
Abstract
During mitotic entry, the centrosomes provide a scaffold for initial activation of the CyclinB/Cdk1 complex, the mitotic kinase Aurora A, and the Aurora A-activating kinase p21-activated kinase (PAK). The activation of PAK at the centrosomes is yet regarded to happen independently of the Rho-GTPases Rac/Cdc42. In this study, Rac1 (but not RhoA or Cdc42) is presented to associate with the centrosomes from early G2 phase until prometaphase in a cell cycle-dependent fashion, as evidenced by western blot analysis of prepared centrosomes and by immunolabeling. PAK associates with the G2/M-phase centrosomes in a Rac1-dependent fashion. Furthermore, specific inhibition of Rac1 by C. difficile toxinB-catalyzed glucosylation or by knockout results in inhibited activation of PAK1/2, Aurora A, and the CyclinB/Cdk1 complex in late G2 phase/prophase and delayed mitotic entry. Inhibition of PAK activation at late G2-phase centrosomes caused by Rac1 inactivation coincides with impeded activation of Aurora A and the CyclinB/Cdk1 complex and delayed mitotic entry.
Introduction
The onset of mitosis is controlled by the activation of the CyclinB/Cdk1 complex.Citation1,Citation2 Expression of CyclinB starts during S phase and reaches its maximum in late G2 phase and early M phase. Cdk1 activity is regulated by the stable association with CyclinB and phosphorylation at various sites. Initial activation of the CyclinB–Cdk1 complex occurs in the cytosol and at centrosomes before the CyclinB–Cdk1 complex is activated in the nucleus during prophase.Citation1,Citation3,Citation4 The centrosomes in late G2 phase/prophase provide a scaffold for components of the mitotic entry regulating signaling cascade, including the mitotic kinases Polo-like kinase-1 (Plk1) and Aurora A, and the mitotic phosphatase Cdc25.Citation5,Citation6 Aurora A phosphorylates (and thereby activates) Plk1, which both contribute to the activation of Cdc25.Citation7,Citation8 Cdc25, in turn, dephosphorylates Cdk1 on inhibitory phosphorylation sites and thereby activates the CyclinB–Cdk1 complex.Citation6
p21-activated kinase (PAK) is a serine/threonine kinase harboring an N-terminal GTPase-binding domain (RBD) and a C-terminal kinase domain. PAK1–3 are downstream effector proteins of the small GTPases Rac1/Cdc42.Citation9,Citation10 Binding of Rac1/Cdc42 in their active GTP-bound state to the RBD abrogates the interaction of the kinase inhibitory domain (KID) with the kinase domain leading to kinase activation. Kinase activity is induced by either autophosphorylation in trans at pT423/402-PAK1/2 within the activation loop of the kinase domain or may involve a third-party kinase such as phosphoinositide-dependent protein kinase-1 (PDK1).Citation9 PAK phosphorylates Plk1 on Ser-49Citation11 and Aurora A on Thr-288 and Ser-342,Citation12 resulting in increased kinase activity of Plk1 and Aurora A. Inhibition of PAK1/2 by ectopic expression of KID results in delayed activation of Aurora A and Plk1 and subsequently the CyclinB–Cdk1 complex.Citation11 Besides PAK, the Rho/Rac effector PRK2/PKN2 seems to contribute to the regulation of mitotic entry, as its RNAi-mediated depletion resulted in delayed mitotic entry.Citation13 Although several effector proteins of Rho-GTPases seem to be involved in the regulation of mitotic entry, the upstream Rho-GTPases regulating them have not yet been identified.
In G2/M phase, a population of PAK2 associates with the centrosomes in a complex with Arf GTPase-activating protein GIT and the PAK-interacting exchange factor (β-PIX). β-PIX binds PAK and functions as a GEF protein for Rac1 and Cdc42.Citation12 In this study, PAK2 and Rac1 are presented to associate with the centrosomes in G2–M phase in a cell cycle-dependent fashion. Thereby, Rac1 recruits PAK to late G2-phase centrosomes. Inhibition of PAK activation at late G2-phase centrosomes by Rac1 inactivation coincides with reduced activation of Aurora A and the CyclinB/Cdk1 complex and delayed mitotic entry. Rac1 is thus identified as a new upstream regulator of the mitotic entry.
Results
Association of Rac1 with the G2 phase centrosomes
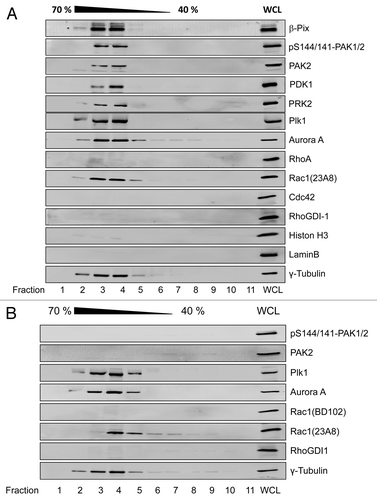
To investigate the presence of PAK and other upstream regulators of mitotic entry at the centrosomes in late G2 phase, HeLa cells, which exhibit a doubling time of about 18 h, were synchronized in early S phase using the thymidine double-block technique. Synchronized HeLa cells entered G2 phase about 8 h after release, as evidenced by the abundance of the 4N peak using FACS analysis of propidium iodide stained cells (). HeLa cells underwent cell division at about 12 h after release from the thymidine block, as evidenced by the re-abundance of the 2N peak (). HeLa cells in late G2 phase were harvested 10 h after release from the block, and the centrosomes were isolated by discontinuous sucrose density gradient ultracentrifugation.Citation14 G2-phase centrosomes were identified by the marker protein γ-tubulin and were free from cytosolic and from nuclear contaminations like RhoGDI-1, Histon-3, or Lamin B (). The mitotic kinases Aurora A and Plk1, the kinases PDK1, PAK2, and PRK2, as well as the PAK-interacting exchange factor (β-Pix) were found to be associated with G2-phase centrosomes (). Interestingly, selectively Rac1, but not RhoA or Cdc42, was present at the G2-phase centrosomes ().
Figure 1. Delayed mitotic entry upon treatment of synchronized Hela cells with variant C. difficile Toxin B. HeLa cells were synchronized using the thymidine double-block technique. HeLa cells were allowed to progress into G2 phase and were treated with variant Toxin B from the C. difficile serotype F strain 1470 (TcdBF) 8 h after release from the second thymidine block or left untreated. Cells were fixed every 2 h at the indicated time points post-release, and cell cycle progression was analyzed by FACS of propidium iodide-stained cells.
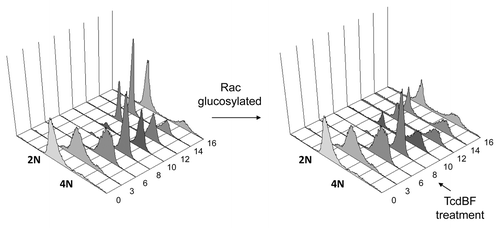
Figure 2. Rac1-dependent association of PAK2 with G2-phase centrosomes HeLa cells were synchronized by the thymidine double block. Cells were allowed to release from block and entered G2 phase 8 h after release from the second block and were left non-treated (A) or treated with ToxinB (30 ng/ml) for 2 h (B). Centrosomes were isolated using sucrose density ultracentrifugation and fractions were analyzed by western blotting for the indicated proteins. Centrosome-enriched fractions were indicated by γ-tubulin and referred to as isolated centrosomes. The γ-tubulin signal was strongest in fractions 3 and 4, corresponding to 60% (w/w) sucrose. Whole cell lysate (WCL) from G2-phase synchronized HeLa cells was applied as positive control, confirming the presence of the respective proteins within the cell lysate.
