
Abstract
Glucocorticoids, such as dexamethasone, enhance protein breakdown via ubiquitin–proteasome system. However, the role of autophagy in organelle and protein turnover in the glucocorticoid-dependent atrophy program remains unknown. Here, we show that dexamethasone stimulates an early activation of autophagy in L6 myotubes depending on protein kinase, AMPK, and glucocorticoid receptor activity. Dexamethasone increases expression of several autophagy genes, including ATG5, LC3, BECN1, and SQSTM1 and triggers AMPK-dependent mitochondrial fragmentation associated with increased DNM1L protein levels. This process is required for mitophagy induced by dexamethasone. Inhibition of mitochondrial fragmentation by Mdivi-1 results in disrupted dexamethasone-induced autophagy/mitophagy. Furthermore, Mdivi-1 increases the expression of genes associated with the atrophy program, suggesting that mitophagy may serve as part of the quality control process in dexamethasone-treated L6 myotubes. Collectively, these data suggest a novel role for dexamethasone-induced autophagy/mitophagy in the regulation of the muscle atrophy program.
Introduction
Glucocorticoids (GC) are steroid hormones that respond to a variety of environmental and physiological stimuli; they have wide-ranging regulatory effects on both development and metabolism.Citation1 Because of their potent anti-inflammatory and immunosuppressive activity, the pharmaceutical industry has developed various synthetic GC for therapeutic use. Although GC are highly effective, they are also associated with adverse effects, including hyperglycemia, weight gain, hypertension, osteoporosis, depression, decreased immunological function, and skeletal muscle weakness.Citation2
The degradation of skeletal muscle proteins is controlled by 2 proteolytic pathways: the ubiquitin–proteasome system and autophagy. Both degradative pathways are activated in several catabolic states, including cancer, AIDS, diabetes, heart failure, and skeletal muscle weakness. Both systems are fine-tuned by the expression of a few critical enzymes.Citation3 Recently, FOXO transcription factors have been identified as the main coordinators of these 2 proteolytic pathways by virtue of their capacity to induce several autophagy-related genes as well as the ubiquitin ligases ATROGIN-1 and MURF1.Citation4,Citation5 In skeletal muscle, the ubiquitin-proteasome system (UPS) seems to control the half-life of sarcomere proteins, and its inhibition has beneficial effects on muscle mass.Citation6 However, the role of autophagy in skeletal muscle homeostasis has only recently been explored.
Accumulating evidence suggests that autophagy activation may exacerbate skeletal muscle mass loss in catabolic states.Citation7 Denervation induces autophagy in skeletal muscle, although at a lower rate than fasting. This effect is mediated by the transcription factor RUNX1, which is upregulated upon denervation and favors the preservation of muscle mass.Citation7 The absence of RUNX1 results in excessive autophagy and skeletal muscle atrophy.Citation7 Moreover, some critical genes associated with autophagy are among the pro-atrophy genes that are also under the control of FOXO3. The expression of FOXO3 is sufficient and necessary for the activation of protein degradation by autophagy both in vitro and in vivo.Citation5 siRNA-induced depletion of ATG5 (a protein required for induction of autophagy) partially prevents loss of skeletal muscle mass.Citation8 In addition, specific expression of the superoxide dismutase 1 gene mutant (SOD1G93A) in skeletal muscle causes muscle wasting and weakness, mainly as a result of autophagy activation.Citation8 The role of autophagy in the atrophic activity of synthetic GC is not well understood. Several studies have shown that GC increases cathepsin L muscle expressionCitation9 and stimulates conversion of LC3-I to LC3-II.Citation10,Citation11
Mitochondria are essential organelles responsible for critical activities in proper cell function.Citation12 Mitochondrial morphology is governed by well-ordered fusion and fission processes. While fusion events are controlled by the large GTPases mitofusins 1/2 (MFN1/2) and optic atrophy protein 1 (OPA1), mitochondrial fission is mediated by fission protein 1 (FIS1) and dynamin1-like (DNM1L).Citation13 Constant renewal is crucial for maintaining healthy mitochondria in skeletal muscle and requires the coordination of mitochondrial biogenesis and selective degradation (mitophagy). Mitochondrial dysfunction leads to impaired skeletal muscle function and development.Citation14,Citation15 Moreover, widespread changes in the mitochondrial network occur in atrophied muscles resulting from denervated mice.Citation13 Expression of the mitochondrial fission machinery, per se, induces muscle wasting by triggering organelle dysfunction and activation of protein kinase, AMP-activated protein kinase (AMPK).Citation13 In addition, FOXO3 overexpression stimulates mitochondrial disruption.Citation14 Interestingly, FOXO3 also activates autophagy and stimulates expression of many autophagy (Atg) genes in myotubes, isolated muscle fiber, and muscle in vivo.Citation5 Lokireddy et al. recently showed that diverse catabolic stimuli, such as starvation and GC, regulate mitochondrial dynamics by an increased FOXO3-dependent expression of the mitochondrial ubiquitin ligase 1 (MUL1), which, in turn, decreases MNF2 protein levels, inducing mitochondrial fission and mitophagy.Citation10 Moreover, GC also modulates mitochondrial function by increasing DNM1L protein levels in hepatoma cells.Citation16
The synthetic GC dexamethasone (Dex) is widely used to treat inflammatory and autoimmune conditions. Dex induces autophagy in lymphocytes and osteocytes.Citation17,Citation18 Although earlier studies have shown that Dex activates skeletal muscle autophagy,Citation10,Citation11 the molecular mechanisms controlling Dex-induced autophagy and their implications for muscle atrophy remain elusive. In this work, we show that Dex stimulates autophagy, mitochondrial fragmentation, and mitophagy in L6 skeletal muscle cells. The inhibition of mitochondrial fragmentation by Mdivi-1 disrupts skeletal muscle autophagy and mitophagy, and increases the expression of the Dex-triggered atrophic program. This latter finding suggests a novel role for Dex-induced autophagy/mitophagy in regulation of the muscle atrophy program.
Results
Dex triggers autophagy in L6 myotubes through GR activation.
To determine whether Dex induces autophagy in L6 rat skeletal muscle cells, we assessed the conversion of LC3-I to LC3-II and the degradation of SQSTM1 by immunoblot. In addition, the formation of endogenous LC3 puncta was determined by immunofluorescence staining and confocal microscopy. Dex induced the lipidation of endogenous LC3, as well as a slight decrease in SQSTM1 levels at 6 h (). Dex-treated myotubes also displayed greater quantities of endogenous LC3 autophagic puncta than untreated controls (). Similar results were obtained in myoblasts of L6 rat cells, myotubes, and myoblasts of C2C12 mouse cells and cultured neonatal cardiomyocytes (Fig. S1A and B). It should be noted that increases in LC3 processing and autophagic puncta can be produced not only from increased autophagosome formation (positive autophagic flux), but also from decreased lysosomal degradation of autophagosomes (impaired autophagic flux). To discriminate between these 2 possibilities, the current validated strategy is measuring autophagic parameters upon addition of lysosomal inhibitors.Citation19 We used bafilomycin-A1 (Baf A1), which inhibits the lysosomal vacuolar type H+-ATPase, increasing lysosomal pH and impairing autophagosome fusion with lysosomes. We found that in the presence of Baf A1, L6 myotubes treated with Dex for 6 h exhibited higher levels of LC3 puncta than untreated cells (). This result indicates that Dex increases autophagic flux.
Figure 1. Western blot analysis of phosphorylated GR, LC3, SQSTM1, and GAPDH in L6 myotubes treated with Dex for indicated time points (A). LC3 and Hoechst 33342 immunofluorescence in L6 cells incubated with Dexamethasone for 6 h (B). Western blot analysis of LC3, SQSTM1, and GAPDH in L6 myotubes incubated with Baf A1 and/or Dex for 6 h (C). Western blot analysis of phophorylated MTOR, total MTOR, phosphorylated AMPK, total AMPK, LC3, SQSTM1, and GAPDH in L6 myotubes incubated with Dexamethasone for 0, 6, and 24 h (D). Western blot analysis of LC3, SQSTM1, BECN1, and GAPDH in LUC and BECN1 knockout L6 myotubes (E). Western blot analysis of GR, LC3, SQSTM1, and GAPDH in control and glucocorticoid receptor knockdown L6 myotubes (F). Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control.
