
Abstract
Polo-like kinases (PLKs) and Aurora kinases (AKs) act as key cell cycle regulators in healthy human cells. In cancer, these protein kinases are often overexpressed and dysregulated, thus contributing to uncontrolled cell proliferation and growth. T-cell acute lymphoblastic leukemia (T-ALL) is a heterogeneous malignancy arising in the thymus from T-cell progenitors. Primary chemoresistant and relapsed T-ALL patients have yet a poor outcome, therefore novel therapies, targeting signaling pathways important for leukemic cell proliferation, are required. Here, we demonstrate the potential therapeutic effects of BI6727, MK-5108, and GSK1070916, three selective inhibitors of PLK1, AK-A, and AK-B/C, respectively, in a panel of T-ALL cell lines and primary cells from T-ALL patients. The drugs were both cytostatic and cytotoxic to T-ALL cells by inducing G2/M-phase arrest and apoptosis. The drugs retained part of their pro-apoptotic activity in the presence of MS-5 bone marrow stromal cells. Moreover, we document for the first time that BI6727 perturbed both the PI3K/Akt/mTORC2 and the MEK/ERK/mTORC1 signaling pathways, and that a combination of BI6727 with specific inhibitors of the aforementioned pathways (MK-2206, CCI-779) displayed significantly synergistic cytotoxic effects. Taken together, our findings indicate that PLK1 and AK inhibitors display the potential for being employed in innovative therapeutic strategies for improving T-ALL patient outcome.
Introduction
The aberrant growth of cancer cells can often be explained also by a dysregulation of the control of both cell cycle and cell division. Cell proliferation encompasses a tightly controlled set of events that are regulated by several nuclear protein kinases. These include checkpoint kinases (CHK), cyclin-dependent kinases (CDK), Polo-like kinases (PLKs), and Aurora kinases (AKs). In cancer, these protein kinases are often dysregulated and cause uncontrolled cell proliferation and growth.Citation1
In humans, 5 members of the PLK family of serine/threonine protein kinases have been identified, and PLK1 is the best characterized.Citation1 PLK1 plays a fundamental role in cell division and its inhibition leads to a failure in completing mitosis.Citation2 Moreover, PLK1 strongly promotes cell cycle progression.Citation3 As PLK1 is overexpressed in a broad range of human tumors, it is currently regarded as a potential target for drug development in cancer therapy.Citation4 The Aurora kinase (AK) family of serine/threonine kinases comprises 3 members referred to as Aurora kinase-A, -B, and -C (AK-A, AK-B and AK-C, respectively). AKs are involved in centrosome function and mitotic spindle assembly, participate in the kinetochore complex, and favor cytokinesis.Citation5 However, both AK-A and AK-B have been associated with tumorigenesis and their overexpression is frequently observed in multiple tumor types, where it is correlated with higher grades of malignancy, higher proliferation rates, and poor prognosis.Citation3,Citation6-Citation8 Unlike AK-A and AK-B that are ubiquitously expressed in all mammalian tissues, AK-C expression is restricted to the testis, where it is involved in meiosis.Citation9
As far as malignant blood disorders are concerned, PLK1 is expressed at high levels in chronic myelogenous leukemia (CML),Citation10 acute myelogenous leukemia (AML),Citation11 and acute lymphoblastic leukemia (ALL).Citation12 Overexpression of AK-A and -B has also been reported in a number of hematological malignancies, which include CML, AML, and ALL.Citation13
Several PLK and AK inhibitors have been synthesized and evaluated in vitro and in animal models, and some of them have reached phase I/II clinical trials for cancer therapy.Citation14 Regarding leukemias, AT9283, an inhibitor of AK-A and -B, has been tested in patients with relapsed/refractory AML,Citation15 while barasertib (AZD1152), an inhibitor of AK-B, has been evaluated in a phase II clinical trial in AML patients aged ≥60 y.Citation16 Moreover, the PLK1 inhibitor BI2536 has been tested in elderly patients with refractory/relapsed AML.Citation11
Regarding ALL, preclinical studies on the efficacy of PLK1 and AK inhibitors have mostly focused on B-cell ALL (B-ALL), which is the most common malignancy in children,Citation17-Citation19 while very little is known about the relevance of PLK1 and AKs as therapeutic targets in T-cell ALL (T-ALL). T-ALL is an aggressive malignant disorder resulting from the occurrence of variable mutations in progenitor cells committed to differentiate into T-cell lineage. Although the prognosis of T-ALL has improved, especially in children, the outcome is still poor for patients refractory to chemotherapy or for those who relapse.Citation20,Citation21 Therefore, it would be very important to identify novel, specific therapeutic targets and strategies for improving T-ALL prognosis in patients who do not respond to current treatment protocols or relapse.Citation22,Citation23
Here, we analyzed the efficacy of BI6727,Citation24 MK-5108,Citation25 and GSK1070916,Citation26 3 selective inhibitors of PLK1, AK-A, and AK-B/C, respectively, in a panel of T-ALL cell lines and primary cells from T-ALL patients. We observed that these drugs were able to induce mitotic arrest and apoptosis of leukemic cells. However, we demonstrated that BI6727 perturbed both the PI3K/Akt/mTORC2 and the MEK/ERK/mTORC1 signaling pathways, and that a combination of BI6727 with either MK-2206 (an Akt inhibitorCitation27) or CCI-779 (temsirolimus, an mTORC1 inhibitorCitation28) displayed a significant synergistic cytotoxic effect. Altogether, our data demonstrate the potential of PLK1 and AK inhibitors in T-ALL treatment.
Results
Inhibitors of PLK1, AK-A, and AK-B affect viability of T-ALL cell lines
We first investigated the expression of PLK1, AK-A, and AK-B in a panel of T-ALL cell lines. Western blot analysis demonstrated that the CCRF-CEM cell line expressed the lowest levels of both PLK1 and AK-A proteins. BE-13, MOLT-4, Jurkat, and HPB-ALL cell lines expressed comparable levels of AK-A. PLK1 was expressed at the highest level in Jurkat cells, whereas the expression levels of AK-B were similar among T-ALL cell lines. PLK1, AK-A, and AK-B were phosphorylated at amino acidic residues indicative of their activation (). The effects of the inhibitors on T-ALL cell viability were evaluated by MTT assays after 48 h treatment, and the IC50 was calculated for each drug. Given that PLK1 and AKs regulate cell cycle and mitosis, we chose to evaluate drug efficacy at 48 h, which is beyond the replication time of all the tested cell lines. Each drug inhibited the phosphorylation of the respective target (). However, the AK-A inhibitor MK-5108 also decreased the levels of p-PLK1, consistently with AK-A being upstream of PLK1.Citation29
Figure 1. Effects of PLK1 and AK-A/-B inhibitors on T-ALL cell viability. (A) BE-13, CCRF-CEM, MOLT-4, Jurkat, and HPB-ALL cell lines were collected, lysed, and analyzed by western blot for the expression of PLK1 and AK-A/-B and of their phosphorylated forms. Molecular weights are indicated on the left. (B) MOLT-4 cells were treated with BI6727, GSK1070916, and MK-2206 at the respective IC50 for 48 h; next they were collected, lysed, and analyzed by western blot. Molecular weights are indicated on the left. CTRL, untreated cells. (C) T-ALL cell lines were treated with the drugs for 48 h. Next the rates of survival were evaluated by MTT assays. Data are representative of 3 independent experiments and SD was less than 10%.