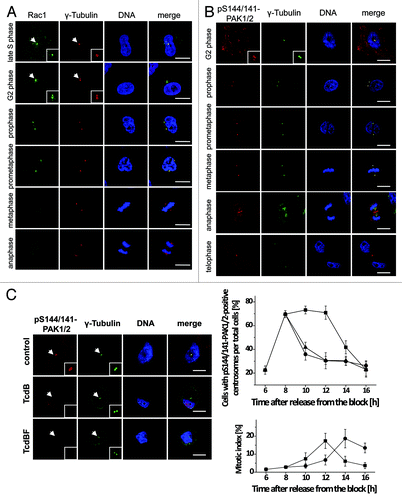
The association of Rac1 with centrosomes was analyzed next in synchronized HeLa cells using immunostaining (). Rac1 associated with the centrosome of HeLa cells beginning in late S phase/early G2 phase (). Rac1 persisted at the centrosomes until prometaphase (). Rac1 was absent from metaphase centrosomes (). At centrosomes, Rac1 clearly co-localized with the centrosomal marker protein γ-tubulin (). Rac1 association with prophase centrosomes was also observed in Rac1fl/fl MEFs (Fig. S1). In corresponding Rac1−/− MEFs, no Rac1 signal was obtained (Fig. S1), excluding non-specific immunostaining. Rac1 thus associates with centrosomes in a cell cycle-dependent manner.
Figure 3. Cell cycle-dependent localization of Rac1 and pS144/141-PAK1/2 at the centrosomes. (A) Synchronized Hela cells were fixed with methanol and stained for Rac1 (green), γ-tubulin (red), and DNA (blue). Scale bar: 15 µm. (B) Synchronized Hela cells were fixed with methanol and stained for pS144/141-PAK1/2 (red), γ-tubulin (green), and DNA (blue). Scale bar: 15 µm. (C) Synchronized HeLa cells were treated with TcdB (30 ng/ml, ●), TcdBF (1 µg/ml, ▲), or buffer (■) 8 h after release from the block. Cells were stained for pS144/141-PAK1/2 (red), γ-tubulin (green), and DNA (blue). The number of cells with pS144/141-PAK1/2-positive centrosomes per total cells and of mitotic per total cells was quantified at the indicated time points by manual counting from 3 independent experiments with >100 cells in each population. Data are mean ± SD.
Rac1-dependent association of PAK1/2 with the G2-phase centrosomes
In HeLa cells, both PAK1 and PAK2 were expressed as analyzed by western blotting (Fig. S2). The activity of PAK1/2 is regulated by phosphorylation at various sites, including positions Ser-144, Ser-199, and Thr-422 in PAK1 and the equivalent positions Ser-141, Ser-192, and Thr-402 in PAK2.Citation9 Thr-422/402 is located in the activation loop of the kinase domain. Phosphorylation at Thr-422/402 results in marked activation of kinase activity. Phosphorylation at Ser-144/141 inhibits a regulatory domain that negatively regulates kinase activity. Ser-199/192 in PAK1/2 is an auto-phosphorylation site. Phosphorylation of either site is indicative of PAK1/2 activation.Citation9 PAK2 has been suggested to be the dominant PAK isoform expressed in HeLa cells.Citation12 This notion was supported by the observation that cellular levels of pS141-PAK2 and pS192-PAK2 were obviously higher than the levels of p144-PAK1 and pS199-PAK1 (Fig. S2). siRNA-mediated knockdown of either PAK1 or PAK2 resulted in efficient protein depletion 48 h after transfection (Fig. S2). Depletion of PAK1 or PAK2 was accompanied by reduced levels of either pS144-PAK1 and pS199-PAK1 or pS141-PAK2 and pS192-PAK2, respectively (Fig S2). In immunocytochemistry, siRNA-mediated knockdown of PAK2 (not of PAK1) resulted in a strong decrease of pS144/141-PAK1/2 staining of HeLa cells (Fig. S3), confirming that PAK2 is the dominant isoform in HeLa cells. Furthermore, these observations confirm the specificity of the phospho-specific antibodies in western blotting and in immunocytochemistry.
PAK2 found at isolated G2-phase centrosomes was active, at least in part, as both pS144/141-PAK1/2 and PAK2 showed positive signals by western blotting (). Immunostaining of synchronized HeLa cells for pS144/141-PAK1/2 () revealed that active PAK1/2 associated with centrosomes beginning in G2 phase and persisted at the spindle poles until anaphase. Later in mitosis, active PAK1/2 localized to the central spindle in telophase and to the midbody during completion of cytokinesis. A comparable profile of cell cycle-dependent PAK localization was obtained upon immunostaining of PAK1/2 using the pT422/402-PAK1/2 antibody (Fig. S4). This observation is in good agreement with published data.Citation15 We also quantified the frequency of association of pS144/141-PAK1/2 to centrosomes and found about 70% of the centrosomes of a synchronized population of G2-phase HeLa cells to be pS144/141-PAK1/2-positive (). In these cells, centrosomes remained PAK-positive until undergoing cell division about 12 h after release from thymidine block ().
For testing whether the centrosomal association of PAK1/2 requires Rac1, we exploited the fact that C. difficile toxin B-catalyzed glucosylation of Rac1 at Thr-35 blocks Rac1–PAK interaction. We employed both the standard Toxin B from the C. difficile strain VPI10463 (TcdB), a broad-spectrum inhibitor of Rho proteins that glucosylates Rho, Rac, and Cdc42 subtype proteins, as well as the variant Toxin B from the C. difficile serotype F strain 1470 (TcdBF).Citation16,Citation17 TcdBF specifically glucosylates and inactivates Rac1 but not RhoA or Cdc42, as confirmed by a pulldown assay using Rhotekin for RhoA or PAK-CRIB for Rac1 and Cdc42 precipitation (Fig. S5).
Evidence for glucosylation of centrosome-associated Rac1 was provided by using the glucosylation-sensitive antibody Rac1-mAb(102) ().Citation18 The apparent loss of Rac1 detection reflected Rac1 glucosylation (not Rac1 degradation), as the alternative (glucosylation-insensitive) Rac1-mAb(23A8) confirmed the presence of Rac1 at G2-phase centrosomes (). Glucosylated Rac1 thus stayed associated with the G2-phase centrosomes. Upon Rac1 glucosylation, PAK1/2 were absent from centrosomes, as analyzed by western blotting of isolated centrosomes using PAK2 and pS144/141-PAK1/2 antibodies (). In contrast, Aurora A and Plk1 stayed associated with G2-phase centrosomes, irrespective of Rac1 glucosylation (), indicating that their centrosomal association was independent of Rac1. Rac1 glucosylation by either TcdB or TcdBF also resulted in a loss of pS144/S141-PAK1/2 from centrosomes of synchronized HeLa cells, as visualized by immunostaining (). The number of pS141/S144-PAK1/2-positive centrosomes strongly decreased upon toxin-catalyzed Rac1 glucosylation in G2-phase/early mitosis at 10–12 h after release from the block (). Together, these results show that Rac1 glucosylation induces the dissociation of PAK1/2 from centrosomes in G2-phase/early mitosis.