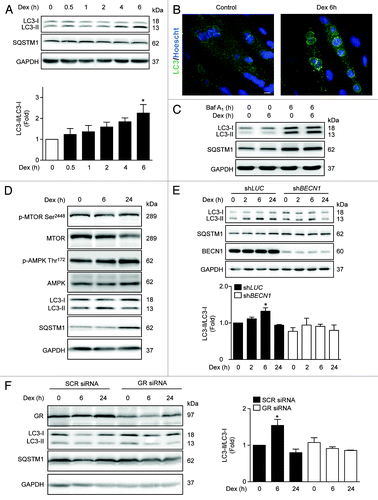
MTOR (mammalian target of rapamycin) and AMPK (protein kinase, AMP activated) are 2 protein kinases that are well known to regulate the early steps of autophagy.Citation20 Therefore, we examined the effect of Dex on the activating phosphorylation of these proteins. Dex treatment stimulated AMPK phosphorylation without changing MTOR phosphorylation, suggesting a role for AMPK in Dex-induced autophagy (). However, 24 h Dex treatment decreased LC3 processing and increased SQSTM1 protein levels (), suggesting that Dex may also increase SQSTM1 protein levels. To determine whether Dex induced LC3 conversion is dependent on autophagy machinery, we generated stable cell lines expressing either a lentiviral vector encoding a BECN1-specific shRNA (shBECN1) construct or an unrelated shRNA (shLUC). shows that Dex-dependent LC3 processing is abolished in shBECN1 cells. GC activity is mainly mediated by the GR; however, studies have also shown that some GC effects seem to be mediated by non-classical receptors and/or non-genomic signaling pathways.Citation21 To evaluate the involvement of GR in Dex-induced autophagy, we transfected myotubes with control or GR siRNA. Decrease in GR levels by transient siRNA transfection blunted the effect of Dex on LC3 processing (). Taken together, these results suggest that Dex induces autophagy in L6 myotubes through GR.
Dex triggers skeletal muscle autophagy through genomic and non-genomic mechanisms.
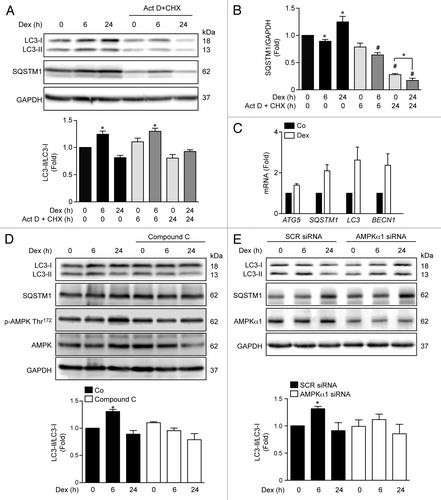
The effects of GC are divided into genomic actions, which depend on GR-mediated gene transcription and de novo protein synthesis; and non-genomic signaling, which does not require nuclear GR-mediated gene transcription.Citation21 These non-genomic actions are thought to be mediated by the activation of signaling pathways, including AMPK.Citation22 Actinomycin D and cycloheximide were used to determine the relationship between Dex-mediated autophagy induction and the effects of GR on gene transcription and/or protein translation. shows the effect of actinomycin D and cycloheximide on Dex-dependent LC3 processing and SQSTM1 levels. Inhibition of protein synthesis with cycloheximide did not prevent Dex-induced LC3 processing, as measured by the ratio LC3-II/LC3-I at 6 h (). However, we found increased expression of SQSTM1 after 24 h of exposure to Dex (). These results suggest that Dex stimulates an early induction of non-genomic autophagy (6 h) followed by an increase in autophagy machinery expression through a genomic mechanism (24 h). In agreement with this conjecture, myotubes exposed to Dex for 24 h exhibited increased mRNA levels for ATG5, SQSTM1, LC3, and BECN1 (). Given the non-genomic induction of autophagy, we tested whether AMPK protein is involved in Dex-dependent early induction of skeletal muscle autophagy. Myotubes treated with the pharmacological inihibitor of AMPK, Compound C, or transfected with an AMPKα1 siRNA showed a decrease in Dex-induced LC3 processing after 6 h of Dex treatment (). Taken together, these results suggest that activation of early autophagy by Dex depends on AMPKα1 activation.
Figure 2. Western blot analysis of LC3, SQSTM1 and GAPDH in Dex, and/or Act D and CHX incubated L6 myotubes for indicated time points (A). Quantification of SQSTM1/GAPDH presented in (A) (B). Quantification of ATG5, SQSTM1, LC3, and BECN1 mRNA levels of control and Dexamethasone incubated L6 myotubules (C). Western blot analysis of LC3-II, SQSTM1, phosphorylated and total AMPK and GAPDH in compound C treated L6 myotubes incubated with Dex for indicated time points (D). Western blot analysis of LC3-II, SQSTM1, AMPKα1, and GAPDH in SCR and AMPKα1 knockdown L6 myotubes incubated with Dex for indicated time points (E). Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control, #P < 0.05 vs. Dex.
Dex induces mitochondrial fragmentation
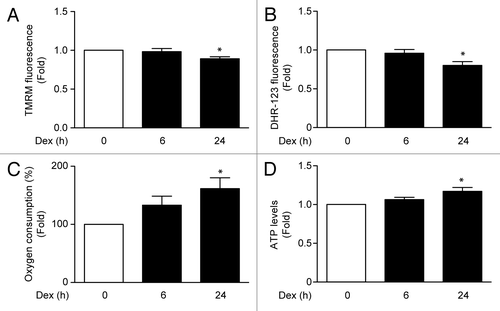
Recent reports in C2C12 skeletal muscle cell line have shown that Dex induces mitochondrial fragmentation, a necessary process for mitophagy.Citation10 To assess whether Dex induces mitochondrial fragmentation in L6 cells, mitochondrial morphology was evaluated by confocal microscopy using Mitotracker Orange dye. Under control conditions, mitochondria showed elongated shapes that changed to a round-shaped morphology after treatment with Dex. The Dex-induced changes in mitochondrial morphology correlated with increased number of mitochondria per cell, and with decreased mean mitochondrial volume (). We also evaluated the effects of Dex on the levels of mitochondrial fusion and fission proteins. Dex increased the expression of the mitochondrial fission protein DNM1L () without changing MNF2 or mtHSP70 levels (; Fig. S2). In a recent work, we showed that mitochondrial fragmentation decreases Ca2+ uptake, which is associated with mitochondrial dysfunction and impaired insulin signaling.Citation23 Therefore, we addressed the question as to whether the Dex-induced morphologic changes in mitochondria would affect mitochondrial Ca2+ uptake. shows that Dex treatment for 6 h followed by histamine incubation enhanced Ca2+ release into the cytosol. However, Dex also reduced histamine-mediated mitochondrial Ca2+ uptake (). Next, we evaluated the effect of Dex on mitochondrial function in L6 myotubes. Mitochondrial membrane potential was reduced by Dex treatment for 24 h (). Moreover, Dex decreased cellular ROS production () and increased oxygen consumption () and ATP production ().
Figure 3. Mitochondria morphology shown by confocal microscopy in L6 myoblast incubated with Dex for 0, 6, and 24h and quantification of mitochondrial volume and number per cell (A). Western blot analysis of DNM1L, MFN2, mtHSP70, and GAPDH in Dex treated L6 myotubes for 0, 6, and 24 h (B). Histamine induced Ca2+ release in control and Dex-treated L6 myotubes (C). Histamine induced Ca2+ uptake by mitochondria in control and Dex incubated L6 myotubes (D). Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control.
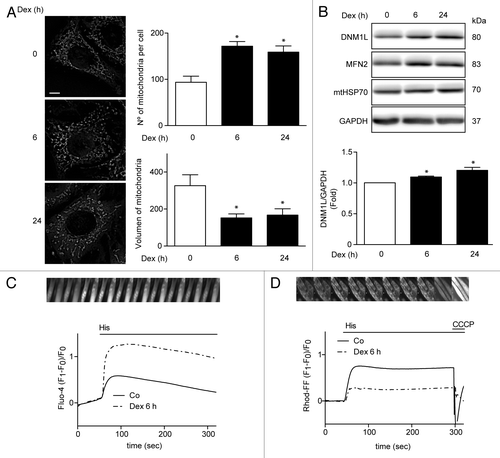
Figure 4. Mitochondria membrane potential (measured by TMRM fluorescence) (A), ROS induction (measured by Dihydrorhodamine-123 fluorescence) (B), oxygen consumption (C), and ATP levels (D) of L6 myotubes incubated with Dex for 0, 6, and 24 h. Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control.
