
Abstract
Several years ago a hypothesis was proposed that the survival of cancer cells depend on elevated expression of molecular chaperones because these cells are prone to proteotoxic stress. A critical prediction of this hypothesis is that depletion of chaperones in cancer cells should lead to proteotoxicity. Here, using the major chaperone Hsp70 as example, we demonstrate that its depletion does not trigger proteotoxic stress, thus refuting the model. Accordingly, other functions of chaperones, e.g., their role in cell signaling, might define the requirements for chaperones in cancer cells, which is critical for rational targeting Hsp70 in cancer treatment.
Introduction
Major molecular chaperones, like Hsp70, Hsp90, or Hsp27, and the transcription factor that regulates their expression, Hsf1, are often expressed in cancer cells at elevated levels.Citation1-Citation3 In 2007, the Lindquist and Mivechi groups discovered that certain types of cancer cannot be developed in Hsf1 knockout mice, thus starting a novel area in cancer research.Citation4,Citation5 More recently several publications reported that Hsf1 is critical for development of a variety of cancers at several stages.Citation6-Citation9 Interestingly, we reported that it is enough to knockout just one of the major chaperones, Hsp70, to similarly block cancer development in the model of Her2-postive breast cancer.Citation10 Overall, it was uncovered that Hsf1 and Hsp70 are critical for the survival and growth of many types of cancer cells, but are dispensable for normal cells.Citation10-Citation12 For example, depletion of either Hsp70 or Hsf1 in untransformed breast epithelial line MCF10A is not harmful. However, similar depletion in cells transformed with Her2 or PIK3CA oncogenes leads to growth arrest and overall senescence.Citation6,Citation11
Following discovery of the essential role of Hsf1 in tumor development, a novel paradigm emerged called “non-oncogene addiction”.Citation13 According to this idea, tumor cells have increased demand for molecular chaperones due to elevated levels of abnormal proteins, which accumulate as a result of the toxic conditions associated with the tumor microenvironment. This argument would not be valid in standard conditions of cell culture, where dependence on chaperones persists. On the other hand, cancer cells that are usually aneuploid have an imbalance in protein complexes, a characteristic which by itself may demand high levels of chaperones. This mechanism was proposed to explain why cancer cells become “addicted” to increased activity of Hsf1 and other chaperones.Citation14 This paradigm is widely accepted in the field and is supported by certain evidence. For example, aneuploid strains of yeast are sensitive to conditions that interfere with protein translation, folding, and degradation.Citation15-Citation17 Also, mammalian cell strains that have certain extra chromosomes demonstrate increased sensitivity to inhibitors of the chaperone Hsp90.Citation18 This data, however, is circuitous, and a direct test of the model has not been performed.
Here, we test the major prediction of the hypothesis that depletion of a major chaperone in cancer cells should reveal hidden proteotoxicity. These experiments were done using Hsp70 as an example.
“Addiction” to Hsp70 may be responsible, at least in part for the phenomenon of “oncogene addiction,” and therefore may define a major avenue in drug design. Indeed, it was reported that certain oncogenes (e.g., Bcr-Abl of Her2), “addiction” to which is targeted by drugs, potently induce Hsp70, which contributes to cancer cell survival.Citation6,Citation19 Understanding why cancer cells become “addicted” to Hsp70 is of fundamental importance. In fact, a number of small molecules that target Hsp70 have been identified, and their further development requires clarity in question what domains and activities of Hsp70 are specifically involved in cancer. If chaperone activity is critical, the peptide-binding domain may be targeted. However, if the chaperone function is irrelevant, other activities (e.g., interaction with co-factors) should be targeted.
Results and Discussion
Previously, we demonstrated that Hsp70 suppresses oncogene-induced senescence (OIS).Citation10-Citation12 Accordingly, depletion of Hsp70 in cancer cells lines led to induction of the cell cycle inhibitor p21 and triggered senescence and permanent growth arrest.Citation10-Citation12 Potentially, these effects could be associated with triggering proteotoxic stress, for example due to aneuploidy-related protein imbalance. The major prediction of this model is that the overall chaperone capacity of cancer cells is limited due to the high levels of abnormal proteins, and thus depletion of Hsp70 should trigger proteotoxic stress. To test this prediction, we probed the proteotoxic effect and the chaperone capacity in cancer cells following knocking down of Hsp70 by monitoring different endpoints of proteotoxicity.
