in red, activators unresponsive to Dicer1 silencing; green, Dicer1 responsive repressors; or gray, constitutively associated proteins.
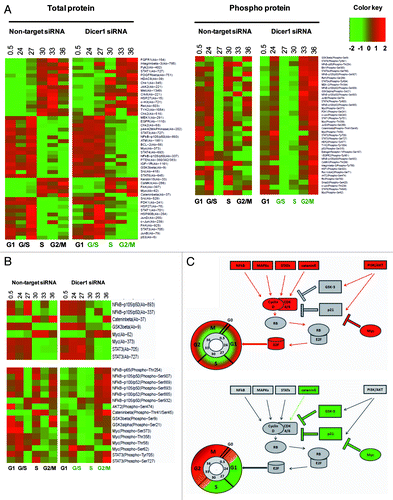
Myc is a transcription factor closely linked to the TGF/SMAD and WNT signaling pathway. Through binding to Enhancer Box (E-box) sequences and by recruiting histone acetyl-transferases, Myc modulates the expression and activity of roughly 15% of all known human genes.Citation4 We found expression and early activation of Myc by phosphorylation on position Ser-62 on schedule in Dicer1-depleted and -competent cells. Phosphorylation at position Ser-62 occurs after mitotic signaling of the Ras/Raf/MAPK module and stabilizes Myc protein.Citation5 Interestingly, however, we detected phosphorylation on positions Thr-58, Thr-358, and Ser-373 early in Dicer1-competent and late in Dicer1-deprived cells. Phosphorylation on Thr-58 is mediated by GSK-3β and leads to Myc degradation.Citation5 Ser-373 is phosphorylated by PAK2 and other kinases and impedes Myc binding to DNA by blocking the requisite Max dimerization site.Citation6 These findings suggest that Myc activation is short lived in Dicer1-competent cells, peaking early after G1 release. In stark contrast, Myc was found activated throughout G1, S, and G2/M in Dicer1-depleted HACAT.
The JAK–STAT (Janus kinase/Signal transducer and activator of transcription) system communicates information from a wide array of cytokines, growth factors, and its receptors to regulate proliferation, differentiation, cell migration, apoptosis, and the epigenetic switch that links inflammation to cancer.Citation7 While STAT1 and STAT6 expression and activation was found unaltered, we detected a substantial delay of STAT3-activating events in Dicer 1-disrupted cells. Upon growth factor-mediated G1 release, STAT3 is phosphorylated on position Ser-727 by RPS6KA5 and at position Tyr-705 by PTK6 or FER-kinases, both regulatory events leading to improved transcriptional activity of STAT3 homo- or STAT1/STAT3 hetero-dimers at interleukin-6 (IL-6)-responsive elements.Citation8 Activated STAT3 induces miR-181b and miR-21 expression in various tumor cells. In turn, miR-21 reduces expression of tumor suppressor genes such as PTEN and others, which ultimately promote activation of AKT and NFκB signaling.Citation9
Mitotic signals leading to apoptosis are fine-tuned and balanced predominantly by p53, AKT, and glycogen synthase kinase-3 (GSK-3) α and β. The substrates of GSK-3β are cell cycle -related molecules, signaling proteins, and transcription factors like D type cyclin, Myc, p53, and others that critically regulate proliferation, cell growth, and cell survival.Citation10,Citation11 GSK-3β is active when phosphorylated at Tyr-216. However, its activity is markedly inhibited following phosphorylation of Ser-21 of GSK-3α and Ser-9 of GSK-3β by AKT and RPS6KA3.Citation12,Citation13 We found GSK-3β and GSK-3α downregulated by phosphorylated Ser-9 and Ser-21 early in Dicer-competent and late in Dicer1-disrupted cells. Upon phosphorylation by GSK-3, the affinity of transcription factor JUN to DNA as well as the gene-regulatory activity of the calcineurin/NFAT system is severely impeded.Citation14 In WNT signaling, GSK-3β complexes with APC, AXIN1, and β-catenin, which leads to phosphorylation at position Thr-41 of β-catenin and its consecutive degradation by the proteasome.Citation15 This is in line with our data, showing delayed phosphorylation of β-catenin at position Thr-41, indicating prolonged mitotic Wnt signaling in Dicer1-disrupted cells. Moreover, GSK-3 is engaged in the anti-apoptotic response of NFκB to TNF-α, leaving mitotic, and thus pro-apoptotic, signals by Myc and other pathways unbalanced in Dicer1-deprived cells early after G1 release.