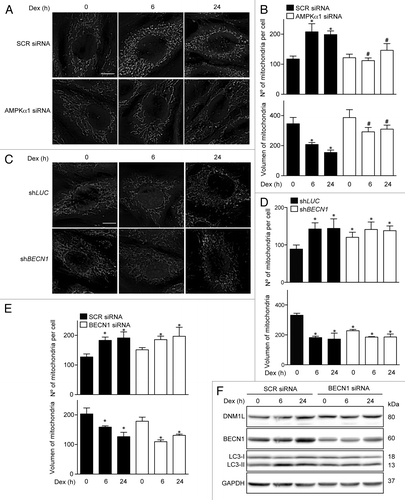
To assess whether mitochondrial fragmentation is associated with the AMPK signaling pathway that regulates Dex-induced early autophagy, the AMPK subunit α1 was knocked down with a specific siRNA. shows that AMPKα1 knockdown prevents Dex-induced mitochondrial fragmentation. In addition, we assessed the impact of autophagy on DEX-modulated changes in mitochondrial dynamics. We observed that stable BECN1 knockout did not prevent Dex-induced mitochondrial fragmentation; in fact, an increase in mitochondrial fragmentation was observed (). Similar results were seen when myotubes were treated with BECN1 siRNA (). Furthermore, the Dex-induced DNM1L increase was not affected by transient knockdown of BECN1 (). These results suggest that mitochondrial fragmentation triggered by Dex depends on AMPKα1 activation but occurs independently from autophagy.
Figure 5. Mitochondria morphology shown by confocal microscopy in SCR and AMPKα1 knockdown L6 myotubes incubated with Dex for 0, 6, and 24 h (A). Quantification of mitochondrial volume and number per cell presented in (A). (B) Mitochondria morphology shown by confocal microscopy in LUC and BECN1 knockout L6 myotubes incubated with Dex for 0, 6, and 24 h (C). Quantification of mitochondrial volume and number per cell presented in (C). (D) Quantification of mitochondrial volume and number per cell in SCR and BECN1 knockdown L6 myotubes incubated with Dex for 0, 6, and 24 h (E). Western blot analysis of DNM1L, BECN1, LC3, and GAPDH in SCR and BECN1 knockdown L6 myotubes incubated with Dex for 0, 6, and 24 h (F). Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. time 0 (SCR siRNA). #P < 0.05 vs. SCR siRNA + Dex.
Mdivi-1 stimulates the Dex-triggered atrophic program.
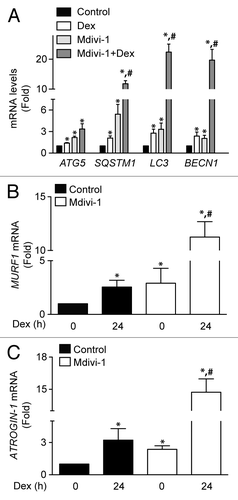
We next studied whether induction of autophagy and mitochondrial fragmentation were associated with the selective mitochondrial degradation process (mitophagy). This process requires mitochondrial fission in order to generate small mitochondria, which are subject to lysosomal degradation.Citation24 Mitophagy is the basis of mitochondrial quality control, and therefore crucial for maintaining a fully functional mitochondrial network. To address the participation of mitophagy in our model, we evaluated whether Dex stimulates mitophagy. Dex significantly increased lysosome–mitochondria colocalization in comparison with untreated cells (), as determined by the Manders’ colocalization index between the lysosomal marker Lysotracker Red (LSTR) and the mitochondria marker Mitotracker Green (MTG) (). The induction of mitophagy is controlled by 2 proteins, the E3 ubiquitin ligase PARK2/parkin and PTEN-induced putative kinase 1 (PINK1). Dex increased the protein levels of PINK1 but had not effect on PARK2 expression (; Fig. S3). Next, the role of mitochondrial fragmentation in Dex-induced mitophagy was studied using the DNM1L inhibitor Mdivi-1 (Mitochondrial division Inhibitor-1). Inhibition of DNM1L by Mdivi-1 decreases Dex-induced mitochondrial fragmentation (Fig. S4). Concomitantly, Mdivi-1 prevented Dex-triggered mitophagy after 6 and 24 h exposure (). Mdivi-1 also decreased oxygen consumption at baseline and in response to Dex (). Mdivi-1 had no effect on the expression of DNM1L, MFN2, and mtHSP70 (). However, Mdivi-1 decreased Dex-induced LC3 processing and increased SQSTM1 protein levels (), suggesting that Mdivi-1 inhibits autophagic flux. Accordingly, Mdivi-1 also reduced the Dex-triggered LC3 processing in the presence of BafA1 (). Mdivi-1 alone increased the expression of ATG5, SQSTM1, LC3, and BECN1, and co-incubation with Dex resulted in a hyperadditive increase in the abundance of the corresponding mRNAs (). Finally, the role of mitochondrial fragmentation/mitophagy in Dex-induced muscle atrophy was assessed. Dex increased mRNA expression of 2 proteins that are commonly associated with muscle atrophy, ATROGIN-1 and MURF1. Mdivi-1 alone increased the expression of these 2 proteins. Dex plus Mdivi-1 caused a much stronger induction of ATROGIN-1 and MURF1 expression in myotubes than either of these 2 compounds alone (), suggesting that inhibition of mitochondrial fragmentation/mitophagy by Mdivi-1 enhances the Dex-stimulated expression of autophagy- and muscle atrophy-related genes. Collectively, these results show that Dex induces autophagy, mitochondrial fragmentation, and mitophagy in L6 muscle cells. This latter process seems to be required for proper Dex-induced autophagy and atrophy.
Figure 6. Mitochondria (MTG) and lysosome (LSTR) colocalization shown by confocal microscopy in control, Dex. and Dex plus Mdivi-1 (DNM1L inhibition) incubated L6 myoblast, quantification of mitochondria and lysosome colocalization expressed by Manders’ coefficient in L6 myoblast incubated with Dex and/or Mdivi-1 for 0, 6, and 24 h (A). Western blot analysis of PINK1, PARK2, and GAPDH in L6 myotubes incubated with Dex and/or Mdivi-1 for 0, 6, and 24 h (B). Oxygen consumption of Dex and/or Mdivi-1 incubated L6 myotubes (C). Western blot analysis of DNM1L, MFN2, mtHSP70, phophorylated AMPK, total AMPK, LC3, SQSTM1, and GAPDH in Dex and/or Mdivi-1 incubated L6 myotubes (D). Western blot analysis of LC3, SQSTM1 and GAPDH in Dex, Mdivi-1, and/or Baf A1 treated L6 myotubes (E). Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control, #P < 0.05 vs. Dex.
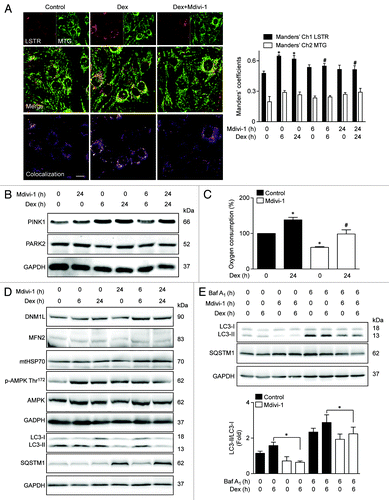
Figure 7. Quantification of ATG5, SQSTM1, LC3, and BECN1 mRNA levels of control, Dex, Mdivi-1, and Dex plus Mdivi-1 incubated L6 myotubes (A). Quantification of MURF1 (B) and ATROGIN-1 (C) mRNA levels in control and Mdivi-1 incubated L6 myotubes. Data: mean ± SEM of at least 3 independent experiments. Statistically significant differences were calculated using ANOVA in combination with a Tukey test for group comparison. *P < 0.05 vs. control, #P < 0.05 vs. Dex.