Delayed mitotic entry in HeLa cells upon Rac1 glucosylation and PAK depletion
Next, we asked whether Rac1 glucosylation and subsequent PAK dissociation from G2-phase centrosomes blocks mitotic entry. Mitotic entry was analyzed in synchronized HeLa cells using mitotic marker proteins.Citation13,Citation15 The mitotic kinase Aurora A is activated by PAK1/2 through phosphorylation at Thr-288. The levels of pT288-Aurora A and its downstream target, the mitotic phosphatase Cdc25, peaked at mitosis entry at 10–12 h (). Cdc25, in turn, dephosphorylates Cdk1 at the inhibitory phosphorylation site Thr-15 and thereby activates the CyclinB–Cdk1 complex.Citation6 Decreasing cellular levels of pY15-Cdk1 were observed 10 to 12 h after release from early S phase (). Furthermore, phosphorylation of Histone H3 at Ser-10, which is indicative of chromatin condensation, also peaked 10 to 12 h after release from early S phase. These observations showed that the cells entered mitosis 10–12 h after release from the block in early S phase. Progression through mitosis resulted in decreasing levels of CyclinB1 12–14 h after release, due to proteasomal degradation (). These observations are consistent with our observations from FACS analysis (), showing that synchronized HeLa cells undergo cell division at about 12 h after release from the thymidine block, as evidenced by the re-abundance of the 2N peak on FACS analysis of propidium iodide-stained cells.
Figure 4. Delayed mitotic entry upon inhibition of Rac1 and PAK1/2. (A) Synchronized HeLa cells were treated with TcdBF (1000 ng/ml) in G2 phase 8 h after release from the second block. Cell lysates were collected at the indicated time points and were analyzed by western blot analysisusing cell cycle marker proteins. β-actin was used as loading control and Rac1 inactivation by glucosylation as well as PAK phosphorylation, indicative for PAK activity, was determined. (B) Activation of the CyclinB/Cdk1 complex was determined by an in vitro kinase assay. The CyclinB–Cdk1 complex was immunoprecipitated by anti-CyclinB1 antibody and incubated with [γ-32P] ATP in the presence of histone H1, as a Cdk1 substrate. Phosphorylation of histone H1 was determined by autoradiography. (C) Synchronized HeLa cells were transfected with siRNA targeting PAK2 (siPAK2a; siPAK2b), or with scrambled control siRNA (scr). Forty-eight h after transfection, mitotic entry was analyzed in synchronized HeLa cells by western blotting using cell cycle marker proteins. (D) Synchronized Rac1fl/fl MEFs were treated with TcdBF (30 ng/ml) in G2 phase 3 h after release from the second block. Cell lysates were collected at the indicated time points and were analyzed by western blot analysis using cell cycle marker proteins. β-actin was used as loading control and Rac1 inactivation by glucosylation as well as PAK phosphorylation, indicative for PAK activity, was determined. (E) Rac1fl/fl and Rac1−/− MEFs were synchronized by the thymidine double-block technique and analyzed for cell cycle progression by western blot analysis of cell cycle marker proteins.
![Figure 4. Delayed mitotic entry upon inhibition of Rac1 and PAK1/2. (A) Synchronized HeLa cells were treated with TcdBF (1000 ng/ml) in G2 phase 8 h after release from the second block. Cell lysates were collected at the indicated time points and were analyzed by western blot analysisusing cell cycle marker proteins. β-actin was used as loading control and Rac1 inactivation by glucosylation as well as PAK phosphorylation, indicative for PAK activity, was determined. (B) Activation of the CyclinB/Cdk1 complex was determined by an in vitro kinase assay. The CyclinB–Cdk1 complex was immunoprecipitated by anti-CyclinB1 antibody and incubated with [γ-32P] ATP in the presence of histone H1, as a Cdk1 substrate. Phosphorylation of histone H1 was determined by autoradiography. (C) Synchronized HeLa cells were transfected with siRNA targeting PAK2 (siPAK2a; siPAK2b), or with scrambled control siRNA (scr). Forty-eight h after transfection, mitotic entry was analyzed in synchronized HeLa cells by western blotting using cell cycle marker proteins. (D) Synchronized Rac1fl/fl MEFs were treated with TcdBF (30 ng/ml) in G2 phase 3 h after release from the second block. Cell lysates were collected at the indicated time points and were analyzed by western blot analysis using cell cycle marker proteins. β-actin was used as loading control and Rac1 inactivation by glucosylation as well as PAK phosphorylation, indicative for PAK activity, was determined. (E) Rac1fl/fl and Rac1−/− MEFs were synchronized by the thymidine double-block technique and analyzed for cell cycle progression by western blot analysis of cell cycle marker proteins.](/cms/asset/322a1606-8a44-46ab-a5ff-3c1c190490a8/kccy_a_10929279_f0004.gif)
Synchronized HeLa cells were treated with TcdBF in G2 phase 8 h after release from the S-phase block. Rac1 was glucosylated within 2 h, as evidenced by the loss of Rac1 detection using the glucosylation-sensitive antibody Rac1-mAb(102)().Citation18 The loss of Rac1 detection upon TcdBF treatment was due to glucosylation and not due to Rac1 degradation, as the cellular level of Rac1 was unchanged if the alternative Rac1-mAb(23A8) was applied (). Rac1 glucosylation by TcdBF resulted in decreased levels of pS199/S192-PAK1/2 and pS144/S141-PAK1/2 (), indicating PAK1/2 inactivation. The cellular levels of PAK1 and PAK2 were constant during G2–M transition and did not change upon TcdBF treatment (). Rac1 glucosylation by TcdBF and subsequent PAK1/2 inactivation prevented Aurora A phosphorylation at Thr-288 in late G2 phase (10 h after release from the S-phase block) (). Instead, the level of pT288-Aurora A increased at later time points and peaked at 14 h after release with a delay of about 2–4 h as compared with non-treated cells (). Rac1 glucosylation prevented Cdk1 dephosphorylation (thus activation) in late G2 phase (10 h after release from the S-phase block) (). Instead, Cdk1 activity peaked at 12–14 h in TcdBF-treated cells, as analyzed by an in vitro kinase assay using histone H1 as a Cdk1 substrate (). Furthermore, pS10-Histone H3 peaked at 12–14 h in TcdBF-treated cells and was also delayed for about 2 h as compared with non-treated cells (). Finally, mitotic degradation of Aurora A, Cdc25, and CyclinB1 did not occur until 16 h after release from the S-phase block and was delayed for at least 2 h (). Hence, Rac1 glucosylation by TcdBF blocked activation of PAK1/2, Aurora A, and the CyclinB/Cdk1 complex in late G2-phase and resulted in delayed mitotic entry. Comparable observations were obtained if synchronized HeLa cells were treated with TcdB, the broad-spectrum inhibitor of Rho, Rac, and Cdc42 subtype GTPases. Glucosylation of Rho GTPases by TcdB results in blocked activation of PAK1/2, Aurora A, and the CyclinB/Cdk1 complex in late G2 phase (10 h after release) (Fig. S6) and delayed activation of Cdk1 (Fig. S7), corroborating published data.Citation15 Treatment with TcdBF or TcdB thus both resulted in a 2-h delay of mitotic entry. Additional inactivation of RhoA and Cdc42 (as in TcdB-treated cells) did not further delay mitotic entry, which correlates with the fact that RhoA and Cdc42 were not present at G2-phase centrosomes () and that RhoA and Cdc42 seemed not to be involved in initial activation of Aurora A and CyclinB/Cdk1.