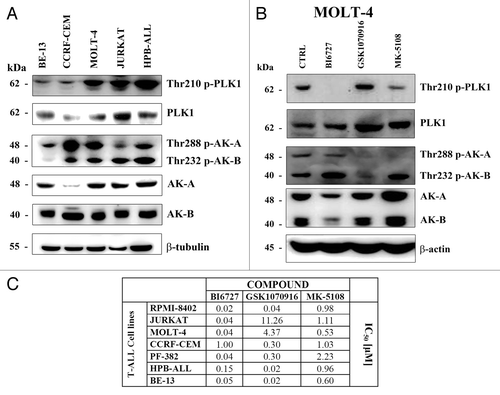
Most cell lines displayed an IC50 for BI6727 around 0.02–0.15 μM, whereas the IC50 of CCRF-CEM cells was 1.0 μM (). As to GSK1070916, the IC50 ranged from 0.02 to 11.26 µM, while the IC50 for MK-5108 was comprised in a range between 0.6 and 2.23 µM. Overall, these findings documented that the PLK1 inhibitor, BI6727, was more efficacious than the other drugs we tested on this panel of T-ALL cell lines.
Inhibitors of PLK1 and AKs block cells in the G2/M phase of the cell cycle
In order to better characterize the drug effects on cell cycle progression, CCRF-CEM, HPB-ALL, and MOLT-4 cell lines were incubated for 48 h with either PLK1 or AK inhibitors at their respective IC50. Flow cytometric analysis of PI-stained samples documented a decrease in the percentage of cells in G0/G1 phase of the cell cycle and a concomitant increase in the G2/M phase (). Of note, there also was the appearance of a sub-G0/G1 peak, which was indicative of DNA fragmentation and apoptosis. Another interesting consequence of the treatment with AK inhibitors was the increase in hyperdiploid cells, as indicated by the presence of peaks with a DNA content greater than 2n (G0/G1) or 4n (G2/M), an observation which was consistent with the findings previously reported for neuroblastoma cells.Citation24
Figure 2. Effects of PLK1 and AK inhibitors on cell cycle progression of T-ALL cell lines. (A) CCRF-CEM, HPB-ALL, and MOLT-4 cell lines were treated for 48 h with BI6727, GSK1070916, and MK-5108. Then, cell cycle analysis was performed by flow cytometry. One representative of 3 separate experiments is shown, which yielded similar results. The percentages of the cells in the various phases of the cell cycle are indicated. (B) MOLT-4 cells were treated for 48 h with BI6727 (0.04 µM). Then, cells were spun down, fixed, permeabilized, and stained with antibody to β-tubulin (green). Nuclei were counterstained with DAPI (blue). (C) T-ALL cell lines were treated with BI6727, GSK1070916, and MK-5108 at their respective IC50s for 48 h, then they were collected, lysed, and analyzed by western blot. Molecular weights are indicated on the left. CTRL, untreated cells.
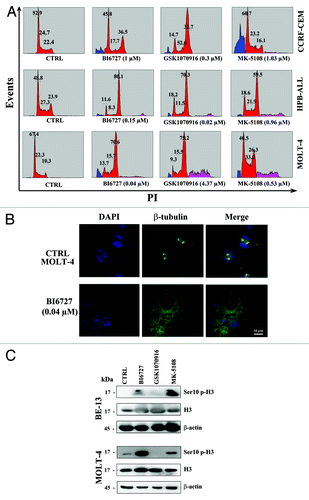
Moreover, BI6727 treatment of MOLT-4 cell line prevented the orderly bipolar mitotic spindle formation, as revealed by immunofluorescence staining with an anti-β-tubulin antibody (). In agreement with recent investigations performed in solid tumors,Citation30,Citation31 we detected an increase in the expression levels of Ser10 p-histone H3 (an established bio-marker for both PLK1 and AK-A activity) in BE-13 and CCRF-CEM cell lines treated with either BI6727 or MK-5108.
Inhibitors of PLK1 and AKs had pro-apoptotic effects on T-ALL cell lines and synergized in reducing cell viability
It was then investigated whether the drug effects on cell viability could be also related to apoptosis.
Flow cytometric analysis of Annexin V/FITC-stained MOLT-4 and BE-13 cells demonstrated an increase in both early (single-positive for Annexin V) and late (double positive for Annexin V and PI) cells in MOLT-4 cells treated with BI6727 and in BE-13 cells treated with GSK1070916 (). We also analyzed caspase-7 and poly (ADP-ribose) polymerase (PARP) cleavage. Western blot analysis documented caspase-7 and PARP cleavage in BE-13, CCRF-CEM, and MOLT-4 cell lines treated with BI6727, GSK1070916, or MK-5108 (). Overall, these findings and those highlighted in demonstrated that inhibition of either PLK1 or AK-A/-B activity decreased viability of T-ALL cell lines by inducing both a cell cycle arrest in the G2/M phase of the cell cycle and apoptosis.
Figure 3. Inhibition of PLK1 and AK-A/-B induced apoptosis in T-ALL cell lines. (A) MOLT-4 cell line was treated with BI6727 (40 nM) for the indicated times, then they were collected and stained with Annexin V-FITC/PI and analyzed by flow-cytometry. (B) BE-13 cells were treated with GSK1070916 (20 nM) for 48 h, then they were collected, stained with Annexin V-FITC/PI, and analyzed by flow-cytometry. (C) T-ALL cell lines were treated for 48 h with BI6727, GSK1070916, and MK-5108 at their respective IC50, then they were collected, lysed, and analyzed by western blot. Molecular weights are indicated on the left. CTRL, untreated cells. (D) HPB-ALL cells were treated for 48 h with BI6727 and MK-5108, used either alone or in combination. Cell viability was analyzed by MTT assays. Results are the mean of at least 3 different experiments ± SD. The combination index (CI) value for each data point was calculated with the appropriate software for dose effect analysis (CalcuSyn).