Discussion
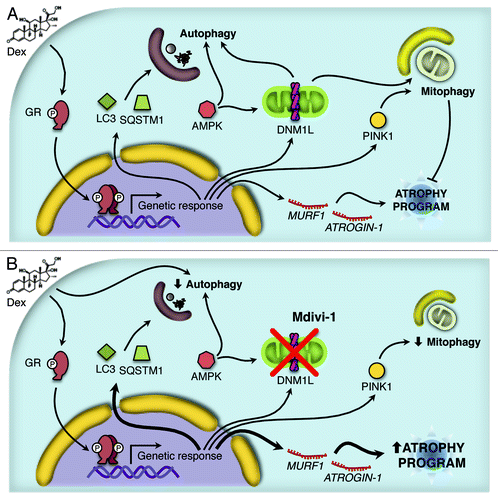
GC have been known to activate the muscular atrophy program through the transcriptional induction of 2 ubiquitin ligases, ATROGIN-1 and MURF1, as well as through the activation of proteasome degradation.Citation25 In the present study, we found evidence that GC stimulation of L6 skeletal muscle cells causes an increase in autophagic flux, mitochondrial fragmentation, and mitophagy. The inhibition of mitochondrial fragmentation by Mdivi-1 disrupted skeletal muscle autophagy and mitophagy and simultaneously amplified the expression of genes that are activated in the course of the Dex-triggered atrophic program. Hence mitochondrial fragmentation occurs independently from and upstream of autophagy functions, and it acts as a negative control instance to reduce the expression of muscle atrophy-relevant genes ().
Figure 8. Working model proposed for the Dex action. Dex induces activation of the autophagy process through the protein AMPK and the expression of several autophagy-related genes and the atrophy program. In addition, Dex triggers mitochondrial fragmentation and consequent mitophagy (A). The inhibition of DNM1L by Mdivi-1 disrupts the mitochondrial fragmentation, autophagy and mitophagy induced by Dex. Furthermore, there is an increment in Dex-induced autophagy-related genes and the atrophy program as consequent of the autophagy/mitophagy reduction (B).
Autophagy is a highly conserved process for degrading cytoplasmic components including damaged organelles, toxic protein aggregates, and intracellular pathogens.Citation26 Basal autophagy plays a key role in eukaryotic cells, degrading long half-lived macromolecules and large supramolecular structures, including organelles such as mitochondria, peroxisomes, and endoplasmic reticulum. Thus, autophagy is a quality control instance that is charged with the task of maintaining essential cellular functions. In addition, autophagy may be upregulated during different types of stress, such as metabolic, genotoxic, and hypoxic, acting as an adaptive mechanism.Citation27
So-called autophagic vacuolar myopathies, including Pompe disease and Danon disease, are characterized by the accumulation of autophagosomes in muscle cells.Citation28,Citation29 Similarly, the “chloroquine myopathy” is marked by the appearance of excessive autophagosomes in response to lysosomal inhibition by the anti-malaria drug chloroquine.Citation30 Basal autophagy in skeletal muscle also plays an important role in the control of muscle mass. Muscle-specific Atg7-deficient mice show profound muscle atrophy and an age-dependent decrease in muscle force.Citation31 The role of autophagy in GC activity is not well understood. Several studies have shown that GC increase cathepsin L muscle expressionCitation9 and conversion of LC3-I to LC3-II.Citation10,Citation11 In this context, our work supports the idea that autophagy is part of the cellular response induced by GC. Indeed, Dex was able to induce LC3 processing, LC3 puncta, and autophagy flux through a largely BECN1-dependent pathway.
GC are lipophilic drugs and act mainly by binding to the cytoplasmic GR. This process induces translocation of this hormone–receptor complex to the nucleus, where it binds to the GC response elements of target genes, and, depending on the presence of co-factors/transcription factors, leads to their transactivation or transrepression.Citation21 This mode of GC action is referred to as the genomic pathway, and because it involves gene transcription and mRNA translation, its effects take hours or even days to become manifest.Citation21 In addition to their genomic effects, GC also mediate rapid actions.Citation32-Citation34 These rapid, non-genomic effects occur within minutes to hours and are relatively insensitive to inhibitors of transcription and translation. Three mechanisms of the non-genomic GC action have been proposed: (1) binding of the GC to the cytoplasmic GR and release of the chaperone molecules; (2) binding of the GC to a plasma membrane GR; and (3) non-specific physicochemical interactions of the GC with the cell membrane.Citation35 GC exert non-genomic actions on the cardiovascular, immune, and neuroendocrine systems.Citation36-Citation38 In skeletal muscle, non-genomic presumably mediated by a plasma membrane GR effects have recently been described.Citation39 Our results support the non-genomic action of GC hypothesis in skeletal muscle, because early Dex-activated autophagy was insensitive to transcription and translation inhibitors but required the presence of GR and AMPKAα1. Nonetheless, the transcriptional induction of the expression of several autophagy genes also supports the activation of genomic pathway in the Dex-triggered autophagic response.
As mentioned above, the FOXO transcription factors seem to be responsible for the genomic action of GC. Several studies have shown that GC stimulate the expression of both FOXO1 and FOXO3 in skeletal muscle.Citation40,Citation41 Chip-sequencing assays have identified several GR binding regions within or in close proximity to the FOXO3 genomic region in C2C12 myotubes and 3T3–L1 adipocytes, thus revealing that FOXO3 is a GR target gene.Citation42,Citation43 FOXO3 also emerged as a master regulator of the atrophy program. In response to pro-cachectic stimuli, FOXO3 activation increases expression of ATROGIN-1 and MURF1.Citation4,Citation44 Moreover, FOXO3 activates several genes involved in autophagy.Citation5,Citation14,Citation44 In this study, we demonstrated that GC stimulates the expression of several autophagy genes, which suggests involvement of FOXO3 in this genomic effect of GC. However, the induction of autophagy was decreased by the pharmacological inhibition and depletion of AMPKα1 suggesting a non-genomic component on autophagy regulation by GC.
Mitochondrial dysfunction underlies various human pathologies, including adult-onset neurodegeneration, cancer, aging, cardiovascular disease, and metabolic disorders, such as obesity and type 2 diabetes.Citation45-Citation48 In the skeletal muscle, mitochondrial dysfunction leads to impaired muscle function and development.Citation14,Citation15 Moreover, there are widespread changes in the mitochondrial network in atrophying muscles from fasted and denervated mice. Expression of the mitochondrial fission machinery per se induces muscle wasting by triggering organelle dysfunction and AMPK activation. FOXO3 overexpression reportedly induces mitochondrial disruption through activation of autophagy,Citation14 which can be attributed to the transactivation of several ATG genes, as observed in myotubes, isolated muscle fibers, and in intact muscle in vivo.Citation5 Recently, diverse catabolic stimuli such as starvation and GC have been found to regulate mitochondrial dynamics through increased DNM1L expression and MNF2 degradation in skeletal muscle.Citation10 Moreover, in hepatoma cells, the increase in mitochondrial respiration and gluconeogenes induced by GC requires DNM1L.Citation16 Our data indicated that Dex induced mitochondrial fragmentation is associated with increased DNM1L protein levels and an altered mitochondrial function, which is in agreement with the aforementioned studies. It should be noted that increased mitochondrial fragmentation has been associated with changes in mitochondrial Ca2+ uptake.Citation23 Constitutive Ca2+ release from ITPR (inositol 1,4,5-trisphosphate receptor) is essential for maintaining mitochondrial bioenergetics. Reduced Ca2+ entry into the mitochondria leads to diminished production of ATP and an increase in the AMP/ATP ratio, activating autophagy through AMPK.Citation49
As discussed above, the induction of mitochondrial fission per se activates AMPK and autophagy. Furthermore, autophagy is a key determinant for mitochondrial health and proper cell function.Citation50 Our results suggest that Dex-triggered autophagy is not responsible for the mitochondrial fragmentation observed in our models. However, AMPK activation seems to be necessary in part for Dex-induced autophagy and mitochondrial fragmentation. Furthermore, the DNM1L inhibitor Mdivi-1 caused reduced mitochondrial fragmentation and disrupted the Dex-induced autophagy process. Indeed, DNM1L inhibition with Mdivi-1 or siRNA prevented the increase in LC3 processing in Cd2+-induced hepatotoxicity.Citation51 Taken together, these data suggest that activation of autophagy is a secondary response to Dex-induced mitochondrial fragmentation.
Mitophagy has a pivotal role in adaptation to stress, by acting as a mitochondrial quality control mechanism.Citation52 Correct incorporation of mitochondria within an autophagosome, which has a diameter of 1 μM, is necessary for a mitochondrial fission event.Citation53 Accordingly, other authors and our own laboratory have demonstrated that mitochondrial fragmentation precedes mitophagy activation in muscle.Citation13 Moreover, inducing mitochondrial fragmentation by knocking down MFN2 increases mitophagy, suggesting that disruption of the network continuity leads to mitochondrial deterioration and increased turnover.Citation23 Accordingly, overexpression of MUL1 induces mitochondrial fragmentation through MNF2 degradation and consequent mitophagy.Citation10 Our data support the notion that mitochondrial fragmentation is a key step in mitochondrial turnover, as the use of Mdivi-1 prevented Dex-induced mitophagy.