AKT critically orchestrates metabolism, proliferation, cell survival, cell growth, and angiogenesis in response to signals communicated via insulin-like growth factor (IGF), platelet-derived growth factor (PDGF), epidermal growth factor (EGF), and many other molecules including the phosphatidyl- inositol 3-kinase (PI3K).Citation16 So far, more than 100 substrates of AKT have been identified. We detected activation of AKT2 through phosphorylation on position Ser-474 by mammalian target of rapamycin (mTOR) immediately after G1 release in wild-type (non-target) and late in Dicer1-deprived cells. AKT regulates cell cycle timing by preventing GSK-3β-mediated degradation of cyclin D1Citation17 and by negatively regulating CDK inhibitors p21Cip1, p27Kip1 Wee1 and Myt1.Citation18-Citation20 In addition to the inhibitory effects on GSK-3 signaling, AKT2 decreases MAP3K5 (apoptosis signal-related kinase) activity induced by oxidative stress, thus supporting cell survival. AKT2 is involved in NFκB- and CREB1 (cyclic AMP-response element binding protein)-dependent gene transcription of pro-survival genes such as BCL2 and MCL1 and inhibition of the pro-apoptotic elements BIM and BAX.Citation16 Moreover, AKT phosphorylates BCL2 family member BAD, causing its dissociation from the BCL-2/BCL-X complex and its subsequent loss of pro-apoptotic function.Citation21
In line with these findings, we observed early activation of NFκB in Dicer1-competent cells and a profound delay after Dicer1 knockdown. The family of nuclear factor κB (NFκB)/Rel transcription factor comprise RelA (p65), c-Rel, RelB, NFκB1 (p105/p50), and NFκB2 (p100/p52). p105 and p100 are proteolytically processed by the proteasome after phosphorylation by NIK and IKKα at Ser-893/Ser-932 or Ser-865/Ser-869 to produce functional p50 and p52 fragments, respectively.Citation22-Citation25 We detected activating phosphorylation events of NFκB p105/50 at position Ser-893, of NFκB p65 at position Thr-254, and of NFκB p100/52 at position Ser-865/Ser-869 immediately after G1 release in Dicer1-competent but very late in Dicer1 knockout HACAT cells. RelA p65 forms homo- or hetero-dimers with p50, p52, and c-Rel and interacts with TP53BP2, GSK-3β, HDAC, and many other key regulatory elements. Complexed with IKB (inhibitors of κB), NFκB/Rel dimers are retained in the cytoplasm in a functionally inactive state and released upon a variety of stimulatory signals to exert pivotal roles in inflammation, immunity, differentiation, cell growth, tumorigenesis, and the inhibition of apoptosis.Citation25 These data, together with the findings reported for GSK-3 and AKT kinase activities, indicate that pro-apoptotic stresses, initiated shortly after G1 release by Myc and other mitotic/pro-apoptotic molecules, might not be sufficiently absorbed by balancing anti-apoptotic mechanisms in Dicer1-deprived HACAT cells ().