Blocked activation of Aurora A and the CyclinB/Cdk1 complex in late G2 phase and delayed mitotic entry were also observed in synchronized HeLa cells upon siRNA-mediated depletion of PAK2. The levels of pS10-Histon H3 peaked at 10–12 h in cells transfected with scrambled siRNA comparable to untreated HeLa cells (). Upon PAK2 knockdown, the levels of pS10-Histon-3 were strongly reduced at the time point 10 h and peaked at 12–14 h (). Knockdown of PAK2 () strongly reduced the level of pT288-Aurora A at 10–12 h. The level of pT288-Aurora A increased at later time point and peaked at 14 h after release (). PAK2 knockdown further resulted in reduced Cdk1 dephosphorylation and CyclinB1 degradation at the time point 10 h (). Cdk1 dephosphorylation and CyclinB1 degradation was not observed until 14 h after release, indicating delayed mitotic entry upon PAK2 knockdown.
The effects of TcdBF on G2–M progression were further analyzed exploiting FACS analysis of propidium iodide-stained cells. Synchronized HeLa cells underwent cell division at about 12 h after release from the thymidine block, as evidenced by the re-abundance of the 2N peak (). In TcdBF-treated cells, the re-abundance of the 2N peak was clearly delayed, indicating delayed G2–M progression (). Rac1 glucosylation by either TcdBF or TcdB thus results in blocked activation of Aurora A and the CyclinB/CDk1 complex in late G2 phase/prophase and in delayed progression through G2–M phase.
Delayed mitotic entry in Rac1−/− MEFs
To further provide evidence on the role of Rac1 and PAK as important upstream regulators of the mitotic entry-regulation signaling cascade, cell cycle progression was further analyzed in SV40 LT-antigen-immortalized Rac1fl/fl MEFs.Citation19 Rac1fl/fl MEFs exhibited a doubling time of about 12 h. Compared with HeLa cells, synchronized Rac1fl/fl MEFs more rapidly progressed through S and G2–M phase. Rac1fl/fl MEFs underwent cell division 4–7 h after release from the thymidine block (), as evidenced by decreasing levels of pY15-Cdk1 (starting from 6 h after release) and the level of pS10-Histone H3 that peaked 4–7 h after release from the pre-S-phase block (). TcdBF treatment of G2-phase Rac1fl/fl MEFs (3 h after release from the block) resulted in Rac1 glucosylation, as detected in terms of decreasing detection of Rac1 using the glucosylation-sensitive antibody Rac1(mAb102) within 2 h (). Rac1 glucosylation was reflected by decreasing levels of pS144/S141-PAK1/2, indicating PAK1/2 inactivation (). The cellular levels of either Rac1 or PAK2 were not affected upon TcdBF treatment (). In TcdBF-treated Rac1fl/fl MEFs, the level of pS10-Histone H3 peaked 7–9 h after release (compared with 5–7 h in non-treated cells) (). Furthermore, dephosphorylation of pY15-Cdk1 and CyclinB1 degradation were not observed within the analyzed time frame (), further indicating delayed G2–M transition.
As opposed to the parental control clone (Rac1fl/fl) and shown previously,Citation19 Rac1−/− MEFs clone 3 did not express Rac1 (). However, in Rac1fl/fl and Rac1−/− MEFs, expression levels of PAK2 were comparable, suggesting that PAK2 expression does not depend on the presence of Rac1 (). In synchronized Rac1−/− MEFs, the level of pS10-Histone H3 did not peak until 8 h after release from the block (), showing that G2–M transition in Rac1−/− MEFs was delayed for about 2 h. Furthermore, decreasing levels of pY15-Cdk1 were not observed within the analyzed time frame (up-to 8 h) (), further indicating delayed G2–M transition. Rac1 inhibition by either toxin-catalyzed glucosylation or genetic depletion resulted in delayed mitotic entry in SV40-transformed MEFs. Interestingly, the cellular level of pS144/S141-PAK1/2 (i.e., active PAK1/2) was comparable in Rac1−/− and Rac1fl/fl MEFs (), suggesting that PAK1/2 was activated by Rac1-independent mechanisms in Rac1−/− MEFs. Rac1-independent activation of PAK1/2, however, seemed not to be sufficient for efficient activation of the CyclinB/Cdk1 complex in late G2 phase/prophase.
Next, synchronized Rac1fl/fl MEFs were stained for pS144/141-PAK1/2 to check if active PAK1/2 was present at the G2-phase centrosomes. Thirty percent of the centrosomes of Rac1fl/fl MEFs were pS144/141-PAK1/2-positive in late G2 phase (4 h after release from the block) (). The centrosomes remained pS144/141-PAK1/2-positive until cells underwent cell division 6–8 h after release from the block (). Evaluation of the mitotic index of Rac1fl/fl MEFs showed that the number of mitotic cells peaked at 6 h after release from the block (). In Rac1−/− MEFs, pS144/141-PAK1/2 was almost absent from G2-phase centrosomes (). In Rac1−/− MEFs, the mitotic index did not peak until 8 h after release from the block and was thus delayed by about 2 h as compared with Rac1fl/fl MEFs (). The latter observations corroborate above observations on delayed mitotic entry, as analyzed exploiting mitotic marker proteins ().
Figure 5. Absence of pS144/141-PAK1/2 from the G2-phase centrosomes of Rac1−/− MEFs. Synchronized Rac1fl/fl and Rac1−/− MEFs and Cdc42fl/- and Cdc42−/− MEFs were synchronized using thymidine double-block technique, fixed at times as indicated and stained for pS144/141-PAK1/2 (red), γ-tubulin (green), and DNA (blue). Scale bar: 15 µm. The number of in Rac1fl/fl (filled symbols) and Rac1−/− (open symbols) MEFs with pS144/141-PAK1/2-positive centrosomes (■) and the mitotic index (▲) were determined by manual counting. Data are mean ± SD from 3 independent experiments with >100 cells in each population.