Both endogenous glucocorticoids and their pharmacological analogs are potent mediators of muscle atrophy.Citation25 The catabolic effects accompanying various pathological conditions, including sepsis, burn injury, diabetes, and inactivity, are at least in part mediated by endogenous GC.Citation54-Citation57 In these catabolic conditions, the atrophied muscles exhibit increased rates of protein degradation, primarily through activation of the ubiquitin–proteasome pathway and a common series of transcriptional adaptations that together constitute the “atrophy program”.Citation58 The proteins induced most dramatically during atrophy are the ubiquitin ligases ATROGIN-1 and MURF1. Accordingly, the simultaneous knockdown of ATROGIN-1 and MURF1 prevented Dex-induced atrophy in cultured myotubes.Citation59 Moreover, expression of the mitochondrial fission machinery per se induces muscle atrophy,Citation13 and the FOXO3-dependent activation of ATROGIN-1 and MURF1 was prevented by expression of a dominant-negative DNM1L mutant.Citation13 The induction of mitochondrial fragmentation and mitophagy by overexpression of MUL1 is sufficient to trigger muscle wasting, while the knockdown of MUL1 prevents starvation-induced muscle wasting. These data suggested a pivotal role for mitochondrial fragmentation in the induction of muscle atrophy. Unexpectedly, however, the inhibition of DNM1L by Mdivi-1 resulted in increased expression of ATROGIN-1 and MURF-1. This latter finding is important, because several reports suggested that Mdivi-1 may be used to ameliorate organ functions in ischemia/reperfusion injury and pressure overload-induced heart failure.Citation60,Citation61 However, Zhang et al. demonstrated that Mdivi-1 aggravated cerebral ischemia-reperfusion injury, suggesting that the protective role of autophagy is attributable to mitophagy-related mitochondrial clearance.Citation62
In conclusion, we propose that Dex-triggered autophagy acts as a repressor of the atrophy program. We surmise that the induction of autophagy operates as a quality control that removes damaged mitochondria, thereby dampening the atrophy program.
Materials and Methods
Antibodies and reagents
The antibodies used in the study were purchased from the following companies. Abcam: MNF2 (ab50838), GR (ab2768), and PINK1 (ab23707). Abnova: SQSTM1 (h00008871). BD Transduction Laboratories: DNM1L (611113). Cell Signaling Technologies: LC3B (2775), BECN1 (3738), MTOR (2983), AMPK (2453), phospho-MTOR (Ser2448, 2971), and phospho-AMPK (Thr172, 2531). Enzo Life Technologies: mtHSP70 (804–077-R100). Millipore: PARK2 (MAB 5512). Santa Cruz Biotechnologies: AMPKα1 (sc-19128). Sigma: GAPDH (G9545). Dexamethasone (Sigma, D2915), bafilomycin A1 (Sigma, B1793), STO609 (Sigma, S1318), Mdivi-1 (Sigma, MO199), GaCl3 (Aldrich, 439770), BAPTA-AM (Invitrogen, B6769), Rutenium Red (Calbiochem, M1510), carbonyl cyanide m chlorophenylhydrazone (CCCP, Sigma, C2759).
Cell culture, siRNA transfection, and production of stable knockdown cells
Rat L6 myoblast cells were cultured in DMEM supplemented with 10% FBS (growth medium, GM). To induce differentiation, subconfluent L6 cells were cultured with media containing 2% FBS (differentiation medium, DM) for 5–7 d to complete differentiation in myotubes. L6 cells were transfected with 100 nM control siRNA or siRNA against BECN1 (Sigma), AMPKα1 (Sigma), and GR (Sigma) using Lipofectamine 2000 (Invitrogen, 11668-019) in Optimem medium (GIBCO, 31985–070) overnight according to the manufacturer’s instructions. To generate a stable cell line that express the shBECN1, L6 cells were transfected in Optimem medium with Lipofectamine 2000 according to the manufacturer’s instructions, using the following plasmids: VSV-G, pMDL, REV and PLKO.1-shBECN1, or PLKO.1-shLUC. After 48 h, the conditioned medium with lentiviral particles was collected and filtered with 0.45-μm pore diameter filters. After 48 h of viral transduction, medium containing 2 μg/ml puromycin was used for the selection process of shBECN1 and shLUC cells.
Western blot
Equal amounts of protein from complete cell extracts were separated by SDS-PAGE (10% polyacrylamide gels) and electrotransferred to nitrocellulose. Membranes were blocked with 5% milk TBS-T. Membranes were incubated with primary antibodies at 4 °C and blotted with horseradish peroxidase-linked secondary antibodies (1:5000 in 1% [w/v] milk in TBS-T). Signals were detected using EZ-ECL (Biological Industries, 20–500–1000A, 20–500–1000B) and quantified by scanning densitometry. Protein content was normalized with GAPDH.
Extraction of RNA and gene expression analysis
Total RNA was obtained from L6 myotubes employing Trizol reagent (Invitrogen, 15596–026) according to manufacturer's protocol. cDNA was prepared from 1 µg of RNA, using SuperScript II enzyme (Invitrogen, A13268), according to manufacturer’s protocol. Real-time PCR was performed using Stratagene Mx3000P (Stratagene) using the Brilliant III Ultra-Fast QPCR and QRT-PCR Master Mix amplification kit (Agilent Technologies, 4309155).
The primers used were: ATROGIN-1: 5′-CCATCAGGAG AAGTGGATCT ATGTT-3′ (sense), 5′- GTTCATGAAG TTCTTTTGGG CGATGC-3′ (antisense); MURF1: 5′-ACAACCTCTG CCGGAAGTGT-3′ (sense), 5′-CGGAAACGAC CTCCAGACAT-3′ (antisense); GADPH: 5′-CCATGGAGAA GGCTGGGG-3′ (sense), 5′-CAAAGTTGTC ATGGATGACC-3′ (antisense); LC3: 5′- AAATAGAAGA GCAGTGTCAGGGG-3′ (sense), 5′-GGGACTGTTT CCAGGGACTT C-3′ (antisense); BECN1: 5′-ACTCTGGAGG TCTCGCTCTG-3′ (sense), 5′-CGGAAACGAC CTCCAGACAT-3′ (antisense); ATG5: 5′-AGTAAACCTCGCTGACAGGC-3′ (sense), 5′-TTCACTGAGCAAAAGCGTGC-3′ (antisense); SQSTM1: 5′-CTGAAGAATGTGGGGGAGAGC-3′ (sense), 5′- CTTCCCTCCA TGTTCCACAT CA-3′ (antisense). All primers used presented optimal amplification efficiency (between 90% and 110%). PCR amplification of the housekeeping gene GADPH was performed as a control. CT value was determined by MXPro software when fluorescence was 25% higher than background. PCR products were verified by melting-curve analysis.
Mitochondrial morphology
L6 cells were incubated with 400 nM MitoTracker Orange (Invitrogen, M7510) in Krebs solution for 30 min. Confocal image stacks of the mitochondrial network were captured with a Zeiss LSM 5 Pascal/Axiovert 200 microscope, using a Plan-Apochromat 63×/1.4 Oil DIC objective, as described elsewhere.Citation63 Z-stacks were deconvolved, thresholded, and 3D-reconstructed using ImageJ software (NIH). Number and volume of individual mitochondria were quantified using the 3D Object Counter plug-in. Each experiment was repeated at least 3 times, and 16–25 cells per condition were quantified.
Cytoplasmic and mitochondrial Ca2+
To determine cytoplasmic and mitochondrial Ca+2 kinetic, time-lapse images were obtained from cultured L6 myotubes preloaded with Fluo-4AM (Invitrogen, F-14201) and Rhod2-FF (Invitrogen, R23983), respectively, using a Zeiss LSM 5 Pascal/Axiovert 200 microscope, as previously described.Citation64
Mitochondrial transmembrane potential (Ψmt) and ROS production
Myotubes were loaded with 200 nM tetramethylrhodamine (TMRM, Invitrogen, T-668)Citation65 or 25 nM dihydrorhodamine-123 (Sigma, D1054),Citation66 respectively, for 30 min. Fluorescence was determined by flow cytometry using a FACScan system (Becton–Dickinson).