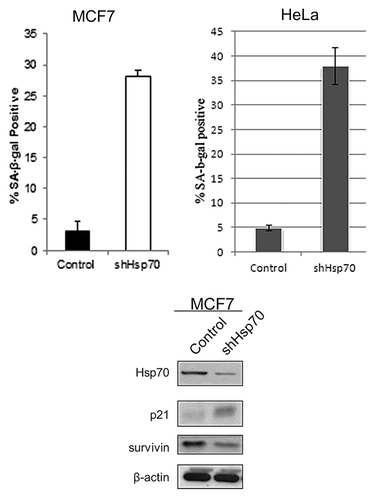
As models, we chose MCF7 and HeLa cells, which have high levels of Hsp70.Citation11,Citation12 Furthermore, we previously used these cells to demonstrate effects of Hsp70 on cell growth and senescence.Citation11,Citation12 When Hsp70 was specifically depleted by retroviral shRNA expression, no changes in levels of the cognate Hsc73 (HspA8) were found in these cells, indicating the specificity of the knockdown.Citation12 Under these conditions in MCF7 and HeLa cells, we observed a dramatic reduction of cell growthCitation12 and massive appearance of SA-β-gal-positive cells, indicating senescence (). As noted previously, these effects were specific to transformed cells and were not seen in untransformed breast epithelial MCF10A cells.Citation10,Citation11
Figure 1. Depletion of Hsp70 results in cancer cell senescence. SA-β-gal-positive cells following Hsp70 depletion in MCF7 and HeLa cells. Results shown are the mean ± SEM of 3 independent experiments. Induction of the cell cycle inhibitor p21 and downregulation of the mitosis factor surviving following Hsp70 depletion in MCF7 cells is also shown.
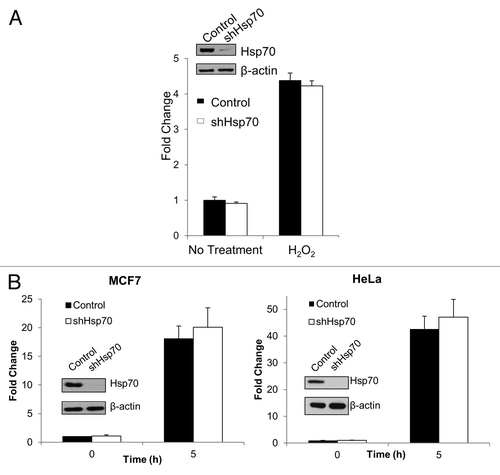
As readout of proteotoxicity, we determined the levels of ubiquitinated proteins. Contrary to the predictions of the hypothesis, depletion of Hsp70 did not significantly alter levels of ubiquitinated proteins (), suggesting minimal buildup of abnormal species. Further, we evaluated activities of stress-responsive transcription factors Hsf1 and Nrf2 in Hsp70 depleted conditions. Hsf1 is activated by the buildup of abnormal proteins, while Nrf2 responds to reduced proteasome activity and oxidative stress. However, depletion of Hsp70 did not cause activation of either Hsf1 or Nrf2 as assessed by corresponding promoter-luciferase reporter constructs in MCF7 or HeLa cells (), further suggesting the lack of proteotoxic stress.
Figure 2. Hsp70 depletion does not alter ubiquitinylated protein levels. Depletion of Hsp70 does not cause apparent change in ubiquitination. MG132 (left panel, MCF7: 5 μM, 4 h; or right panel, HeLa: 10 μM, 4 h) was used to inhibit the proteasome (positive ubiquitination control). All samples prepared in lysis buffer containing N-ethylmaleimide (NEM, 10 mM). Samples were immunoblotted using an anti-ubiquitin antibody (lower panels). Upper panels: efficiency of Hsp70 depletion. Representative blots of triplicate experiments are shown. Quantification of levels of ubiquitinated proteins in HeLa cells is shown at the bottom.
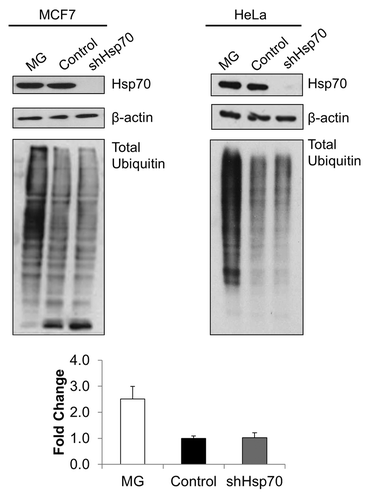
Figure 3. Hsp70 knockdown does not activate stress-sensitive transcription factors, NRF2 or HSF1. (A) Depletion of Hsp70 does not activate NRF2 luciferase reporter. Left panel: luciferase activity was measured at baseline or following hydrogen peroxide treatment (300 μM, 6 h) in MCF7 cells to mimic increased oxidative stress and normalized to baseline control. The means and ± SEM of 3 experiments are shown. (B) Depletion of Hsp70 does not activate Hsf1 luciferase reporter. Luciferase activity was measured at baseline (0 h) or 5 h after heat shock (45 °C, 10 min) and normalized to baseline control. Left panel: MCF7 cells. Right panel: HeLa cells. The means and ± SEM of 3 experiments are shown.