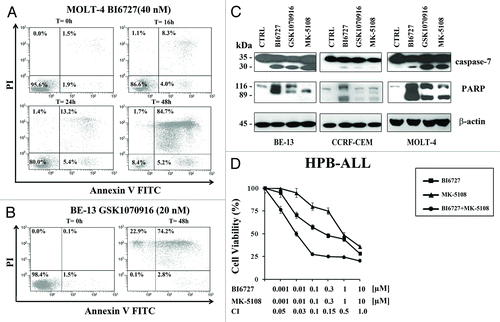
PLK1 and AKs directly interact at the molecular level in the regulation of cell cycle progression, and more effective antileukemic effects may thus be achieved when targeting of both enzymes is combined.Citation32 This could allow the use of lower drug concentrations resulting in less severe side effects. We, therefore, investigated whether the combination of PLK1 and AK-A inhibitors could synergize in T-ALL cells. HPB-ALL cells were treated with increasing concentrations of BI6727 and MK-5108 for 48 h, either alone or in combination. MTT assays documented the existence of a strong synergism (CI < 0.3) at drug concentrations lower than their respective IC50.
BI6727 also displays pro-apoptotic effects in the presence of mouse stromal cells
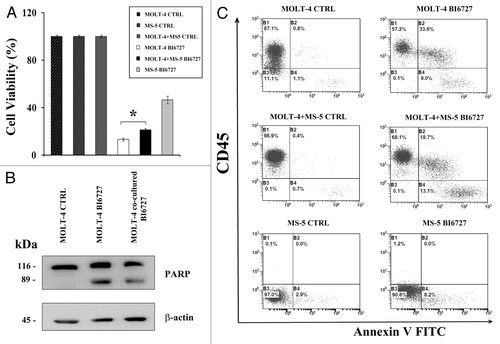
It is well documented how leukemic cells interact with cells of the bone marrow microenvironment that provides a protection from chemotherapy treatment and induces a drug-resistant phenotype.Citation33 MS-5 stromal cells have been reported to secrete a variety of growth factors and cytokines as well as extracellular matrix proteinsCitation34 that could recreate in vitro most of the in vivo interactions between leukemic cells and the bone marrow microenvironment.Citation35 We used a Transwell system that allowed a cross-talk between MOLT-4 cells and the mouse stromal cells MS-5 by the diffusion of small molecules. We analyzed the difference of viability of MOLT-4 and MS-5 cells treated with BI6727 when grown alone or co-cultured. Cell viability, as assessed by MTT assays, decreased (P = 0.0087) when MOLT-4 cells were cultured alone if compared with MOLT-4 cells co-cultured with MS-5 cells (). However, the drug retained part of its pro-apoptotic activity, as indicated by PARP cleavage, which was detected by western blot (). Moreover, flow cytometric analysis on CD45+-gated cells confirmed that BI6727 retained its pro-apoptotic effect even when MOLT-4 cells were directly added to MS-5 monolayers. ().
Figure 4. BI6727 retains pro-apoptotic effects also in the presence of a microenvironment of bone marrow stromal cells. (A) The MS-5 cell line was grown in the lower chamber of Transwell® 6-well plates, than MOLT-4 cells were added to the upper chamber containing a 0.4-µm-polyester membrane and treated with BI6727 (40 nM) for 48 h. The viability of treated cell lines grown alone and co-coltured was then evaluated by MTT assays. CTRL, untreated cells. (B) The MS-5 cell line was grown in the lower chamber of Transwell® 6-well plates, then MOLT-4 cells were added to the upper chamber containing a 0.4-µm-polyester membrane and treated with BI6727 (40 nM) for 48 h. Then, cells were separately collected, lysed, and analyzed by western blot for PARP cleavage. Molecular weights are indicated on the left. CTRL, untreated cells. (C) MOLT-4 cells were directly seeded on top of MS-5 cells and treated with BI6727 (40 nM). After 48 h, cells were harvested with trypsin/EDTA, washed, and resuspended in binding buffer containing Annexin V-FITC. Cells were counterstained with either a PE-conjugated anti-CD45 antibody or with an irrelevant isotypic control antibody and analyzed by flow cytometry after electronic gating on CD45. CTRL, untreated cells.
PLK1 inhibition influences both PI3K/Akt/mTORC2 and MEK/ERK /mTORC1 signaling pathways in T-ALL cells
It is now emerging that PLK1 functions could be intertwined with both PI3K/Akt and MEK/ERK signaling.Citation36,Citation37 Therefore, we tested whether BI6727 could modulate either of these signaling pathways. BI6727 increased the phosphorylation levels of Ser473 p-Akt (an mTORC2 substrate) as well as those of both Thr389 p-p70S6K and Ser235/236 p-S6RP, two mTORC1 down-stream substrates. In contrast, the total expression levels of these proteins were unaffected by the drug (). Similar results were detected with CCFR-CEM cells (not shown). Therefore, we treated MOLT-4 cells with CCI-779 used either as a single agent or in combination with BI6727. Although CCI-779 is mainly considered to be an mTORC1 inhibitor, it is also capable of inhibiting mTORC2 activity, especially when used in cells of hematopoietic lineage.Citation38 Consistently, CCI-779, when used either as single agent or in combination with BI6727, blocked the upregulation of p-Akt, whereas total levels of expression were unaffected by the drugs (). Overall, these results suggested that inhibition of PLK1 may led to upregulated mTORC1/mTORC2 signaling. Nevertheless, it should be considered that mTORC1 activity could also be regulated through MEK/ERK signaling.Citation39 Accordingly, treatment of MOLT-4 cells with BI6727 resulted in increased phosphorylation levels of Thr202/Tyr204 p-ERK and of its downstream substrate, Thr573 p-p90RSK (; refs. Citation40–Citation42). Treatment with the MEK inhibitor U0126 blunted the phosphorylation of both ERK and p90RSK. Intriguingly, U0126 did not affect the basal levels of Ser235/236 p-S6RP; however, it completely blocked S6RP phosphorylation induced by BI6727. Overall, these findings demonstrated that increased S6RP phosphorylation, which was detected in MOLT-4 in response to PLK1 inhibition, was dependent on aberrantly activated MEK/ERK signaling.
Figure 5. BI6727 upregulates PI3K/Akt/mTORC2 and MEK/ERK/mTORC1 signaling in MOLT-4 cells. (A) Cells were treated for 48 h with BI6727 (0.04 µM), CCI-779 (0.1 µM), and the combination of the 2 drugs, then they were collected, lysed, and analyzed by western blot. Molecular weights are indicated on the left. CTRL, untreated cells. (B) Cells were treated for 48 h with BI6727 (0.04 µM), U0126 (10 µM), or the 2 drugs combined, then they were collected, lysed, and analyzed by western blot. Molecular weights are indicated on the left. CTRL, untreated cells. (C) Cells were treated for 48 h with BI6727 and CCI-779, used either alone or in combination. The combination index (CI) value for each data point was calculated with the appropriate software for dose effect analysis (CalcuSyn). Results are the mean of at least 3 different experiments ± SD. (D) Cells were treated for 48 h with BI6727 and MK-2206, used either alone or in combination. The combination index (CI) value for each data point was calculated with the appropriate software for dose effect analysis (CalcuSyn). Results are the mean of at least 3 different experiments ± SD.