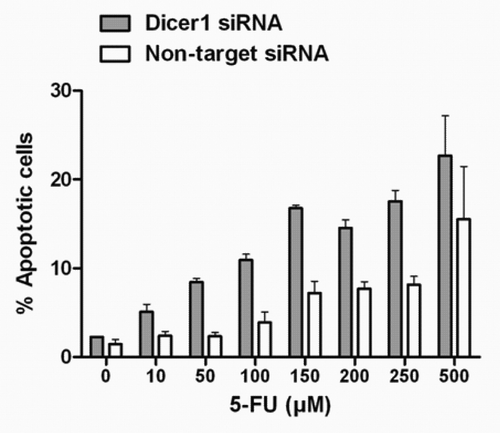
Having obtained this information, we next set out to exploit the temporal dissociation of pro- and anti-apoptotic signals after Dicer1 depletion in order to amplify the sensitivity to chemotherapy in p53-mutated tumor cells. To this end, Dicer1-silenced and Dicer1-competent (non-target) HACAT cells were subjected to increasing doses of fluorouracil (5-FU), an anti-nucleoside cytotoxic drug routinely used in the treatment of epithelial cancers. The main target of 5-FU is thymidylate synthase, an enzyme that is essential for DNA synthesis and DNA repair. However, 5-FU also impairs mRNA and consequently protein synthesis, leading to S-phase arrest of exposed cells.Citation26 As shown in , silencing of Dicer1 alone and in the absence of 5-FU had no apoptotic effect. However, we observed a significant higher rate of apoptosis occurring in Dicer1-depleted cells as compared with HACAT controls in a dose-dependent manner. Importantly, we documented synthetic lethality of Dicer1 and low-dose 5-FU against the background of p53 deficiency.
Figure 3. Synergistic effect of Dicer1 knockdown and 5-FU on p53-mutated HACAT cells. HACAT cells were transiently transfected with Dicer1 siRNA (50 nM) or non-target control siRNA, synchronized for 48 h, and treated with increasing doses of 5-FU (10–500 µM) or vehicle for additional 48 h after cell cycle release. Results show the percentage of Annexin-FITC/PI-positive cells by flow cytometry. The data presented are means ± SD of 3 experiments.
Recently, a series of landmark reports indicate that p53 deficiency can be exploited for targeted tumor therapy.Citation27-Citation29 Cyclotherapy is a strategy that uses activators of p53 to arrest normal cells in the G1/S phase of the cell cycle.Citation30-Citation32 Tumor cells with defective p53 signaling continue to replicate and remain sensitive for subsequent conventional chemotherapy. This therapeutic effect can be further enhanced by rapamycin, an inhibitor of the nutrition-sensing mTOR pathway, and metformin, an anti-diabetic drug. The 2 substances arrest normal cells in G1 and G2, thus protecting them from subsequent cell cycle-specific chemotherapy.Citation33 We have demonstrated that silencing of Dicer1 by siRNA techniques temporally separates pro- and anti-apoptotic signals. The combination of the 2 strategies, sensitizing p53-defective cells by inhibiting Dicer1 function and simultaneously protecting normal cells with p53 activators, may synergistically improve the therapeutic effort to efficiently and selectively target p53-defective tumor cells.
Materials and Methods
Cells and cell culture
The p53-mutated human keratinocyte cell line HACAT was purchased from CLS and maintained in DMEM (PAA Laboratories), supplemented with 10% heat-inactivated FBS (PAN-biotech).
Reagents and Abs
Abs used were polyclonal rabbit anti-Dicer1 (Cell Signaling Technology), monoclonal mouse anti-tubulin Ab-2 (Thermo Scientific), and monoclonal mouse anti-cip1/p21 (BD Transduction Laboratories™). HRP-conjugated secondary Abs used were polyclonal Goat Anti-Rabbit Immunoglobulins/HRP and Polyclonal Rabbit Anti-Mouse Immunoglobulins/HRP, both Dako (Glostrup, Denmark). 5-Fluorouracil (5-FU) was purchased from Sigma-Aldrich, dissolved in DMSO, and stored at −20 °C. For the experiments, the drug was diluted to the appropriate concentrations, and the final concentration of DMSO was max 0.01% in medium.