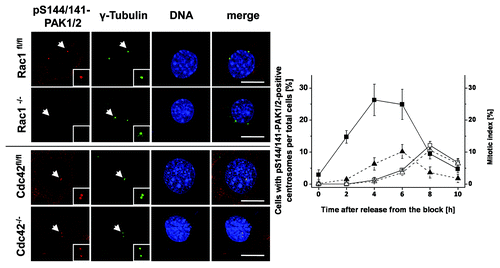
To investigate a possible role of Cdc42 in the regulation of mitotic entry, cell cycle progression was further analyzed in SV40 LT-antigen-immortalized Cdc42−/− and their parental control line Cdc42fl/- Citation20. The level of pS10-Histone H3 peaked in both control and Cdc42-knockout MEFs 5–7 h after release from the S-phase block (Fig. S8). Furthermore, dephosphorylation of pY15-Cdk1 and CyclinB1 degradation started 5–7 h after release from the block in both cell lines (Fig. S8). G2–M progression of Cdc42fl/− and Cdc42−/− MEFs was thus comparable, likely excluding a significant role of Cdc42 in the regulation of mitotic entry. Furthermore, G2-phase centrosomes of Cdc42fl/− and Cdc42−/− MEFs stained positive for pS144/S141-PAK1/2 (), excluding a role for Cdc42 in PAK recruitment to G2-phase centrosomes. Taken together, these data suggest that the recruitment of PAK1/2 to the G2-phase centrosomes specifically requires Rac1. Rac1 inhibition by either toxin-catalyzed glucosylation or genetic depletion blocks PAK1/2 recruitment to G2-phase centrosomes and results in blocked activation of Aurora A and the CyclinD/Cdk1 complex in late G2-phase/prophase and delayed mitotic entry.
Discussion
This study shows for the first time that Rac1, but not RhoA or Cdc42, is present at isolated G2-phase centrosomes (). Cell cycle-dependent analysis by immunocytochemistry reveals that Rac1 associates with the centrosomes from late S phase on up until early mitosis (). These observations partly corroborate a recent report describing association of EGFP-tagged Rac1 with the centrosomes of MDCK cells in prophase.Citation21
Rac1 associates with G2-phase centrosomes in its non-glucosylated () as well as glucosylated form (). Glucosylation of Rho/Ras-GTPases at Thr-35 within switch I prevents binding of Rac1 to all those proteins the interaction of which depends on this region, including effector proteins as well as GAP and GDI proteins.Citation22-Citation24 However, glucosylated Rho/Ras-GTPases still interact with their GEF proteins,Citation22 which is plausible, as initial GEF interaction involves strands β2, β3, and portions of switch II (rather than switch I) of Rho/Ras-GTPases.Citation25 RacGEF proteins thus appear to be plausible candidates as centrosomal binding proteins of glucosylated and non-glucosylated Rac1. In particular, the RacGEF β-PIX could be detected here at isolated G2-phase centrosomes ().
PAK2 associates with the centrosomes from G2 phase up to metaphase, as analyzed by western blotting of isolated G2-phase centrosomes () and by immunocytochemistry using 2 distinct phospho-specific antibodies (; Fig. S4). The immunolabeling data are fully consistent with reports from several other groups.Citation11,Citation12,Citation15,Citation26 The observations of this study provide several lines of evidence for a crucial role of Rac1 in the recruitment of PAK1/2 to G2-phase centrosomes: (1) Rac1 and PAK1/2 co-localize at the G2-phase centrosomes (); (2) Rac glucosylation results in PAK dissociation from G2-phase centrosomes (); and (3) PAK recruitment to G2-phase centrosomes is prevented in Rac1−/− MEFs (). Rac1, likely activated by β-PIX, thus recruits PAK to the G2-phase centrosomes. Mutant PAK1-S76P (devoid of Rho protein binding) fused to the centrosome-targeting sequences of GIT1 or AKAP450 was previously reported to be activated at centrosomes.Citation12 This led to the conclusion that PAK1/2 activation does not require Rac1 once recruited to the centrosomes. PAK may be activated by autophosphorylation or by a PAK upstream kinase, such as PDK1, that is also present at G2-phase centrosomes (). However, based on our results, we conclude that Rac1 accumulation at centrosomes constitutes a prerequisite for efficient recruitment and activation of PAK1/2 at the G2-phase centrosomes.
Upon activation at centrosomes, PAK has been shown to dissociate from the GIT/PIX complex and to subsequently bind to and phosphorylate (and thereby activate) Aurora ACitation12,Citation15 and Plk1.Citation11 Aurora A and Plk1 subsequently facilitate Cdk1 activation in mitotic entry through activation of the phosphatase Cdc25.Citation6 siRNA-mediated depletion of PAK2 (representing the predominant PAK isoform in HeLa cells [Fig. S2]) inhibited activation of Aurora A and the CyclinB/Cdk1 complex in G2 phase and delayed G2–M transition (). These observations are consistent with the finding that inhibition of PAK by ectopic expression of the kinase-inhibitory domain (KID) delays progression in G2–M phase.Citation11
A role for a specific Rho-GTPases in the regulation of mitotic entry has not yet been established. Inactivation of Rho-GTPases by TcdB, a broad-spectrum inhibitor of Rho-GTPases, has formerly been shown to delay mitotic entry of synchronized Hela cells.Citation12,Citation15 In this study, Rac1 has been identified as an upstream regulator of mitotic entry, as Rac1 inactivation by either toxin-catalyzed glucosylation () or genetic deletion () inhibits activation of Aurora A and the CyclinB/Cdk1 complex in G2 phase. A significant role for Cdc42 could be excluded based on the observation that its deletion does neither affect the activation of CyclinB/Cdk1 complex in G2 phase nor mitotic entry (Fig. S8). Thus, in terms of the regulation of mitotic entry, PAK1/2 is specifically regulated by Rac1 (not by Cdc42). Besides PAK, the regulation of mitotic entry by Rac1 may involve further effector proteins, which need to be established in future investigations. An appropriate candidate is the Rho/Rac effector protein PRK2/PKN2, which is present at isolated G2-phase centrosomes () and has also been shown to contribute to the regulation of mitotic entry.Citation13
Finally, inhibition of Rac1/PAK signaling by various approaches delays mitotic entry but does not result in a G2–M arrest. After the delay, G2–M progression occurred apparently normally, and particularly Rac1−/− MEFs progressed through mitosis. A Rac1/PAK-dependent pathway apparently initiates activation of Aurora A/Plk1 and, subsequently, the CyclinB/Cdk1 complex under usual culture conditions. When it fails, other, Rac1-independent pathways leading to the initial activation of Aurora A and Plk1 at G2-phase centrosomes seems to be activated to compensate for the loss of Rac1. This view is supported by the observation that Aurora A and Plk1 are present at G2-phase centrosomes of cells with Rho-GTPases being inactivated ().
The results provided here support the concept that the G2-phase centrosomes provide a scaffold for components of mitotic entry-regulating signaling pathways, including the interaction of Rac1 and PAK and subsequent interaction of PAK with Aurora A/Plk1. PAK2 is specifically recruited to centrosomes by Rac1, with Rac1 likely being activated by β-PIX. The GIT/β-PIX/PAK/Rac1/Cdc42 complex is well-characterized and established in the regulation of focal adhesion (FA) turnover.Citation27 Translocation of focal adhesion proteins to centrosomes has been hypothesized to provide a link between cell cycle progression and cytoskeletal reorganization during G2–M phase.Citation28,Citation29 Rac1 and PAK appear to represent new players in linking cell cycle and cytoskeletal signaling.