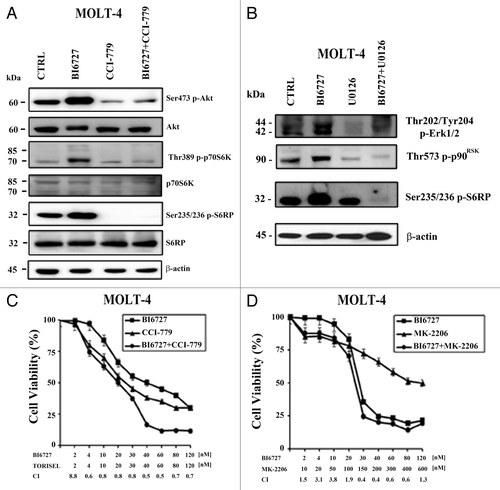
The relevance of both Akt and mTORC1 activation induced by BI6727 was investigated using MTT assays in MOLT-4 cells treated with BI6727 and either MK-2206 (an Akt inhibitor) or CCI-779, used as single agents or in combination. Both of these drugs are now being tested in phase I/II clinical trials for patients with leukemias (ClinicalTrials.gov: NCT01369849; NCT01403415). Result analysis documented that both the drug combinations were synergistic ().
Inhibitors of PLK1 and AKs displayed cytotoxic effects on T-ALL patient samples
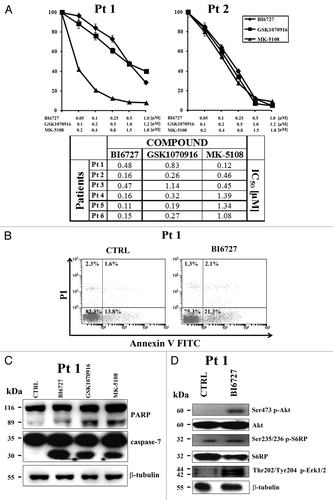
To better assess the effectiveness of PLK1 and AK inhibitors as a potential therapeutic agents in T-ALL, we examined 6 pediatric T-ALL patient samples isolated from bone marrow or peripheral blood. Samples expressed variable amount of PLK1 and AK-A/-B, as documented by western blot analysis (data not shown). The effects of the inhibitors on T-ALL lymphoblast samples were evaluated first by treating the cells with increasing concentrations of the inhibitors and then analyzing the rates of viability by MTT assays after 96 h. A marked reduction in leukemia cell viability was detected. The patient IC50 for BI6727 ranged between 0.11 and 0.48 μM, while for the AK inhibitors, it ranged between 0.12 and 1.39 μM (). Induction of apoptosis in primary T-ALL cells treated with BI6727 (0.5 µM) was suggested by flow cytometric analysis of Annexin V–FITC/PI-stained samples (). Moreover, western blot analysis of samples treated for 48 h with BI6727, GSK1070916, and MK-5108, documented a significant accumulation of the caspase-7 cleaved form, as well as of cleaved PARP (). Interestingly, and similarly to the MOLT-4 cell line, western blotting analysis on patient samples treated with BI6727 confirmed an increase in Ser473 p-Akt, Ser235/236 p-S6RP, and Thr202/Tyr204 p-ERK 1/2 levels ().
Figure 6. PLK1 and AK-A/-B inhibitors are cytotoxic to T-ALL primary cells. (A) Lymphoblasts from T-ALL patients were cultured in RPMI1640 medium supplemented with 20% FBS, insulin-transferrin-sodium selenite, and 10 ng/ml interleukin-7. MTT assays were performed on samples treated for 96 h with BI6727, GSK1070916, and MK-5108. Results are the mean of at least 2 different experiments ± SD. (B) Patient samples were treated with BI6727 (0.5 µM) for 48 h, then cells were collected, stained with Annexin V-FITC/PI, and analyzed by flow-cytometry for apoptosis. A representative sample is shown. CTRL, untreated cells. (C) Western blot analysis of a patient sample treated for 48 h with 0.5 µM BI6727, 0.5 µM GSK1070916, and 0.8 µM MK-5108. Thirty micrograms (30 μg) of protein/lane were electrophoresed on SDS-PAGE, transferred to nitrocellulose membrane, and probed with the appropriate antibodies. One representative of 2 different experiments is shown. Molecular weights are indicated on the left. CTRL, untreated cells. (D) Western blot analysis of a patient sample treated for 48 h with 0.5 µM BI6727. Thirty micrograms (30 μg) of protein/lane were electrophoresed on SDS-PAGE, transferred to nitrocellulose membrane, and probed with the appropriate antibodies. One representative of 2 different experiments is shown. Molecular weights are indicated on the left. CTRL, untreated cells.
Discussion
Primary chemoresistant and relapsed T-ALL patients have yet a poor outcome; therefore novel therapies, capable of hitting signaling pathways critical for T-ALL proliferation, are required. Traditional anti-cancer therapies rely on drugs that interfere with cell cycle progression. In particular, vinca alkaloids that interact with tubulin, preventing the microtubules polymerization in mitotic spindles,Citation43 and taxanes, which bind to tubulin molecules and impair the formation of the mitotic spindle,Citation44 block cells in the G2/M phase of the cell cycle. However, it is well known that these compounds cause significant side effects, including neurotoxicity.Citation45 Therefore, more specific mitosis-targeting drugs with enhanced therapeutic efficacy and fewer side effects should be developed. Mitosis-specific kinases, such as members of PLK and AK families, were identified as potential targets for cancer treatment. Accordingly, several PLK and AK inhibitors have been developed over the last years.Citation14 Promising results were obtained by targeting PLK1, as its inhibition preferably kills cancer cells compared with normal cells.Citation46 The aim of this study was to test the therapeutic potential of PLK1 and AK inhibitors (BI6727, MK-5108, and GSK1070916) on T-ALL cell lines and primary T-ALL lymphoblasts. All these inhibitors are able to interfere with cell cycle progression, reducing drastically the cellular proliferation followed by the induction of apoptosis.
Indeed, we have documented that, even if they displayed a different efficacy, the PLK1 and AK inhibitors that we tested decreased the viability of T-ALL cell lines and primary T-ALL lymphoblasts. Among the drugs we tested, BI6727 proved to be the most efficacious in terms of IC50. Only the CCRF-CEM cell line seemed to be less sensitive to BI6727 (IC50 = 1 μM). Interestingly, western blotting analysis documented that this cell line expressed lower levels of PLK1. Overall, our findings suggested that PLK and AK inhibitors reduced viability of most of the T-ALL cell lines and primary samples we studied, acting at submicromolar concentrations. Reduction in cell viability was due to both a block in the G2/M phase of the cell cycle and induction of apoptosis. BI6727 and MK-5108 could be employed together, and the combined treatment resulted in a synergistic reduction of cell viability.