Transient transfection of siRNA
HACAT cells were transiently transfected with Dicer1 siRNA (Cell Signaling Technology) using the TransIT-siQUEST transfection reagent (Mirus Bio) according to the manufacturer’s instructions. Non-target smart pool siRNA (Dharmacon) was used as control. Briefly, 2.10Citation5 HACAT cell were transfected with a solution containing 250 µl Opti-MEM medium (Invitrogen), 3.5 µl TransIT-siQUEST transfection reagent, and 50 nM Dicer1 siRNA or non-target siRNA. Transfected cells were synchronyzed in serum-free medium for 48 h and released into G1 by serum supplemented medium. Transfection efficiency was assessed by western blot.
Cell viability: Annexin-V apoptosis assay
After exposure to 5-FU for 48 h in the presence or absence of Dicer1 siRNA, apoptotis was assessed using the Annexin-FITC/PI apoptosis detection kit (Invitrogen). Briefly, HACAT cells were harvested and washed twice with PBS. 1.106 cells/ml were stained with 100 µg/ml propidium iodide in 50 µl of annexin-binding buffer. Cells were incubated at room temperature for 15 min, resuspended in 400 µL of binding buffer, and immediately analyzed using a FACScalibur flow cytometer (Becton Dickinson); 10 000 events were analyzed using CellQuest software (BD Biosciences).
Cell cycle analysis
The effect of Dicer knockdown on cell cycle progression was determined by propidium iodide staining. In brief, 2.105 HACAT cells were synchronized in G1 by serum starvation for 48 h. DNA content for each time point after G1 release in serum supplemented medium was determined by FACS analysis. Knockdown experiments were performed using Dicer1 siRNA and non-target siRNA (50 nM) according to the manufacturer's instructions. Cells were washed in PBS, fixed in 70% ethanol overnight, resuspended in 500 µl of PI/RNASE staining solution (BD PharMingen), and incubated for 15 min at room temperature in the dark. Samples were analyzed by FACSCalibur flow cytometry (Becton Dickinson); 10 000 events were analyzed with CellQuest software (BD Biosciences).
Western blot analysis
Whole-cell lysates were collected in RIPA buffer (Sigma-Aldrich), containing Protease- and Phosphatase Inhibitor Cocktail (Roche). Samples were denatured at 95 °C for 5 min. Equal amounts of total protein were loaded in each well for electrophoresis in NuPAGER SDS-PAGE Gel system and transferred to nitrocellulose membranes (Whatman). Membranes were incubated with primary antibody followed by incubation with HRP-linked secondary antibodies. Chemiluminiscence was detected using the Amersham ECL Plus™ Western Blotting Detection Reagent (GE Healthcare). Densitometric analyses were performed using Quantity One software (Bio-Rad).
Phosphorylation antibody array analysis
Phosphorylation-specific antibody microarrays (Fullmoon Biosystems) were used to monitor up- and downregulated proteins in HACAT cells. Two hundred forty-eight (248) antibodies related to proteins involved in cell cycle, signal transduction, and cancer were investigated. The array layout consisted of antibodies against phosphorylated and total proteins using actin and GAPDH as controls. Microarrays were hybridized with biotinylated proteins from HACAT cell lysates transiently transfected with Dicer1 siRNA or non-target siRNA and harvested at the indicated time points after release from starvation. Following protein conjugation to an antibody, Cy3-streptavidin was added, and slides were processed following manufacturer’s instructions (Biosystems FM. Antibody Microarray User’s Guide; http://www.fullmoonbiosystems.com/protocols/AntibodyArrayUserGuide.pdf) and scanned using an Axon GenePix 4000B microarray scanner (Molecular Devices). Averaged and normalized intensity data were Log2 transformed and filtered to identify proteins that show periodicity and Dicer-dependent changes during cell cycle progression. The analysis was performed by using R software (available from: http://www.r-project.org) and R packages developed by the BioConductor project (available from: http://www.bioconductor.org).