ATP measurement
Intracellular ATP content was determined using Cell Titer-Glow Luminescent Cell Viability Assay (Promega, G7571) following manufacturer’s instructions. Signals were measured in a TopCount NXT microplate luminescence counter (PerkinElmer).Citation64
Oxygen consumption
Cells were plated on 100-mm dishes, and when they reached confluence, the cells were treated according to experimental design. After trypsinization, oxygen levels of the cell suspension in PBS were measured polarographically at 25 °C using a #5331 Clark electrode (Yellow Springs Instruments).Citation64,Citation67
Mitophagy
Cells were incubated for 30 min with Lysotracker Red (LSTR, 100 nM) (Invitrogen, L7528) and MitoTracker Green (MTG, 400 nM) (Invitrogen, M-7514) and maintained in Krebs solution. Confocal images were captured with a Leica TCS SP5 confocal microscope using a Plan-Apochromat 63×/1.4 Oil DIC objective.Citation23 For colocalization analysis only one focal plane was analyzed. Images were deconvolved, and the background was subtracted using ImageJ software (NIH). Signal colocalization was quantified using Manders’ algorithm, as previously described.Citation63
Statistical analysis
Data are mean ± SEM of the indicated sample size (n). Multiple groups were analyzed using one-way ANOVA followed by a protected Tukey test. Statistical significance was defined as P < 0.05.
| Abbreviations: | ||
| Act D | = | actinomycin D |
| ATG5 | = | autophagy-related 5 |
| Baf A1 | = | bafilomycin A1 |
| BECN1 | = | Beclin 1, autophagy related |
| CHX | = | cycloheximide |
| Dex | = | dexamethasone |
| DNM1L | = | dynamin 1-like/dynamin-related protein 1 |
| FIS1 | = | fission protein 1 |
| FOXO | = | forkhead box O |
| GAPDH | = | glyceraldehyde 3-phosphate dehydrogenase |
| GC | = | glucocorticoids |
| GR | = | glucocorticoid receptor |
| LC3 | = | microtubule-associated protein 1 light chain 3 |
| LSTR | = | lisotracker red |
| Mdivi-1 | = | mitochondrial division inhibitor-1 |
| MFN2 | = | mitofusin 2 |
| MUL1 | = | mitochondrial ubiquitin ligase 1 |
| MTG | = | mitotracker green |
| MURF1 | = | muscle ring finger-1, MTOR, mechanistic target of rapamycin |
| LUC | = | luciferase |
| PARK2 | = | parkin |
| AMPK | = | AMP-activated protein kinase |
| PINK1 | = | PTEN induced putative kinase 1 |
| RuRed | = | ruthenium red |
| SCR | = | scrambled |
| SOD | = | superoxide dismutase |
| SQSTM1 | = | sequestosome 1 |
Additional material
Download Zip (2.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
This research was supported by FONDECYT (grant 1120212 to S.L., 3120220 to C.Q., and 11130285 to R.T.), CONICYT (grant Anillo ACT1111 to S.L.; FONDAP 15130011 to S.L. and R.T.). We are thankful for the PhD or MSc fellowships from CONICYT Chile to F.P., A.E.R., D.G., C.V.T., and C.L.C. V.P. thanks to the International Postdoctoral Bicentennial Program from CONICYT, Chile and the American Heart Association. G.K. is financed by the Ligue contre le Cancer (équipe labelisée); Agence National de la Recherche (ANR); Association pour la recherche sur le cancer (ARC); Cancéropôle Ile-de-France; Institut National du Cancer (INCa); Fondation Bettencourt-Schueller; Fondation de France; Fondation pour la Recherche Médicale (FRM); the European Commission (ArtForce); the European Research Council (ERC); the LabEx Immuno-Oncology; and the Paris Alliance of Cancer Research Institutes (PACRI). We also thank Fidel Albornoz and Gindra Latorre for their excellent technical assistance.
Related Research Data
References
- Revollo JR, Cidlowski JA. Mechanisms generating diversity in glucocorticoid receptor signaling. Ann N Y Acad Sci 2009; 1179:167 - 78; http://dx.doi.org/10.1111/j.1749-6632.2009.04986.x; PMID: 19906239
- Ramamoorthy S, Cidlowski JA. Exploring the molecular mechanisms of glucocorticoid receptor action from sensitivity to resistance. Endocr Dev 2013; 24:41 - 56; http://dx.doi.org/10.1159/000342502; PMID: 23392094
- Sandri M. Signaling in muscle atrophy and hypertrophy. Physiology (Bethesda) 2008; 23:160 - 70; http://dx.doi.org/10.1152/physiol.00041.2007; PMID: 18556469
- Sandri M, Sandri C, Gilbert A, Skurk C, Calabria E, Picard A, Walsh K, Schiaffino S, Lecker SH, Goldberg AL. Foxo transcription factors induce the atrophy-related ubiquitin ligase atrogin-1 and cause skeletal muscle atrophy. Cell 2004; 117:399 - 412; http://dx.doi.org/10.1016/S0092-8674(04)00400-3; PMID: 15109499
- Mammucari C, Schiaffino S, Sandri M. Downstream of Akt: FoxO3 and mTOR in the regulation of autophagy in skeletal muscle. Autophagy 2008; 4:524 - 6; PMID: 18367868
- Lynch GS, Schertzer JD, Ryall JG. Therapeutic approaches for muscle wasting disorders. Pharmacol Ther 2007; 113:461 - 87; http://dx.doi.org/10.1016/j.pharmthera.2006.11.004; PMID: 17258813
- Wang X, Blagden C, Fan J, Nowak SJ, Taniuchi I, Littman DR, Burden SJ. Runx1 prevents wasting, myofibrillar disorganization, and autophagy of skeletal muscle. Genes Dev 2005; 19:1715 - 22; http://dx.doi.org/10.1101/gad.1318305; PMID: 16024660
- Dobrowolny G, Aucello M, Rizzuto E, Beccafico S, Mammucari C, Boncompagni S, Belia S, Wannenes F, Nicoletti C, Del Prete Z, et al. Skeletal muscle is a primary target of SOD1G93A-mediated toxicity. Cell Metab 2008; 8:425 - 36; http://dx.doi.org/10.1016/j.cmet.2008.09.002; PMID: 19046573
- Deval C, Mordier S, Obled C, Bechet D, Combaret L, Attaix D, Ferrara M. Identification of cathepsin L as a differentially expressed message associated with skeletal muscle wasting. Biochem J 2001; 360:143 - 50; http://dx.doi.org/10.1042/0264-6021:3600143; PMID: 11696001
- Lokireddy S, Wijesoma IW, Teng S, Bonala S, Gluckman PD, McFarlane C, Sharma M, Kambadur R. The ubiquitin ligase Mul1 induces mitophagy in skeletal muscle in response to muscle-wasting stimuli. Cell Metab 2012; 16:613 - 24; http://dx.doi.org/10.1016/j.cmet.2012.10.005; PMID: 23140641
- Yamamoto D, Maki T, Herningtyas EH, Ikeshita N, Shibahara H, Sugiyama Y, Nakanishi S, Iida K, Iguchi G, Takahashi Y, et al. Branched-chain amino acids protect against dexamethasone-induced soleus muscle atrophy in rats. Muscle Nerve 2010; 41:819 - 27; http://dx.doi.org/10.1002/mus.21621; PMID: 20169591
- Lee SR, Kim HK, Song IS, Youm J, Dizon LA, Jeong SH, Ko TH, Heo HJ, Ko KS, Rhee BD, et al. Glucocorticoids and their receptors: insights into specific roles in mitochondria. Prog Biophys Mol Biol 2013; 112:44 - 54; http://dx.doi.org/10.1016/j.pbiomolbio.2013.04.001; PMID: 23603102
- Verdejo HE, del Campo A, Troncoso R, Gutierrez T, Toro B, Quiroga C, Pedrozo Z, Munoz JP, Garcia L, Castro PF, et al. Mitochondria, myocardial remodeling, and cardiovascular disease. Curr Hypertens Rep 2012; 14:532 - 9; http://dx.doi.org/10.1007/s11906-012-0305-4; PMID: 22972531