Materials and Methods
Reagents and antibodies
DAPI (40.6-diamidino-2-phenylindole) was from Serva (EDTA, rhodamine-conjugated phalloidin, GTP[γS], RNase, and propidium iodide were from Sigma). Nocodazole, cytochalasin D, histone H1, were from Calbiochem; A/G-agarose beads were from Santa Cruz. GST-sepharose and [γ-32P]ATP were from Amersham. Protease inhibitor Complete and phosphatase inhibitor Phosstop were from Roche. Transfection reagent jetPRIME was from PeqLab. Super West Signal Femto was from Pierce. Methanol and paraformaldehyde were from J.T. Baker. Fetal calf serum (FCS) was from PAN. Penicillin/Streptomycin was from PAA Laboratories. Non-essential amino acids and cell culture medium were from Gibco/Invitrogene. Sodium pyruvate was from Biochrom. Toxin B from C. difficile strain VPI10463 (TcdB) and the variant Toxin B serotyp F from C. difficile strain 1470 (TcdBF) were purified as described.Citation16
Anti-Aurora A (#4718), anti-pT288-Aurora A (#3079), anti-PAK1 (#2602), anti-PAK2 (#2608), anti-phospho-T423/402-PAK1/2 (#2601), anti-phospho-S199/S192-PAK1/2 (#2605), anti-phospho-T15-Cdk1 (#9111), anti-PDK1 (#3062), anti-Cdk1 (#9112), anti-Histone H3 (#9715) were from Cell Signaling Technology. Anti-phospho-S10-histone H3 (3H10, 05-806), anti-CyclinB1 (05-373), anti-β-PIX (07-1450), and anti-Rac1 (23A8) were from Millipore. Anti-phospho-S144/141-PAK1/2 (EP656Y, ab40795), anti-γ-tubulin (GTU-88, ab11316), and anti-γ-tubulin (ab11320) were from Abcam. Anti-α-tubulin (YL 1/2) was from Linaris. Anti-RhoGDI (K-21), anti-Cdc25B (C-20), anti-RhoA (26C4), anti-Rac1 (23A8), anti-Lamin B (M-20), and anti-CyclinB1 were from Santa Cruz. Anti-Rac1 (102) and anti-Cdc42 (610929) were from BD Transduction Laboratories. Anti-β-actin (AC-40) was from Sigma. Horseradish peroxidase-conjugated secondary antibodies were from Rockland. Anti-rat IgG Alexa Fluor 488 goat secondary antibody, anti-mouse IgG Alexa Fluor 488 goat secondary antibody and anti-rabbit IgG Alexa Fluor 555 goat secondary antibody were from Invitrogen. siRNAs were from Dharmacon/ Thermo Fisher Scientific: PAK1a (D-003521-05; CAACAAAGAA CAAUCACUA); PAK1b (D-003521-07; AGAAAUACCA GCACUAUGA; PAK2a (D-003597-05; AGAAGGAACU GAUCAUUAA); PAK2b (D-003597-07; GAGCAGAGCA AACGCAGUA).
Cell culture and synchronization
SV40 LT-antigen-immortalized Rac1−/− (clone 3) and Rac1fl/fl Citation19 and LT-antigen-immortalized Cdc42−/− (clone 3–9-9) and Cdc42fl/− (clone 3–9) mouse embryonic fibroblastsCitation20 were cultivated in Dulbecco modified Essential medium supplemented with 10% FCS, 100 µM penicillin, 100 U/ml streptomycin, non-essential amino acids, and 1 mM sodium pyruvate at 37 °C and 5% CO2. HeLa cells were cultivated in Dulbecco modified Essential medium (Biochrom) supplemented with 10% FCS, 100 µM penicillin, 100 U/ml streptomycin and 1 mM sodium pyruvate at 37 °C and 5% CO2. Upon confluence, cells were passaged. Synchronisation of the cell cycle was performed by the thymidine double-block technique. Exponentially growing HeLa cells were exposed to 2 mM 20-deoxy-thymidine in full-growth medium at 37 °C and 5% CO2 for 16 h. The medium was then removed and replaced by full-growth medium without thymidine. After 9 h incubation, the medium was again replaced by full-growth medium containing thymidine. Cells were incubated for 16 h. Subsequently, the medium was again replaced by thymidine-free full-growth medium and released into the cell cycle. Synchronization was confirmed using FACS analysis. MEFs were exposed to 2 mM 20-deoxy-thymidine and the medium was replaced for 7 h with full-growth medium without thymidine between both thymidine blocks.
Determination of DNA content by flow cytometry
Cells were seeded onto a 30 mm dish and synchronized using the thymidine double-block technique. At the indicated time points, cells were harvested by trypsinization, washed in phosphate-buffered saline, and fixed in 70% ethanol at −20 °C for 30 min. The cells were then centrifuged at 1000 rpm for 5 min. RNA was removed by incubation with 30 µg/ml RNase for 1 h at room temperature. Subsequently, cells were stained with propidium iodide (50 µg/ml). Cells were analyzed using a FACScan flow cytometer (Becton Dickinson) and CellQuest Pro software (Becton Dickinson). Quantitative analysis was performed using WinMDI 2.8.
Western blot analysis
Cells were washed with PBS and were scraped in Laemmli lysis buffer containing SDS and DTT supplemented with 5 mM NaF and 1 mM Na3VO4. Cells were lysed by sonification and proteins denaturated at 95 °C for 10 min. Proteins of the whole-cell lysate were separated by SDS-PAGE and transferred onto nitrocellulose membranes. The membranes were blocked with 5% non-fat dried milk for 60 min. Subsequently, the membrane was incubated with primary antibody at 4 °C overnight and secondary antibody conjugated with horseradish peroxidase at room temperature for 1 h. Blots were analyzed by chemiluminescence reaction of ECL Femto using Kodak Image Station 440 CF. Depicted blots show representative results from one of 3 independent experiments.
Pull-down assay of Rho GTPases
Intracellular Rho GTPase inactivation by glucosylation was determined by pull-down assay of RhoA, Rac1 and Cdc42 using GST-RBD (Rho binding domain of Rhotekin) and GST-PAK-CRIB (p21-binding domain of Pak1). The RBD domain, encoding the N-terminal 90 amino acids of Rhotekin, or the PAK-CRIP domain, encoding amino acids 56–272 of PAK1, were expressed as GST fusion proteins in E. coli and immobilized on glutathione sepharose. Cells were scraped in binding buffer (50 mM Tris pH 7.4, 150 mM NaCl, 5 mM MgCl2, 5 mM DTT, 1 mM PMSF, protease inhibitor Complete [Roche], 1% NP-40) at 4 °C and lysed by sonification. The lysate was centrifuged at 13200 × g for 10 min. The supernatant was incubated with 10 mM EDTA and 100 µM GTP[γS] at 30 °C for 15 min to exchange the nucleotide. The complex of GTPase and GTP[γS] was stabilized by the addition of 60 mM MgCl2. The samples were incubated for 60 min with immobilized GST-RBD (RhoA) and GST-PAK-CRIB (Rac1/Cdc42), respectively, at 4 °C. Beads were washed with binding buffer. Bound Rho GTPases were eluted by incubation in Laemmli lysis buffer at 95 °C for 10 min and subjected to SDS–PAGE western blot analysis.