Remarkably, BI6727 retained at least part of its pro-apoptotic activity also when MOLT-4 cells were co-cultured with MS-5 stromal cells, which partly mimic the leukemic bone marrow microenvironment that is known to promote leukemic cell proliferation and survival.Citation47
An entirely novel finding that emerged from our study is that PLK1 inhibition interferes with both PI3K/Akt/mTORC2 and MEK/ERK/mTORC1, which are 2 signaling pathways frequently hyperactivated in T-ALL.Citation48 Indeed, we documented that when MOLT-4 cells were treated with BI6727, Ser473 p-Akt levels increased, which is indicative of upregulated mTORC2 activity. There also was an increase in the phosphorylation levels of p-70S6K and p-S6RP, two downstream targets of mTORC1. Co-treatment with CCI-779 blunted increased phosphorylation of Akt, p70S6K, and S6RP. It should be recalled here that CCI-779, besides blocking mTORC1 activity, acts as an inhibitor of mTORC2, especially when administered for extended periods of time (48 h in our case) in cells of hematopoietic lineage.Citation38
Previous studies indicated that MEK/ERK signaling was upregulated in PLK1-depleted cells.Citation49 Accordingly, in response to BI6727, we detected increased phosphorylation of ERK and of its downstream target p90RSK, an activator of mTORC1.Citation50 Interestingly, treatment with the MEK inhibitor U0126, besides reducing ERK and p90RSK phosphorylation levels, prevented the BI6727-induced, but not the basal, S6RP phosphorylation. This indicated that in cells treated with BI6727, mTORC1 was activated by the MEK/ERK/ p90RSK axis. Similar findings were also observed with T-ALL primary samples.
Both CCI-779 and MK-2206, an Akt inhibitor,Citation27 synergized with BI6727 in reducing the viability of MOLT-4 cells. Interestingly, a functional link between PLK1 and mTOR signaling pathway has been recently reported,Citation51 as a combination of BI2536 (a PLK1 inhibitor) and PI3K-mTOR dual inhibitors eradicated colon cancer stem cells, which overcome conventional cancer therapies and are involved in relapse and in tumor regeneration by switching off c-Myc protein.
Therefore, also in T-ALL, the efficacy of PLK1 inhibition could be limited by the presence of multiple feedback loops and/or cross-talk with alternative oncogenic signaling pathways.Citation52
In summary, we characterized the effects of PLK1 and AK inhibitors on T-ALL cells, confirming that they are potentially useful drugs in the treatment of this disorder. The identification of a link between PLK1 and PI3K/Akt/mTORC2 and PLK1 and MEK/ERK/mTORC1 signaling pathways supports the use of a combined therapy. Hence, our data not only provided insight into the effects of PLK1 and AK inhibitors, but may also be important in the design of therapeutic protocols that involve targeting of both PLK1 and other pathways.
Materials and Methods
Materials
BI6727, GSK1070916, MK-5108, CCI-779, and UO126 were from Selleck Chemicals. For western blotting and fluorescence microscopy, primary antibodies were bought from Cell Signaling Technology. A FITC-conjugated anti-rabbit IgG antibody was purchased from Sigma-Aldrich. Anti-CD45-APC-conjugated antibody for flow cytometry was obtained from Becton-Dickinson.
Cell culture and primary T-ALL samples
The T-ALL cell lines RPMI-8402, Jurkat, MOLT-4, CCRF-CEM, BE-13, PF-382, and HPB-ALL were grown in RPMI 1640, supplemented with either 10% or 20% fetal bovine serum (FBS), L-glutamine, and penicillin-streptomycin. Samples from T-ALL pediatric patients, obtained after informed consent according to the Ethics Committee of Human Experimentation guidelines, were isolated using Ficoll-Paque (GE Healthcare Bio-Sciences AB), and grown as previously described.Citation53 MS-5 mouse stromal cells were grown in MEM Alpha medium supplemented with 10% FBS.
Cell viability analysis
MTT (3-[4,5-Dimethylthythiazol-2-yl]-2,5-Diphenyltetrazolium Bromide) assays were performed to assess the sensitivity of cells to drugs, as previously described.Citation54,Citation55
Cell cycle and apoptosis analysis
Flow cytometric analysis of the cell cycle was performed using a propidium iodide (PI)/RNase A staining according to standard procedures, as described previously.Citation56 Samples were analyzed on a FC500 flow cytometer (Beckman Coulter) with the appropriate software (CXP, Beckman Coulter). For analysis of apoptosis induction, T-ALL cell lines grown alone or co-cultured with MS-5 cells were washed twice in PBS, labeled with Annexin V/FITC in binding buffer, and then analyzed on an FC500 flow cytometer after electronic gating on CD45+ leukemic cells.Citation53
Western blot analysis
This was performed by standard methods, as previously reported.Citation57 Analysis with an antibody to either β-actin or β-tubulin demonstrated equal protein loading.
Fluorescence microscope analysis
Cells were cytocentrifuged to coverslips (200 g, 5 min), fixed, and permeabilized with 100% methanol for 3 min on ice. Samples were stained with an anti-β-tubulin antibody (1:100), followed by FITC-conjugated anti-rabbit IgG (1:500). Slides were then treated with ProLong® Gold antifade reagent (Life Technologies Italia), containing 4',6-diamidino-2-phenylindole (DAPI). The analysis was performed on an Axio Imager Microscope (Zeiss) equipped with an Apotome module. Images were acquired with AxioVision software (Zeiss).
Combined drug effect analysis
The combination effect and potential synergy were evaluated from quantitative analysis of dose-effect relationships, as described previously.Citation58 For each combination experiment, a combination index (CI) number was calculated using the CalcuSyn software (Biosoft). This method of analysis generally defines CI values of 0.9 to 1.1 as additive, 0.3 to 0.9 as synergistic, and <0.3 as strongly synergistic, whereas values >1.1 are considered antagonistic.
T-ALL cell co-culture with MS-5 mouse stromal cells
MOLT-4 cells were seeded at 2.5 × 105/ml and, after an overnight incubation, cell suspension was transferred on the top of MS-5 mouse stromal cells (at 70% confluence) and treated with BI6727 (40 nM). After 48 h, MOLT-4 cells were collected, washed, and incubated with an APC-conjugated anti-CD45 antibody or with an irrelevant isotypic control antibody. After a 30-min incubation, cells were resuspended in binding buffer containing Annexin V-FITC and analyzed by flow cytometry after electronic gating on CD45+ leukemic cells. In another set of experiments, MS-5 cells were grown in the lower chamber of Transwell® 6-well plates (Corning) containing a 0.4-µm polyester membrane, then MOLT-4 cells were added to the upper chamber and treated with BI6727 (40 nM). After 48 h, the viability of treated cell lines grown either alone or co-cultured was evaluated. Furthermore, cells were collected, lysed, and analyzed by western blot.