Statistical analysis
All data presented are derived from at least 3 independent determinations unless otherwise noted. Statistical analysis was done using Graph Pad Prism 5.0 software. Mean of ± SD is displayed in the figures.
| Abbreviations: | ||
| BAX | = | BCL2-associated X protein |
| BCL2 | = | B-cell CLL/lymphoma 2 |
| BIM | = | BH3-only protein from the Bcl-2 family |
| CDK | = | cyclin-dependent kinase |
| c-JUN/JNK | = | N-terminal kinase |
| CREB1 | = | cyclic AMP-response element binding protein |
| EGF | = | epidermal growth factor |
| Elk1 | = | ETS domain-containing protein Elk-1 |
| FACS | = | fluorescence-activated cell sorting |
| FBS | = | fetal bovine serum |
| FER | = | Fer (fps/fes related) tyrosine kinase |
| FITC/PI | = | fluorescein/propidium iodide |
| 5-FU | = | 5-Fluorouracil |
| GSK-3β | = | glycogen synthase kinase-3 beta |
| HACAT | = | human immortalized keratinocyte |
| HDAC | = | histone deacetylase |
| IGF | = | insulin-like growth factor |
| IKB | = | inhibitors of κB |
| IKKα | = | inhibitor of kappa light polypeptide gene enhancer in B-cells |
| IL-6 | = | Interleukin-6 |
| JAK-STAT | = | Janus kinase/Signal transducer and activator of transcription |
| MAPK | = | mitogen-activated protein kinase |
| MAP2K2 (MEK-2) | = | mitogen-activated protein kinase kinase 2 |
| MAP3K5 | = | mitogen-activated protein kinase kinase kinase 5 |
| MCL1 | = | myeloid cell leukemia sequence 1 |
| miRNAs | = | microRNAs |
| Myc | = | v-myc avian myelocytomatosis viral oncogene homolog |
| Myt1 | = | myelin transcription factor 1 |
| NFAT | = | nuclear factor of activated T-cells |
| NFκB | = | nuclear factor kappa B |
| NIK | = | NF-κB-inducing kinase |
| PAK2 | = | p21 protein (Cdc42/Rac)-activated kinase 2 |
| PDGF | = | platelet-derived growth factor |
| PTEN | = | phosphatase and tensin homolog |
| PTK6 | = | protein tyrosine kinase 6 |
| Raf1 | = | RAF proto-oncogene serine/threonine-protein kinase |
| Ras | = | rat sarcoma |
| Rb | = | retinoblastoma tumor suppressor |
| RPS6KA5 | = | ribosomal protein S6 kinase, 90kDa, polypeptide 5 |
| siRNA | = | small interfering RNA |
| STAT | = | signal transducer and activator of transcription |
| TGF | = | transforming growth factor |
| TS | = | thymidylate synthase |
| Wee1 | = | WEE1 homolog (S. pombe) |
| WNT | = | wingless-type MMTV integration site family, member 3 |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
We gratefully acknowledge financial support from the Deutsche José Carreras Leukämie-Stiftung, project DJCLS R 10/07 to G.S.
Authorship
T.S.N. performed the laboratory work. S.K. and T.S.N. did analyses and interpretation of data. T.S.N. and G.S. wrote the paper. H.E. made substantial contributions to conception and design.