Centrosome isolation
Centrosome isolation was based on a method by Moudjou and Bornens.Citation14 5 × 107 HeLa cells were incubated with 1 µM nocodazole and 1 µg/ml cytochalasin D at 37 °C for 1 h. Cells were washed with PBS and 0.1 × PBS and rinsed with hypotonic lysis buffer (1 mM TRIS-HCl at pH 7.4, 0.5 mM MgCl2, 0.4 mM DTT, Protease inhibitor Complete [Roche], phosphatase inhibitor phosstop [Roche], 0.5% NP-40) on ice with agitation for 5 min. The cell lysate was centrifuged at 2000× g for 10 min to remove cell debris and nuclei. The supernatant was adjusted with 50× PIPES buffer (500 mM PIPES at pH 7.2). Two units/ml DNase was added and the lysate incubated on ice for 30 min. The lysate was then underlaid with 60% sucrose solution (60% sucrose in 10 mM PIPES, pH 7.2, 0.1%, Triton X-100, 0.4 mM DTT), and centrosomes were sedimented into the sucrose cushion by centrifugation at 10 000 × g at 4 °C for 30 min. 1.5 ml of the crude centrosome preparation were layered onto a discontinuous sucrose gradient, consisting of 750 µl of 70% sucrose, 450 µl of 50% sucrose, and 450 µl of 40% sucrose solution (10 mM PIPES at pH 7.2, 0.1% Triton X-100 and 0.4 mM DTT) and centrifuged at 120 000 × g at 4 °C for 1 h using a Beckman Optima Max ultracentrifuge and a Beckman MLS-50 swinging bucket rotor. Fractions were collected from the bottom, 200 µl per fraction from fractions 1–9; 450 µl per fraction from fractions 10–11. Centrosomes were recovered in 1 ml of 10 mM PIPES buffer, pH 7.2, and sedimented at 16 000 × g for 10 min in a microfuge. The centrosomes were denaturated in Laemmli lysis buffer at 95 °C for 10 min. Samples were subjected to SDS-PAGE and western blot analysis.
CyclinB–Cdk1 kinase assay
1 × 107 HeLa cells were seeded onto a 100-mm dish, synchronized using the thymidine double block and treated as indicated. HeLa cells were scraped in a lysis buffer containing 50 mM TRIS-HCl (pH 7.5), 250 mM NaCl, 1 mM EDTA, 1 mM EGTA, 0.1% NP40, 0.8 mM DTT, 50 mM β-glycerol-phosphate, 25 mM α-naphthyl-phosphate, 1 mM Na3VO4, 1 mM NaF, 100 µM PMSF, Complete protease inhibitor (Roche) and sonicated (3×, 5 s). Cell lysates were centrifuged at 13 000 × g at 4 °C for 10 min. The supernatants were pre-cleared with 20 µl protein A/G- conjugated agarose at 4 °C for 1 h. The supernatants were then incubated with primary antibody to CyclinB1 at 1:50 dilution for 3 h and with 20 µl protein A/G-conjugated agarose at 4 °C for 1 h, subsequently. Immunoprecipitated samples were washed and recovered in a reaction buffer containing 50 mM TRIS-HCl (pH 7.5), 12 mM MgCl2, 0.8 mM DTT, 50 mM β-glycerol-phosphate, 25 mM α-naphthyl-phosphate, 1 mM Na3VO4. The samples were incubated with 10 µg of histone H1 and [γ-32P] ATP (1 µCi) in reaction buffer supplemented with 100 µM ATP in a total volume of 40 µl at 30 °C for 30 min. Reactions were terminated by the addition of 10 µl 5 × Laemmli lysis buffer. The samples were incubated for 5 min at 95 °C, subjected to SDS-PAGE and analyzed by autoradiography using Packard Phosphoimager Cyclone.
RNA interference
siRNA-mediated knockdown of PAK was conducted in HeLa cells using jetPRIME transfection reagent. HeLa cells were seeded at a density of 2.5 × 105 overnight and were transfected with 20 pmol siRNAs using 2 µl jetPRIME according to manufacturer’s protocol. The medium was changed 24 h after transfection, and cells were analyzed 48 h after transfection. siRNA-mediated knockdown was determined by western blot and immunofluorescence.
Immunocytochemistry
For immunocytochemistry, cells were seeded at a density of 3 × 105 on coverslips and treated as indicated. Cells were fixed either with ice-cold methanol for 20 min at −20 °C or with 3.7% formaldehyde for 10 min at room temperature. Fixation for Rac1 staining was conducted using 3.7% formaldehyde/0.5% Triton in PBS at 37 °C for 7.5 min. Coverslips were washed 2 times with PBS and cells were subsequently incubated in 3.7% formaldehyde in PBS at room temperature for an additional 7.5 min. Samples were blocked with 5% BSA in PBS at room temperature for 1 h. Samples were incubated with primary antibodies diluted in 5% BSA in PBS for 1 h at room temperature. Primary antibodies were applied in 1:100 dilution for antibodies to Rac1 (23A8) and γ-tubulin (GTU-88); 1:200 dilutions for antibodies to pT422/402-PAK1/2, pS144/141-PAK1/2; 1:500 dilution to γ-tubulin (ab11320). Subsequently, cells were incubated with Alexa Fluor 488, 555, 594 conjugated secondary antibodies diluted to 1:1000 in PBS and DAPI (1 µg/ml) for 1 h at room temperature. Coverslips were embedded on slides using prolong Antifade (Invitrogen) and analyzed by fluorescence microscopy using Leica confocal microscope Inverted-2. Depicted pictures were representative images.
Additional material
Download Zip (1.5 MB)Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Funding
This work is supported by Deutsche Forschungsgemeinschaft Priority program 1150 to H.G. and K.R.