- Romanello V, Guadagnin E, Gomes L, Roder I, Sandri C, Petersen Y, Milan G, Masiero E, Del Piccolo P, Foretz M, et al. Mitochondrial fission and remodelling contributes to muscle atrophy. EMBO J 2010; 29:1774 - 85; http://dx.doi.org/10.1038/emboj.2010.60; PMID: 20400940
- Chen H, Vermulst M, Wang YE, Chomyn A, Prolla TA, McCaffery JM, Chan DC. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell 2010; 141:280 - 9; http://dx.doi.org/10.1016/j.cell.2010.02.026; PMID: 20403324
- Hernández-Alvarez MI, Paz JC, Sebastián D, Muñoz JP, Liesa M, Segalés J, Palacín M, Zorzano A. Glucocorticoid modulation of mitochondrial function in hepatoma cells requires the mitochondrial fission protein Drp1. Antioxid Redox Signal 2013; 19:366 - 78; http://dx.doi.org/10.1089/ars.2011.4269; PMID: 22703557
- Xia X, Kar R, Gluhak-Heinrich J, Yao W, Lane NE, Bonewald LF, Biswas SK, Lo WK, Jiang JX. Glucocorticoid-induced autophagy in osteocytes. J Bone Miner Res 2010; 25:2479 - 88; http://dx.doi.org/10.1002/jbmr.160; PMID: 20564240
- Grandér D, Kharaziha P, Laane E, Pokrovskaja K, Panaretakis T. Autophagy as the main means of cytotoxicity by glucocorticoids in hematological malignancies. Autophagy 2009; 5:1198 - 200; http://dx.doi.org/10.4161/auto.5.8.10122; PMID: 19855186
- Klionsky DJ, Abdalla FC, Abeliovich H, Abraham RT, Acevedo-Arozena A, Adeli K, Agholme L, Agnello M, Agostinis P, Aguirre-Ghiso JA, et al. Guidelines for the use and interpretation of assays for monitoring autophagy. Autophagy 2012; 8:445 - 544; http://dx.doi.org/10.4161/auto.19496; PMID: 22966490
- Lavandero S, Troncoso R, Rothermel BA, Martinet W, Sadoshima J, Hill JA. Cardiovascular autophagy: concepts, controversies, and perspectives. Autophagy 2013; 9:1455 - 66; http://dx.doi.org/10.4161/auto.25969; PMID: 23959233
- Kadmiel M, Cidlowski JA. Glucocorticoid receptor signaling in health and disease. Trends Pharmacol Sci 2013; 34:518 - 30; http://dx.doi.org/10.1016/j.tips.2013.07.003; PMID: 23953592
- Kewalramani G, Puthanveetil P, Wang F, Kim MS, Deppe S, Abrahani A, Luciani DS, Johnson JD, Rodrigues B. AMP-activated protein kinase confers protection against TNF-alpha-induced cardiac cell death. Cardiovasc Res 2009; 84:42 - 53; http://dx.doi.org/10.1093/cvr/cvp166; PMID: 19477967
- del Campo A, Parra V, Vásquez-Trincado C, Gutiérrez T, Morales PE, López-Crisosto C, Bravo-Sagua R, Navarro-Marquez MF, Verdejo HE, Contreras-Ferrat A, et al. Mitochondrial fragmentation impairs insulin-dependent glucose uptake by modulating Akt activity through mitochondrial Ca2+ uptake. Am J Physiol Endocrinol Metab 2014; 306:E1 - 13; PMID: 24085037
- Chan DC. Fusion and fission: interlinked processes critical for mitochondrial health. Annu Rev Genet 2012; 46:265 - 87; http://dx.doi.org/10.1146/annurev-genet-110410-132529; PMID: 22934639
- Schakman O, Kalista S, Barbé C, Loumaye A, Thissen JP. Glucocorticoid-induced skeletal muscle atrophy. Int J Biochem Cell Biol 2013; 45:2163 - 72; http://dx.doi.org/10.1016/j.biocel.2013.05.036; PMID: 23806868
- Mizushima N, Levine B, Cuervo AM, Klionsky DJ. Autophagy fights disease through cellular self-digestion. Nature 2008; 451:1069 - 75; http://dx.doi.org/10.1038/nature06639; PMID: 18305538
- Levine B, Kroemer G. Autophagy in aging, disease and death: the true identity of a cell death impostor. Cell Death Differ 2009; 16:1 - 2; http://dx.doi.org/10.1038/cdd.2008.139; PMID: 19079285
- Raben N, Wong A, Ralston E, Myerowitz R. Autophagy and mitochondria in Pompe disease: nothing is so new as what has long been forgotten. Am J Med Genet C Semin Med Genet 2012; 160C:13 - 21; http://dx.doi.org/10.1002/ajmg.c.31317; PMID: 22253254
- Nishino I, Fu J, Tanji K, Yamada T, Shimojo S, Koori T, Mora M, Riggs JE, Oh SJ, Koga Y, et al. Primary LAMP-2 deficiency causes X-linked vacuolar cardiomyopathy and myopathy (Danon disease). Nature 2000; 406:906 - 10; http://dx.doi.org/10.1038/35022604; PMID: 10972294
- Suzuki T, Nakagawa M, Yoshikawa A, Sasagawa N, Yoshimori T, Ohsumi Y, Nishino I, Ishiura S, Nonaka I. The first molecular evidence that autophagy relates rimmed vacuole formation in chloroquine myopathy. J Biochem 2002; 131:647 - 51; http://dx.doi.org/10.1093/oxfordjournals.jbchem.a003147; PMID: 11983070
- Masiero E, Agatea L, Mammucari C, Blaauw B, Loro E, Komatsu M, Metzger D, Reggiani C, Schiaffino S, Sandri M. Autophagy is required to maintain muscle mass. Cell Metab 2009; 10:507 - 15; http://dx.doi.org/10.1016/j.cmet.2009.10.008; PMID: 19945408
- Buttgereit F, Krauss S, Brand MD. Methylprednisolone inhibits uptake of Ca2+ and Na+ ions into concanavalin A-stimulated thymocytes. Biochem J 1997; 326:329 - 32; PMID: 9291100
- Croxtall JD, Choudhury Q, Flower RJ. Glucocorticoids act within minutes to inhibit recruitment of signalling factors to activated EGF receptors through a receptor-dependent, transcription-independent mechanism. Br J Pharmacol 2000; 130:289 - 98; http://dx.doi.org/10.1038/sj.bjp.0703272; PMID: 10807665
- Buttgereit F, Scheffold A. Rapid glucocorticoid effects on immune cells. Steroids 2002; 67:529 - 34; http://dx.doi.org/10.1016/S0039-128X(01)00171-4; PMID: 11960631
- Buttgereit F, Wehling M, Burmester GR. A new hypothesis of modular glucocorticoid actions: steroid treatment of rheumatic diseases revisited. Arthritis Rheum 1998; 41:761 - 7; http://dx.doi.org/10.1002/1529-0131(199805)41:5<761::AID-ART2>3.0.CO;2-M; PMID: 9588727
- Ayroldi E, Cannarile L, Migliorati G, Nocentini G, Delfino DV, Riccardi C. Mechanisms of the anti-inflammatory effects of glucocorticoids: genomic and nongenomic interference with MAPK signaling pathways. FASEB J 2012; 26:4805 - 20; http://dx.doi.org/10.1096/fj.12-216382; PMID: 22954589
- Lee SR, Kim HK, Youm JB, Dizon LA, Song IS, Jeong SH, Seo DY, Ko KS, Rhee BD, Kim N, et al. Non-genomic effect of glucocorticoids on cardiovascular system. Pflugers Arch 2012; 464:549 - 59; http://dx.doi.org/10.1007/s00424-012-1155-2; PMID: 23001133
- Bellavance MA, Rivest S. The neuroendocrine control of the innate immune system in health and brain diseases. Immunol Rev 2012; 248:36 - 55; http://dx.doi.org/10.1111/j.1600-065X.2012.01129.x; PMID: 22725953
- Pérez MH, Cormack J, Mallinson D, Mutungi G. A membrane glucocorticoid receptor mediates the rapid/non-genomic actions of glucocorticoids in mammalian skeletal muscle fibres. J Physiol 2013; 591:5171 - 85; http://dx.doi.org/10.1113/jphysiol.2013.256586; PMID: 23878367
- Nishimura M, Mikura M, Hirasaka K, Okumura Y, Nikawa T, Kawano Y, Nakayama M, Ikeda M. Effects of dimethyl sulphoxide and dexamethasone on mRNA expression of myogenesis- and muscle proteolytic system-related genes in mouse myoblastic C2C12 cells. J Biochem 2008; 144:717 - 24; http://dx.doi.org/10.1093/jb/mvn126; PMID: 18835828