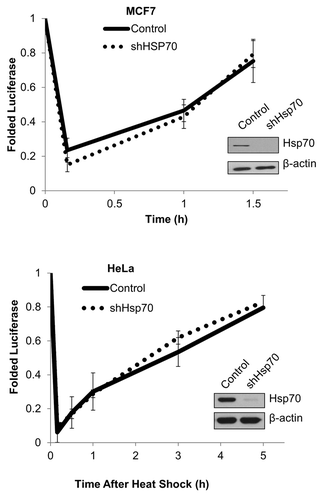
Finally, we evaluated the ability of cells to refold luciferase, a folding substrate of Hsp70 family members. Control and Hsp70-depleted cells expressing luciferase were exposed to brief heat shock followed by recovery at 37 °C. Under these conditions, luciferase becomes unfolded and inactivated followed by refolding and resumption of catalytic activity. Hsp70 depletion did not affect either the degree of luciferase refolding or its kinetics in either MCF7 or HeLa cells (). Therefore, the chaperone capacity of cells appears to be not markedly reduced by depletion of Hsp70. This data indicates that in Hsp70-depleted cells, other chaperones (e.g., Hsc70 or other Hsp70 family members) can substitute for Hsp70, and there is no significant proteotoxicity.
Figure 4. Cancer cell chaperone capacity is not perturbed by Hsp70 depletion. Hsp70 knockdown does not affect luciferase refolding. Cells were heat shocked at 45 °C for 10 min, and luciferase activity was measured over time (left panel, MCF7; right panel, HeLa). Plot shows mean and ± SEM (n = 3).
Interestingly, we previously reported that proteotoxic stress is not seen upon depletion in cancer cells of another chaperone Hsp27.Citation20 This example with Hsp27 did not represent a very strong argument against the model simply because the Hsp27 may have a relatively narrow substrate specificity, which may not be revealed by general proteotoxic measurements. With Hsp70, however, the situation is different, since it is known to have a very wide range of substrates, and lack of evidence of proteotoxicity under the conditions that trigger senescence indicates that cancer cell growth is not associated with internal proteotoxicity.
Accordingly, the reason for the dependence of cancer cells on Hsp70 must be different. It may be related to the function of Hsp70 in cancer signaling. In fact, we and others have reported that depletion of Hsp70 leads to dramatic changes in multiple signaling pathways involved in cancer. For example, Hsp70 has been implicated in control of JNK and ERK pathways, and chaperone function was dispensable for these regulations.Citation21,Citation22 Communication with cancer-related signaling pathways may be mediated by various co-factors, like DnaJ or Bag family members. Therefore, drug design efforts should be directed toward these interactions of Hsp70.
These data, while suggesting targeting the signaling function of Hsp70, do not negate possible beneficial effects of targeting the chaperone capacity of cells, e.g., by inhibiting the folding function of multiple members of the Hsp70 family. Indeed, aptamers that inhibit the peptide-binding domain of Hsp70 demonstrated anti-cancer effects,Citation23 and cancer cells were specifically sensitive to treatments that enhance proteotoxicity, like puromycin.Citation24 Therefore, the possibility is that inhibition of the signaling function should be combined with inhibition of the chaperone function. Furthermore, inhibition of Hsp70 may be beneficial in combination with certain conventional therapies, like doxorubicin or bortezomib.Citation25,Citation26
Materials and Methods
Cell cultures
HEK293T, MCF-7, and HeLa were from ATCC. MCF7 and HeLa cells were cultivated in Dulbecco modified Eagle medium (DMEM) supplemented with 10% fetal bovine serum. HEK293T cells were maintained in DMEM supplemented with 10% heat-inactivated FBS. All cultures contained 1% penicillin/streptomycin. For heat shock at 45 °C, a circulating water bath was used.
Compounds and antibodies
MG132 was purchased from Biomol and puromycin from Sigma. Antibodies: anti-Hsp70 was from Stressgen, anti-β-actin was from Cell Signaling, and anti-multi-ubiquitin (FK2) was from Enzo Life Sciences.