Related Research Data
Reference
- Gavet O, Pines J. Activation of cyclin B1-Cdk1 synchronizes events in the nucleus and the cytoplasm at mitosis. J Cell Biol 2010; 189:247 - 59; http://dx.doi.org/10.1083/jcb.200909144; PMID: 20404109
- Santos SD, Wollman R, Meyer T, Ferrell JE Jr.. Spatial positive feedback at the onset of mitosis. Cell 2012; 149:1500 - 13; http://dx.doi.org/10.1016/j.cell.2012.05.028; PMID: 22726437
- Jackman M, Lindon C, Nigg EA, Pines J. Active cyclin B1-Cdk1 first appears on centrosomes in prophase. Nat Cell Biol 2003; 5:143 - 8; http://dx.doi.org/10.1038/ncb918; PMID: 12524548
- Gavet O, Pines J. Progressive activation of CyclinB1-Cdk1 coordinates entry to mitosis. Dev Cell 2010; 18:533 - 43; http://dx.doi.org/10.1016/j.devcel.2010.02.013; PMID: 20412769
- Doxsey S, Zimmerman W, Mikule K. Centrosome control of the cell cycle. Trends Cell Biol 2005; 15:303 - 11; http://dx.doi.org/10.1016/j.tcb.2005.04.008; PMID: 15953548
- Lindqvist A, Rodríguez-Bravo V, Medema RH. The decision to enter mitosis: feedback and redundancy in the mitotic entry network. J Cell Biol 2009; 185:193 - 202; http://dx.doi.org/10.1083/jcb.200812045; PMID: 19364923
- Macůrek L, Lindqvist A, Lim D, Lampson MA, Klompmaker R, Freire R, Clouin C, Taylor SS, Yaffe MB, Medema RH. Polo-like kinase-1 is activated by aurora A to promote checkpoint recovery. Nature 2008; 455:119 - 23; http://dx.doi.org/10.1038/nature07185; PMID: 18615013
- Seki A, Coppinger JA, Jang CY, Yates JR, Fang G. Bora and the kinase Aurora a cooperatively activate the kinase Plk1 and control mitotic entry. Science 2008; 320:1655 - 8; http://dx.doi.org/10.1126/science.1157425; PMID: 18566290
- Bokoch GM. Biology of the p21-activated kinases. Annu Rev Biochem 2003; 72:743 - 81; http://dx.doi.org/10.1146/annurev.biochem.72.121801.161742; PMID: 12676796
- Dummler B, Ohshiro K, Kumar R, Field J. Pak protein kinases and their role in cancer. Cancer Metastasis Rev 2009; 28:51 - 63; http://dx.doi.org/10.1007/s10555-008-9168-1; PMID: 19165420
- Maroto B, Ye MB, von Lohneysen K, Schnelzer A, Knaus UG. P21-activated kinase is required for mitotic progression and regulates Plk1. Oncogene 2008; 27:4900 - 8; http://dx.doi.org/10.1038/onc.2008.131; PMID: 18427546
- Zhao ZS, Lim JP, Ng YW, Lim L, Manser E. The GIT-associated kinase PAK targets to the centrosome and regulates Aurora-A. Mol Cell 2005; 20:237 - 49; http://dx.doi.org/10.1016/j.molcel.2005.08.035; PMID: 16246726
- Schmidt A, Durgan J, Magalhaes A, Hall A. Rho GTPases regulate PRK2/PKN2 to control entry into mitosis and exit from cytokinesis. EMBO J 2007; 26:1624 - 36; http://dx.doi.org/10.1038/sj.emboj.7601637; PMID: 17332740
- Bornens M, Moudjou M. Studying the composition and function of centrosomes in vertebrates. Methods Cell Biol 1999; 61:13 - 34; http://dx.doi.org/10.1016/S0091-679X(08)61973-1; PMID: 9891307
- Ando Y, Yasuda S, Oceguera-Yanez F, Narumiya S. Inactivation of Rho GTPases with Clostridium difficile toxin B impairs centrosomal activation of Aurora-A in G2/M transition of HeLa cells. Mol Biol Cell 2007; 18:3752 - 63; http://dx.doi.org/10.1091/mbc.E07-03-0281; PMID: 17634283
- Huelsenbeck J, Dreger S, Gerhard R, Barth H, Just I, Genth H. Difference in the cytotoxic effects of toxin B from Clostridium difficile strain VPI 10463 and toxin B from variant Clostridium difficile strain 1470. Infect Immun 2007; 75:801 - 9; http://dx.doi.org/10.1128/IAI.01705-06; PMID: 17145947
- Genth H, Dreger SC, Huelsenbeck J, Just I. Clostridium difficile toxins: more than mere inhibitors of Rho proteins. Int J Biochem Cell Biol 2008; 40:592 - 7; http://dx.doi.org/10.1016/j.biocel.2007.12.014; PMID: 18289919
- Genth H, Huelsenbeck J, Hartmann B, Hofmann F, Just I, Gerhard R. Cellular stability of Rho-GTPases glucosylated by Clostridium difficile toxin B. FEBS Lett 2006; 580:3565 - 9; http://dx.doi.org/10.1016/j.febslet.2006.04.100; PMID: 16730714
- Steffen A, Ladwein M, Dimchev GA, Hein A, Schwenkmezger L, Arens S, Ladwein KI, Margit Holleboom J, Schur F, Victor Small J, et al. Rac function is crucial for cell migration but is not required for spreading and focal adhesion formation. J Cell Sci 2013; 126:4572 - 88; http://dx.doi.org/10.1242/jcs.118232; PMID: 23902686
- Czuchra A, Wu X, Meyer H, van Hengel J, Schroeder T, Geffers R, Rottner K, Brakebusch C. Cdc42 is not essential for filopodium formation, directed migration, cell polarization, and mitosis in fibroblastoid cells. Mol Biol Cell 2005; 16:4473 - 84; http://dx.doi.org/10.1091/mbc.E05-01-0061; PMID: 16014609
- Woodcock SA, Rushton HJ, Castañeda-Saucedo E, Myant K, White GR, Blyth K, Sansom OJ, Malliri A. Tiam1-Rac signaling counteracts Eg5 during bipolar spindle assembly to facilitate chromosome congression. Curr Biol 2010; 20:669 - 75; http://dx.doi.org/10.1016/j.cub.2010.02.033; PMID: 20346677
- Herrmann C, Ahmadian MR, Hofmann F, Just I. Functional consequences of monoglucosylation of Ha-Ras at effector domain amino acid threonine 35. J Biol Chem 1998; 273:16134 - 9; http://dx.doi.org/10.1074/jbc.273.26.16134; PMID: 9632667
- Sehr P, Joseph G, Genth H, Just I, Pick E, Aktories K. Glucosylation and ADP ribosylation of rho proteins: effects on nucleotide binding, GTPase activity, and effector coupling. Biochemistry 1998; 37:5296 - 304; http://dx.doi.org/10.1021/bi972592c; PMID: 9548761
- Genth H, Aktories K, Just I. Monoglucosylation of RhoA at threonine 37 blocks cytosol-membrane cycling. J Biol Chem 1999; 274:29050 - 6; http://dx.doi.org/10.1074/jbc.274.41.29050; PMID: 10506156
- Rossman KL, Der CJ, Sondek J. GEF means go: turning on RHO GTPases with guanine nucleotide-exchange factors. Nat Rev Mol Cell Biol 2005; 6:167 - 80; http://dx.doi.org/10.1038/nrm1587; PMID: 15688002
- Banerjee M, Worth D, Prowse DM, Nikolic M. Pak1 phosphorylation on t212 affects microtubules in cells undergoing mitosis. Curr Biol 2002; 12:1233 - 9; http://dx.doi.org/10.1016/S0960-9822(02)00956-9; PMID: 12176334
- ten Klooster JP, Jaffer ZM, Chernoff J, Hordijk PL. Targeting and activation of Rac1 are mediated by the exchange factor beta-Pix. J Cell Biol 2006; 172:759 - 69; http://dx.doi.org/10.1083/jcb.200509096; PMID: 16492808
- Cotteret S, Chernoff J. Pak GITs to Aurora-A. Dev Cell 2005; 9:573 - 4; http://dx.doi.org/10.1016/j.devcel.2005.10.005; PMID: 16256730
- Pugacheva EN, Roegiers F, Golemis EA. Interdependence of cell attachment and cell cycle signaling. Curr Opin Cell Biol 2006; 18:507 - 15; http://dx.doi.org/10.1016/j.ceb.2006.08.014; PMID: 16919436