Statistical analysis
The data are presented as mean values from 3 separate experiments ± SD. Data were statistically analyzed by a Dunnet test after one-way analysis of variance (ANOVA) at a level of significance of P < 0.05 vs. control samples.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This work was supported by a grant from MIUR FIRB 2010 (RBAP10447J_003) to A.M.M. F.L. was supported by Special Project AIRC 5x1000 n. 9962 and MIUR Progetto di Rilevante Interesse Nazionale, PRIN 2010.
References
- Pitts TM, Davis SL, Eckhardt SG, Bradshaw-Pierce EL. Targeting nuclear kinases in cancer: development of cell cycle kinase inhibitors. Pharmacol Ther 2014; 142:258 - 69; http://dx.doi.org/10.1016/j.pharmthera.2013.12.010; PMID: 24362082
- Petronczki M, Lénárt P, Peters JM. Polo on the Rise-from Mitotic Entry to Cytokinesis with Plk1. Dev Cell 2008; 14:646 - 59; http://dx.doi.org/10.1016/j.devcel.2008.04.014; PMID: 18477449
- Lens SM, Voest EE, Medema RH. Shared and separate functions of polo-like kinases and Aurora kinases in cancer. Nat Rev Cancer 2010; 10:825 - 41; http://dx.doi.org/10.1038/nrc2964; PMID: 21102634
- Eckerdt F, Yuan J, Strebhardt K. Polo-like kinases and oncogenesis. Oncogene 2005; 24:267 - 76; http://dx.doi.org/10.1038/sj.onc.1208273; PMID: 15640842
- Carmena M, Earnshaw WC. The cellular geography of Aurora kinases. Nat Rev Mol Cell Biol 2003; 4:842 - 54; http://dx.doi.org/10.1038/nrm1245; PMID: 14625535
- Tatsuka M, Sato S, Kitajima S, Suto S, Kawai H, Miyauchi M, Ogawa I, Maeda M, Ota T, Takata T. Overexpression of Aurora-A potentiates HRAS-mediated oncogenic transformation and is implicated in oral carcinogenesis. Oncogene 2005; 24:1122 - 7; http://dx.doi.org/10.1038/sj.onc.1208293; PMID: 15592510
- Gautschi O, Heighway J, Mack PC, Purnell PR, Lara PN Jr., Gandara DR. Aurora kinases as anticancer drug targets. Clin Cancer Res 2008; 14:1639 - 48; http://dx.doi.org/10.1158/1078-0432.CCR-07-2179; PMID: 18347165
- Kelly KR, Ecsedy J, Mahalingam D, Nawrocki ST, Padmanabhan S, Giles FJ, Carew JS. Targeting Aurora kinases in cancer treatment. Curr Drug Targets 2011; 12:2067 - 78; http://dx.doi.org/10.2174/138945011798829410; PMID: 21777198
- Tang CJ, Lin CY, Tang TK. Dynamic localization and functional implications of Aurora-C kinase during male mouse meiosis. Dev Biol 2006; 290:398 - 410; http://dx.doi.org/10.1016/j.ydbio.2005.11.036; PMID: 16386730
- Gleixner KV, Ferenc V, Peter B, Gruze A, Meyer RA, Hadzijusufovic E, Cerny-Reiterer S, Mayerhofer M, Pickl WF, Sillaber C, et al. Polo-like kinase 1 (Plk1) as a novel drug target in chronic myeloid leukemia: overriding imatinib resistance with the Plk1 inhibitor BI 2536. Cancer Res 2010; 70:1513 - 23; http://dx.doi.org/10.1158/0008-5472.CAN-09-2181; PMID: 20145140
- Müller-Tidow C, Bug G, Lübbert M, Krämer A, Krauter J, Valent P, Nachbaur D, Berdel WE, Ottmann OG, Fritsch H, et al. A randomized, open-label, phase I/II trial to investigate the maximum tolerated dose of the Polo-like kinase inhibitor BI 2536 in elderly patients with refractory/relapsed acute myeloid leukaemia. Br J Haematol 2013; 163:214 - 22; PMID: 24033250
- Hartsink-Segers SA, Exalto C, Allen M, Williamson D, Clifford SC, Horstmann M, Caron HN, Pieters R, Den Boer ML. Inhibiting Polo-like kinase 1 causes growth reduction and apoptosis in pediatric acute lymphoblastic leukemia cells. Haematologica 2013; 98:1539 - 46; http://dx.doi.org/10.3324/haematol.2013.084434; PMID: 23753023
- Farag SS. The potential role of Aurora kinase inhibitors in haematological malignancies. Br J Haematol 2011; 155:561 - 79; http://dx.doi.org/10.1111/j.1365-2141.2011.08898.x; PMID: 21980926
- Marzo I, Naval J. Antimitotic drugs in cancer chemotherapy: promises and pitfalls. Biochem Pharmacol 2013; 86:703 - 10; http://dx.doi.org/10.1016/j.bcp.2013.07.010; PMID: 23886991
- Foran J, Ravandi F, Wierda W, Garcia-Manero G, Verstovsek S, Kadia T, Burger J, Yule M, Langford G, Lyons J, et al. A Phase I and Pharmacodynamic Study of AT9283, a Small-Molecule Inhibitor of Aurora Kinases in Patients With Relapsed/Refractory Leukemia or Myelofibrosis. Clin Lymphoma Myeloma Leuk 2013. doi: http://dx.doi.org/10.1016/j.clml.2013.11.001.