References
- Hatfield SD, Shcherbata HR, Fischer KA, Nakahara K, Carthew RW, Ruohola-Baker H. Stem cell division is regulated by the microRNA pathway. Nature 2005; 435:974 - 8; http://dx.doi.org/10.1038/nature03816; PMID: 15944714
- Jafarnejad SM, Li G. Regulation of p53 by ING family members in suppression of tumor initiation and progression. Cancer Metastasis Rev 2012; 31:55 - 73; http://dx.doi.org/10.1007/s10555-011-9329-5; PMID: 22095030
- Orlando DA, Lin CY, Bernard A, Wang JY, Socolar JES, Iversen ES, Hartemink AJ, Haase SB. Global control of cell-cycle transcription by coupled CDK and network oscillators. Nature 2008; 453:944 - 7; http://dx.doi.org/10.1038/nature06955; PMID: 18463633
- Conacci-Sorrell M, McFerrin L, Eisenman RN. An overview of MYC and its interactome. Cold Spring Harb Perspect Med 2014; 4:a014357; http://dx.doi.org/10.1101/cshperspect.a014357; PMID: 24384812
- Sears R, Nuckolls F, Haura E, Taya Y, Tamai K, Nevins JR. Multiple Ras-dependent phosphorylation pathways regulate Myc protein stability. Genes Dev 2000; 14:2501 - 14; http://dx.doi.org/10.1101/gad.836800; PMID: 11018017
- Huang Z, Traugh JA, Bishop JM. Negative control of the Myc protein by the stress-responsive kinase Pak2. Mol Cell Biol 2004; 24:1582 - 94; http://dx.doi.org/10.1128/MCB.24.4.1582-1594.2004; PMID: 14749374
- Schindler C, Plumlee C. Inteferons pen the JAK-STAT pathway. Semin Cell Dev Biol 2008; 19:311 - 8; http://dx.doi.org/10.1016/j.semcdb.2008.08.010; PMID: 18765289
- STAT3 Gene - GeneCards |P40763. Available at: http://www.genecards.org/cgi-bin/carddisp.pl?gene=STAT3&search=STAT3
- Haghikia A, Hoch M, Stapel B, Hilfiker-Kleiner D. STAT3 regulation of and by microRNAs in development and disease. JAKSTAT 2012; 1:143 - 50; http://dx.doi.org/10.4161/jkst.19573; PMID: 24058763
- Frame S, Cohen P. GSK3 takes centre stage more than 20 years after its discovery. Biochem J 2001; 359:1 - 16; http://dx.doi.org/10.1042/0264-6021:3590001; PMID: 11563964
- Rayasam GV, Tulasi VK, Sodhi R, Davis JA, Ray A. Glycogen synthase kinase 3: more than a namesake. Br J Pharmacol 2009; 156:885 - 98; http://dx.doi.org/10.1111/j.1476-5381.2008.00085.x; PMID: 19366350
- Cross DA, Alessi DR, Cohen P, Andjelkovich M, Hemmings BA. Inhibition of glycogen synthase kinase-3 by insulin mediated by protein kinase B. Nature 1995; 378:785 - 9; http://dx.doi.org/10.1038/378785a0; PMID: 8524413
- King TD, Gandy JC, Bijur GN. The protein phosphatase-1/inhibitor-2 complex differentially regulates GSK3 dephosphorylation and increases sarcoplasmic/endoplasmic reticulum calcium ATPase 2 levels. Exp Cell Res 2006; 312:3693 - 700; http://dx.doi.org/10.1016/j.yexcr.2006.08.010; PMID: 16987514
- Neal JW, Clipstone NA. Glycogen synthase kinase-3 inhibits the DNA binding activity of NFATc. J Biol Chem 2001; 276:3666 - 73; http://dx.doi.org/10.1074/jbc.M004888200; PMID: 11063740
- Hagen T, Vidal-Puig A. Characterisation of the phosphorylation of beta-catenin at the GSK-3 priming site Ser45. Biochem Biophys Res Commun 2002; 294:324 - 8; http://dx.doi.org/10.1016/S0006-291X(02)00485-0; PMID: 12051714
- AKT2 Gene - GeneCards |P31751. Available at: http://www.genecards.org/cgi-bin/carddisp.pl?gene=AKT2