- Waddell DS, Baehr LM, van den Brandt J, Johnsen SA, Reichardt HM, Furlow JD, Bodine SC. The glucocorticoid receptor and FOXO1 synergistically activate the skeletal muscle atrophy-associated MuRF1 gene. Am J Physiol Endocrinol Metab 2008; 295:E785 - 97; http://dx.doi.org/10.1152/ajpendo.00646.2007; PMID: 18612045
- Kuo T, Lew MJ, Mayba O, Harris CA, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor-binding sites in myotubes identifies gene networks modulating insulin signaling. Proc Natl Acad Sci U S A 2012; 109:11160 - 5; http://dx.doi.org/10.1073/pnas.1111334109; PMID: 22733784
- Yu CY, Mayba O, Lee JV, Tran J, Harris C, Speed TP, Wang JC. Genome-wide analysis of glucocorticoid receptor binding regions in adipocytes reveal gene network involved in triglyceride homeostasis. PLoS One 2010; 5:e15188; http://dx.doi.org/10.1371/journal.pone.0015188; PMID: 21187916
- Zhao J, Brault JJ, Schild A, Cao P, Sandri M, Schiaffino S, Lecker SH, Goldberg AL. FoxO3 coordinately activates protein degradation by the autophagic/lysosomal and proteasomal pathways in atrophying muscle cells. Cell Metab 2007; 6:472 - 83; http://dx.doi.org/10.1016/j.cmet.2007.11.004; PMID: 18054316
- Dodson S, Baracos VE, Jatoi A, Evans WJ, Cella D, Dalton JT, Steiner MS. Muscle wasting in cancer cachexia: clinical implications, diagnosis, and emerging treatment strategies. Annu Rev Med 2011; 62:265 - 79; http://dx.doi.org/10.1146/annurev-med-061509-131248; PMID: 20731602
- Emery AE. The muscular dystrophies. Lancet 2002; 359:687 - 95; http://dx.doi.org/10.1016/S0140-6736(02)07815-7; PMID: 11879882
- Evans WJ. Skeletal muscle loss: cachexia, sarcopenia, and inactivity. Am J Clin Nutr 2010; 91:1123S - 7S; http://dx.doi.org/10.3945/ajcn.2010.28608A; PMID: 20164314
- Lecker SH. Ubiquitin-protein ligases in muscle wasting: multiple parallel pathways?. Curr Opin Clin Nutr Metab Care 2003; 6:271 - 5; http://dx.doi.org/10.1097/01.mco.0000068963.34812.e5; PMID: 12690258
- Cárdenas C, Miller RA, Smith I, Bui T, Molgó J, Müller M, Vais H, Cheung KH, Yang J, Parker I, et al. Essential regulation of cell bioenergetics by constitutive InsP3 receptor Ca2+ transfer to mitochondria. Cell 2010; 142:270 - 83; http://dx.doi.org/10.1016/j.cell.2010.06.007; PMID: 20655468
- Rambold AS, Lippincott-Schwartz J. Mechanisms of mitochondria and autophagy crosstalk. Cell Cycle 2011; 10:4032 - 8; http://dx.doi.org/10.4161/cc.10.23.18384; PMID: 22101267
- Pi H, Xu S, Zhang L, Guo P, Li Y, Xie J, Tian L, He M, Lu Y, Li M, et al. Dynamin 1-like-dependent mitochondrial fission initiates overactive mitophagy in the hepatotoxicity of cadmium. Autophagy 2013; 9:1780 - 800; http://dx.doi.org/10.4161/auto.25665; PMID: 24121705
- Kroemer G, Mariño G, Levine B. Autophagy and the integrated stress response. Mol Cell 2010; 40:280 - 93; http://dx.doi.org/10.1016/j.molcel.2010.09.023; PMID: 20965422
- East DA, Campanella M. Ca2+ in quality control: an unresolved riddle critical to autophagy and mitophagy. Autophagy 2013; 9:1710 - 9; http://dx.doi.org/10.4161/auto.25367; PMID: 24121708
- Tiao G, Fagan J, Roegner V, Lieberman M, Wang JJ, Fischer JE, Hasselgren PO. Energy-ubiquitin-dependent muscle proteolysis during sepsis in rats is regulated by glucocorticoids. J Clin Invest 1996; 97:339 - 48; http://dx.doi.org/10.1172/JCI118421; PMID: 8567953
- Fang CH, James HJ, Ogle C, Fischer JE, Hasselgren PO. Influence of burn injury on protein metabolism in different types of skeletal muscle and the role of glucocorticoids. J Am Coll Surg 1995; 180:33 - 42; PMID: 8000653
- Hu Z, Wang H, Lee IH, Du J, Mitch WE. Endogenous glucocorticoids and impaired insulin signaling are both required to stimulate muscle wasting under pathophysiological conditions in mice. J Clin Invest 2009; 119:3059 - 69; PMID: 19759515
- Fitts RH, Romatowski JG, Peters JR, Paddon-Jones D, Wolfe RR, Ferrando AA. The deleterious effects of bed rest on human skeletal muscle fibers are exacerbated by hypercortisolemia and ameliorated by dietary supplementation. Am J Physiol Cell Physiol 2007; 293:C313 - 20; http://dx.doi.org/10.1152/ajpcell.00573.2006; PMID: 17409123
- Lecker SH, Goldberg AL, Mitch WE. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J Am Soc Nephrol 2006; 17:1807 - 19; http://dx.doi.org/10.1681/ASN.2006010083; PMID: 16738015
- Castillero E, Alamdari N, Lecker SH, Hasselgren PO. Suppression of atrogin-1 and MuRF1 prevents dexamethasone-induced atrophy of cultured myotubes. Metabolism 2013; 62:1495 - 502; http://dx.doi.org/10.1016/j.metabol.2013.05.018; PMID: 23866982
- Givvimani S, Munjal C, Tyagi N, Sen U, Metreveli N, Tyagi SC. Mitochondrial division/mitophagy inhibitor (Mdivi) ameliorates pressure overload induced heart failure. PLoS One 2012; 7:e32388; http://dx.doi.org/10.1371/journal.pone.0032388; PMID: 22479323
- Ong SB, Subrayan S, Lim SY, Yellon DM, Davidson SM, Hausenloy DJ. Inhibiting mitochondrial fission protects the heart against ischemia/reperfusion injury. Circulation 2010; 121:2012 - 22; http://dx.doi.org/10.1161/CIRCULATIONAHA.109.906610; PMID: 20421521
- Zhang X, Yan H, Yuan Y, Gao J, Shen Z, Cheng Y, Shen Y, Wang RR, Wang X, Hu WW, et al. Cerebral ischemia-reperfusion-induced autophagy protects against neuronal injury by mitochondrial clearance. Autophagy 2013; 9:1321 - 33; http://dx.doi.org/10.4161/auto.25132; PMID: 23800795
- Parra V, Eisner V, Chiong M, Criollo A, Moraga F, Garcia A, Härtel S, Jaimovich E, Zorzano A, Hidalgo C, et al. Changes in mitochondrial dynamics during ceramide-induced cardiomyocyte early apoptosis. Cardiovasc Res 2008; 77:387 - 97; http://dx.doi.org/10.1093/cvr/cvm029; PMID: 18006463
- Troncoso R, Vicencio JM, Parra V, Nemchenko A, Kawashima Y, Del Campo A, Toro B, Battiprolu PK, Aranguiz P, Chiong M, et al. Energy-preserving effects of IGF-1 antagonize starvation-induced cardiac autophagy. Cardiovasc Res 2012; 93:320 - 9; http://dx.doi.org/10.1093/cvr/cvr321; PMID: 22135164
- Munoz JP, Chiong M, García L, Troncoso R, Toro B, Pedrozo Z, Diaz-Elizondo J, Salas D, Parra V, Núñez MT, et al. Iron induces protection and necrosis in cultured cardiomyocytes: Role of reactive oxygen species and nitric oxide. Free Radic Biol Med 2010; 48:526 - 34; http://dx.doi.org/10.1016/j.freeradbiomed.2009.11.017; PMID: 19969068
- Marambio P, Toro B, Sanhueza C, Troncoso R, Parra V, Verdejo H, García L, Quiroga C, Munafo D, Díaz-Elizondo J, et al. Glucose deprivation causes oxidative stress and stimulates aggresome formation and autophagy in cultured cardiac myocytes. Biochim Biophys Acta 2010; 1802:509 - 18; http://dx.doi.org/10.1016/j.bbadis.2010.02.002; PMID: 20176105
- Bravo R, Vicencio JM, Parra V, Troncoso R, Munoz JP, Bui M, Quiroga C, Rodriguez AE, Verdejo HE, Ferreira J, et al. Increased ER-mitochondrial coupling promotes mitochondrial respiration and bioenergetics during early phases of ER stress. J Cell Sci 2011; 124:2143 - 52; http://dx.doi.org/10.1242/jcs.080762; PMID: 21628424