- Kantarjian HM, Martinelli G, Jabbour EJ, Quintás-Cardama A, Ando K, Bay JO, Wei A, Gröpper S, Papayannidis C, Owen K, et al, SPARK-AML1 Investigators. Stage I of a phase 2 study assessing the efficacy, safety, and tolerability of barasertib (AZD1152) versus low-dose cytosine arabinoside in elderly patients with acute myeloid leukemia. Cancer 2013; 119:2611 - 9; http://dx.doi.org/10.1002/cncr.28113; PMID: 23605952
- Chen YP, Lin HJ, Chen JS, Tsai MY, Hsieh HP, Chang JY, Chen NF, Chang KC, Huang WT, Su WC, et al. CDKN1A-mediated responsiveness of MLL-AF4-positive acute lymphoblastic leukemia to Aurora kinase-A inhibitors. Int J Cancer 2013; PMID: 24382688
- Fei F, Lim M, Schmidhuber S, Moll J, Groffen J, Heisterkamp N. Treatment of human pre-B acute lymphoblastic leukemia with the Aurora kinase inhibitor PHA-739358 (Danusertib). Mol Cancer 2012; 11:42; http://dx.doi.org/10.1186/1476-4598-11-42; PMID: 22721004
- Uckun FM, Ozer Z, Qazi S, Tuel-Ahlgren L, Mao C. Polo-like-kinase 1 (PLK1) as a molecular target to overcome SYK-mediated resistance of B-lineage acute lymphoblastic leukaemia cells to oxidative stress. Br J Haematol 2010; 148:714 - 25; http://dx.doi.org/10.1111/j.1365-2141.2009.07983.x; PMID: 19912216
- Pui CH, Mullighan CG, Evans WE, Relling MV. Pediatric acute lymphoblastic leukemia: where are we going and how do we get there?. Blood 2012; 120:1165 - 74; http://dx.doi.org/10.1182/blood-2012-05-378943; PMID: 22730540
- Bhojwani D, Pui CH. Relapsed childhood acute lymphoblastic leukaemia. Lancet Oncol 2013; 14:e205 - 17; http://dx.doi.org/10.1016/S1470-2045(12)70580-6; PMID: 23639321
- Van Vlierberghe P, Ferrando A. The molecular basis of T cell acute lymphoblastic leukemia. J Clin Invest 2012; 122:3398 - 406; http://dx.doi.org/10.1172/JCI61269; PMID: 23023710
- Martelli AM, Chiarini F, Evangelisti C, Cappellini A, Buontempo F, Bressanin D, Fini M, McCubrey JA. Two hits are better than one: targeting both phosphatidylinositol 3-kinase and mammalian target of rapamycin as a therapeutic strategy for acute leukemia treatment. Oncotarget 2012; 3:371 - 94; PMID: 22564882
- Grinshtein N, Datti A, Fujitani M, Uehling D, Prakesch M, Isaac M, Irwin MS, Wrana JL, Al-Awar R, Kaplan DR. Small molecule kinase inhibitor screen identifies polo-like kinase 1 as a target for neuroblastoma tumor-initiating cells. Cancer Res 2011; 71:1385 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-10-2484; PMID: 21303981
- Shan W, Akinfenwa PY, Savannah KB, Kolomeyevskaya N, Laucirica R, Thomas DG, Odunsi K, Creighton CJ, Lev DC, Anderson ML. A small-molecule inhibitor targeting the mitotic spindle checkpoint impairs the growth of uterine leiomyosarcoma. Clin Cancer Res 2012; 18:3352 - 65; http://dx.doi.org/10.1158/1078-0432.CCR-11-3058; PMID: 22535157
- Adams ND, Adams JL, Burgess JL, Chaudhari AM, Copeland RA, Donatelli CA, Drewry DH, Fisher KE, Hamajima T, Hardwicke MA, et al. Discovery of GSK1070916, a potent and selective inhibitor of Aurora B/C kinase. J Med Chem 2010; 53:3973 - 4001; http://dx.doi.org/10.1021/jm901870q; PMID: 20420387
- Simioni C, Neri LM, Tabellini G, Ricci F, Bressanin D, Chiarini F, Evangelisti C, Cani A, Tazzari PL, Melchionda F, et al. Cytotoxic activity of the novel Akt inhibitor, MK-2206, in T-cell acute lymphoblastic leukemia. Leukemia 2012; 26:2336 - 42; http://dx.doi.org/10.1038/leu.2012.136; PMID: 22614243
- Malaguti P, Vari S, Cognetti F, Fabi A. The Mammalian target of rapamycin inhibitors in breast cancer: current evidence and future directions. Anticancer Res 2013; 33:21 - 8; PMID: 23267124
- Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by Aurora A to promote checkpoint recovery. Nature 2008; 455:119 - 23; http://dx.doi.org/10.1038/nature07185; PMID: 18615013
- Hikichi Y, Honda K, Hikami K, Miyashita H, Kaieda I, Murai S, Uchiyama N, Hasegawa M, Kawamoto T, Sato T, et al. TAK-960, a novel, orally available, selective inhibitor of polo-like kinase 1, shows broad-spectrum preclinical antitumor activity in multiple dosing regimens. Mol Cancer Ther 2012; 11:700 - 9; http://dx.doi.org/10.1158/1535-7163.MCT-11-0762; PMID: 22188812
- Farrell P, Shi L, Matuszkiewicz J, Balakrishna D, Hoshino T, Zhang L, Elliott S, Fabrey R, Lee B, Halkowycz P, et al. Biological characterization of TAK-901, an investigational, novel, multitargeted Aurora B kinase inhibitor. Mol Cancer Ther 2013; 12:460 - 70; http://dx.doi.org/10.1158/1535-7163.MCT-12-0657; PMID: 23358665
- Tsykunova G, Reikvam H, Ahmed AB, Nepstad I, Gjertsen BT, Bruserud Ø. Targeting of polo-like kinases and their cross talk with Aurora kinases--possible therapeutic strategies in human acute myeloid leukemia?. Expert Opin Investig Drugs 2012; 21:587 - 603; http://dx.doi.org/10.1517/13543784.2012.668525; PMID: 22424119
- Konopleva M, Tabe Y, Zeng Z, Andreeff M. Therapeutic targeting of microenvironmental interactions in leukemia: mechanisms and approaches. Drug Resist Updat 2009; 12:103 - 13; http://dx.doi.org/10.1016/j.drup.2009.06.001; PMID: 19632887
- Suzuki J, Fujita J, Taniguchi S, Sugimoto K, Mori KJ. Characterization of murine hemopoietic-supportive (MS-1 and MS-5) and non-supportive (MS-K) cell lines. Leukemia 1992; 6:452 - 8; PMID: 1375698
- Tabe Y, Konopleva M. Advances in understanding the leukaemia microenvironment. Br J Haematol 2014; 164:767 - 78; http://dx.doi.org/10.1111/bjh.12725; PMID: 24405087
- Leonard MK, Hill NT, Bubulya PA, Kadakia MP. The PTEN-Akt pathway impacts the integrity and composition of mitotic centrosomes. Cell Cycle 2013; 12:1406 - 15; http://dx.doi.org/10.4161/cc.24516; PMID: 23574721
- Jalili A, Moser A, Pashenkov M, Wagner C, Pathria G, Borgdorff V, Gschaider M, Stingl G, Ramaswamy S, Wagner SN. Polo-like kinase 1 is a potential therapeutic target in human melanoma. J Invest Dermatol 2011; 131:1886 - 95; http://dx.doi.org/10.1038/jid.2011.136; PMID: 21654832