- Takahashi-Yanaga F, Sasaguri T. GSK-3beta regulates cyclin D1 expression: a new target for chemotherapy. Cell Signal 2008; 20:581 - 9; http://dx.doi.org/10.1016/j.cellsig.2007.10.018; PMID: 18023328
- Zhou BP, Liao Y, Xia W, Spohn B, Lee MH, Hung MC. Cytoplasmic localization of p21Cip1/WAF1 by Akt-induced phosphorylation in HER-2/neu-overexpressing cells. Nat Cell Biol 2001; 3:245 - 52; http://dx.doi.org/10.1038/35060032; PMID: 11231573
- Motti ML, Califano D, Troncone G, De Marco C, Migliaccio I, Palmieri E, Pezzullo L, Palombini L, Fusco A, Viglietto G. Complex regulation of the cyclin-dependent kinase inhibitor p27kip1 in thyroid cancer cells by the PI3K/AKT pathway: regulation of p27kip1 expression and localization. Am J Pathol 2005; 166:737 - 49; http://dx.doi.org/10.1016/S0002-9440(10)62295-X; PMID: 15743786
- Katayama K, Fujita N, Tsuruo T. Akt/protein kinase B-dependent phosphorylation and inactivation of WEE1Hu promote cell cycle progression at G2/M transition. Mol Cell Biol 2005; 25:5725 - 37; http://dx.doi.org/10.1128/MCB.25.13.5725-5737.2005; PMID: 15964826
- Datta SR, Dudek H, Tao X, Masters S, Fu H, Gotoh Y, Greenberg ME. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997; 91:231 - 41; http://dx.doi.org/10.1016/S0092-8674(00)80405-5; PMID: 9346240
- Sun S-C. The noncanonical NF-κB pathway. Immunol Rev 2012; 246:125 - 40; http://dx.doi.org/10.1111/j.1600-065X.2011.01088.x; PMID: 22435551
- NFKB2 Gene - GeneCards | Q00653. Available at: http://www.genecards.org/cgi-bin/carddisp.pl?gene=NFKB2&search=NFKB
- Nuclear factor NF-kappa-B p105 subunit - Homo sapiens (Human). Available at: http://www.uniprot.org/uniprot/P19838
- Transcription factor p65 - Homo sapiens (Human). Available at: http://www.uniprot.org/uniprot/Q04206
- Guo X, Goessl E, Jin G, Collie-Duguid ES, Cassidy J, Wang W, O’Brien V. Cell cycle perturbation and acquired 5-fluorouracil chemoresistance. Anticancer Res 2008; 28:1A 9 - 14; PMID: 18383818
- Leeuwen IMM Van. Cyclotherapy : opening a therapeutic window in cancer treatment Oncotarget 2012; 3:596–600.
- Blagosklonny MV. Wt p53 impairs response to chemotherapy: make lemonade to spare normal cells. Oncotarget 2012; 3:601 - 7; PMID: 22802145
- Blagosklonny MV. “Targeting the absence” and therapeutic engineering for cancer therapy. Cell Cycle 2008; 7:1307 - 12; http://dx.doi.org/10.4161/cc.7.10.6250; PMID: 18487952
- Blagosklonny MV, Robey R, Bates S, Fojo T. Pretreatment with DNA-damaging agents permits selective killing of checkpoint-deficient cells by microtubule-active drugs. J Clin Invest 2000; 105:533 - 9; http://dx.doi.org/10.1172/JCI8625; PMID: 10683383
- Rao B, van Leeuwen IMM, Higgins M, Campbel J, Thompson AM, Lane DP, Lain S. Evaluation of an Actinomycin D/VX-680 aurora kinase inhibitor combination in p53-based cyclotherapy. Oncotarget 2010; 1:639 - 50; PMID: 21317459
- Choong ML, Yang H, Lee MA, Lane DP. Specific activation of the p53 pathway by low dose actinomycin D: a new route to p53 based cyclotherapy. Cell Cycle 2009; 8:2810 - 8; http://dx.doi.org/10.4161/cc.8.17.9503; PMID: 19657224
- Apontes P, Leontieva OV, Demidenko ZN, Li F, Blagosklonny MV. Exploring long-term protection of normal human fibroblasts and epithelial cells from chemotherapy in cell culture. Oncotarget 2011; 2:222 - 33; PMID: 21447859