- Zeng Z, Sarbassov D, Samudio IJ, Yee KW, Munsell MF, Ellen Jackson C, Giles FJ, Sabatini DM, Andreeff M, Konopleva M. Rapamycin derivatives reduce mTORC2 signaling and inhibit AKT activation in AML. Blood 2007; 109:3509 - 12; http://dx.doi.org/10.1182/blood-2006-06-030833; PMID: 17179228
- Sengupta S, Peterson TR, Sabatini DM. Regulation of the mTOR complex 1 pathway by nutrients, growth factors, and stress. Mol Cell 2010; 40:310 - 22; http://dx.doi.org/10.1016/j.molcel.2010.09.026; PMID: 20965424
- Kang NJ, Lee KW, Rogozin EA, Cho YY, Heo YS, Bode AM, Lee HJ, Dong Z. Equol, a metabolite of the soybean isoflavone daidzein, inhibits neoplastic cell transformation by targeting the MEK/ERK/p90RSK/activator protein-1 pathway. J Biol Chem 2007; 282:32856 - 66; http://dx.doi.org/10.1074/jbc.M701459200; PMID: 17724030
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Franklin RA, Montalto G, Cervello M, Libra M, Candido S, Malaponte G, et al. Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascade inhibitors: how mutations can result in therapy resistance and how to overcome resistance. Oncotarget 2012; 3:1068 - 111; PMID: 23085539
- McCubrey JA, Steelman LS, Chappell WH, Abrams SL, Montalto G, Cervello M, Nicoletti F, Fagone P, Malaponte G, Mazzarino MC, et al. Mutations and deregulation of Ras/Raf/MEK/ERK and PI3K/PTEN/Akt/mTOR cascades which alter therapy response. Oncotarget 2012; 3:954 - 87; PMID: 23006971
- Ali R, Mirza Z, Ashraf GM, Kamal MA, Ansari SA, Damanhouri GA, Abuzenadah AM, Chaudhary AG, Sheikh IA. New anticancer agents: recent developments in tumor therapy. Anticancer Res 2012; 32:2999 - 3005; PMID: 22753764
- Rao CV, Kurkjian CD, Yamada HY. Mitosis-targeting natural products for cancer prevention and therapy. Curr Drug Targets 2012; 13:1820 - 30; http://dx.doi.org/10.2174/138945012804545533; PMID: 23140292
- Schloss JM, Colosimo M, Airey C, Masci PP, Linnane AW, Vitetta L. Nutraceuticals and chemotherapy induced peripheral neuropathy (CIPN): a systematic review. Clin Nutr 2013; 32:888 - 93; http://dx.doi.org/10.1016/j.clnu.2013.04.007; PMID: 23647723
- Degenhardt Y, Lampkin T. Targeting Polo-like kinase in cancer therapy. Clin Cancer Res 2010; 16:384 - 9; http://dx.doi.org/10.1158/1078-0432.CCR-09-1380; PMID: 20068088
- Reikvam H, Nepstad I, Bruserud Ø, Hatfield KJ. Pharmacological targeting of the PI3K/mTOR pathway alters the release of angioregulatory mediators both from primary human acute myeloid leukemia cells and their neighboring stromal cells. Oncotarget 2013; 4:830 - 43; PMID: 23919981
- Martelli AM, Tabellini G, Ricci F, Evangelisti C, Chiarini F, Bortul R, McCubrey JA, Manzoli FA. PI3K/AKT/mTORC1 and MEK/ERK signaling in T-cell acute lymphoblastic leukemia: new options for targeted therapy. Adv Biol Regul 2012; 52:214 - 27; http://dx.doi.org/10.1016/j.advenzreg.2011.09.019; PMID: 21983557
- Li R, Chen DF, Zhou R, Jia SN, Yang JS, Clegg JS, Yang WJ. Involvement of polo-like kinase 1 (Plk1) in mitotic arrest by inhibition of mitogen-activated protein kinase-extracellular signal-regulated kinase-ribosomal S6 kinase 1 (MEK-ERK-RSK1) cascade. J Biol Chem 2012; 287:15923 - 34; http://dx.doi.org/10.1074/jbc.M111.312413; PMID: 22427657
- Gangarossa G, Valjent E. Regulation of the ERK pathway in the dentate gyrus by in vivo dopamine D1 receptor stimulation requires glutamatergic transmission. Neuropharmacology 2012; 63:1107 - 17; http://dx.doi.org/10.1016/j.neuropharm.2012.06.062; PMID: 22796106
- Tan J, Li Z, Lee PL, Guan P, Aau MY, Lee ST, Feng M, Lim CZ, Lee EY, Wee ZN, et al. PDK1 signaling toward PLK1-MYC activation confers oncogenic transformation, tumor-initiating cell activation, and resistance to mTOR-targeted therapy. Cancer Discov 2013; 3:1156 - 71; http://dx.doi.org/10.1158/2159-8290.CD-12-0595; PMID: 23887393
- Cunningham JT, Ruggero D. New connections between old pathways: PDK1 signaling promotes cellular transformation through PLK1-dependent MYC stabilization. Cancer Discov 2013; 3:1099 - 102; http://dx.doi.org/10.1158/2159-8290.CD-13-0581; PMID: 24124229
- Lonetti A, Antunes IL, Chiarini F, Orsini E, Buontempo F, Ricci F, Tazzari PL, Pagliaro P, Melchionda F, Pession A, et al. Activity of the pan-class I phosphoinositide 3-kinase inhibitor NVP-BKM120 in T-cell acute lymphoblastic leukemia. Leukemia 2013; •••; http://dx.doi.org/10.1038/leu.2013.369; PMID: 24310736
- Buontempo F, Chiarini F, Bressanin D, Tabellini G, Melchionda F, Pession A, Fini M, Neri LM, McCubrey JA, Martelli AM. Activity of the selective IκB kinase inhibitor BMS-345541 against T-cell acute lymphoblastic leukemia: involvement of FOXO3a. Cell Cycle 2012; 11:2467 - 75; http://dx.doi.org/10.4161/cc.20859; PMID: 22713244
- Chiarini F, Lonetti A, Teti G, Orsini E, Bressanin D, Cappellini A, Ricci F, Tazzari PL, Ognibene A, Falconi M, et al. A combination of temsirolimus, an allosteric mTOR inhibitor, with clofarabine as a new therapeutic option for patients with acute myeloid leukemia. Oncotarget 2012; 3:1615 - 28; PMID: 23271044
- Bressanin D, Evangelisti C, Ricci F, Tabellini G, Chiarini F, Tazzari PL, Melchionda F, Buontempo F, Pagliaro P, Pession A, et al. Harnessing the PI3K/Akt/mTOR pathway in T-cell acute lymphoblastic leukemia: eliminating activity by targeting at different levels. Oncotarget 2012; 3:811 - 23; PMID: 22885370
- Buontempo F, Orsini E, Martins LR, Antunes I, Lonetti A, Chiarini F, Tabellini G, Evangelisti C, Evangelisti C, Melchionda F, et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia 2014; 28:543 - 53; http://dx.doi.org/10.1038/leu.2013.349; PMID: 24253024
- Chou TC, Talalay P. Quantitative analysis of dose-effect relationships: the combined effects of multiple drugs or enzyme inhibitors. Adv Enzyme Regul 1984; 22:27 - 55; http://dx.doi.org/10.1016/0065-2571(84)90007-4; PMID: 6382953