Plasmids, shRNAs, and virus production
Retroviral and lentiviral vectors, and infection: RNAi-Ready pSIREN-RetroQ vector from BD
Biosciences retroviral delivery system was used for knockdown of Hsp70. The sequence of human Hsp70 gene was selected as reported before:Citation12 5′TATGGACTCC AACCTGGATA A-3′. Retroviruses expressing shRNAs or control enhanced green fluorescent protein (EGFP) were produced by co-transfection of HEK293T cells via Lipofectamine 2000 with plasmids expressing retroviral genomes, packaging proteins Gag-Pol, and the VSV-G protein. Lentiviral vectors were produced by co-transfection of HEK293T cells with plasmids lentiviral packaging plasmid, psPAX2, VSV-G expression plasmid, pMD2.G, and either control EGFP or alternative transgene-expressing constructs. At 48 h after transfection, retrovirus or lentivirus containing supernatants were collected and frozen at −80 °C. Cells were infected with twice diluted supernatant and 10 μg/mL polybrene overnight, washed, and selected with puromycin (10 μg/mL) 48 h after infection. Retroviral or lentiviral vectors expressing EGFP were used as an infection efficiency indicator: usually about 75% of cells were fluorescent 2 d after infection.
Senescence-associated β-galactosidase assay
SA-β-gal assay was performed using X-gal (pH 6.0) as described previously.Citation12 Breifly, cells were plated at low density and fixed with 4% formaldehyde. The β-gal activity was determined by incubation with 1 mg/mL of X-gal in 40 mM sodium citrate, 5 mM K3FeCN6, 5 mM K4FeCN6, 150 mM NaCl, and 2 mM MgCl2 diluted in PBS (pH 6.0). Cells were incubated for 48 h, and the number of stained cells was counted under microscope from 5 different fields, and the proportion of stained cells was calculated. The results were expressed as mean values ± SEM on 3 independent experiments.
Reporter viruses
HSE-luc lentiviral vector reported previouslyCitation27 using the pGreenFire lentiviral vector (System Biosciences): 6 tandem repeats of heat shock elements (HSE)- 5′-GAACCTTCGC GAATTTTCAA GAATGTTCAG GAATATTCTA GAACTTTCCT GAACCTT-3′at the ClaI and SpeI sites.
NRF2-luc lentiviral vector was constructed using the pGreenFire Lentiviral vector (System Biosciences): 6 tandem repeats of the NRF2 consensus sequence- 5′-TCACAGTGAC TCAGCAAAAT T-3′ between the Cla1 and Spe1 sties.
Luciferase reporter assays
Cells were infected initially with HSE-luc or NRF2-luc lentivirus, and then infected again with control or shHsp70 retrovirus, followed by puromycin selection. Three days after selection, cells were either heat shocked at 45 °C for 10 min (for HSF1) followed by a 3 h incubation at 37 °C, or treated with 300 μM hydrogen peroxide for 5 h (for NRF2). After incubation period, the medium was aspirated, cells were washed twice with PBS, lysis buffer was added directly to the plate, and the plate was frozen at −80 °C. Upon thawing, lysates were plated into 96-well plate. Fifty microliters (50 μl) of luciferase assay reagent (Promega) was injected into each well, and luminescence was read by luminometer. In parallel, lysates were plated into black 96-well plates, and GFP fluorescence was read by the same luminometer. All measurements were done in triplicates, and the assays were repeated 3 times.
Luciferase refolding assay
Cells were co-infected with both EGFP and retroviral reporter encoding luciferase under the cytomegalovirus (CMV) promoter.Citation28 For refolding assay, cells were subjected to heat shock at 45 °C for 10 min, and then allowed to recover for the indicated time periods. At each time point, cells were washed in twice with ice-cold PBS, lysed with cell lysis reagent (Promega), and frozen at −80 °C. Upon thawing, lysates were plated into 96-well plate. Fifty microliters (50 μl) of luciferase assay reagent (Promega) was injected into each well, and luminescence was read by luminometer. In parallel, lysates were plated into black 96-well plate, and GFP fluorescence was read by the same luminometer. All measurements were done in triplicates, and the assays were repeated 3 times.
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
References
- Ciocca DR, Calderwood SK. Heat shock proteins in cancer: diagnostic, prognostic, predictive, and treatment implications. Cell Stress Chaperones 2005; 10:86 - 103; http://dx.doi.org/10.1379/CSC-99r.1; PMID: 16038406
- Calderwood SK, Khaleque MA, Sawyer DB, Ciocca DR. Heat shock proteins in cancer: chaperones of tumorigenesis. Trends Biochem Sci 2006; 31:164 - 72; http://dx.doi.org/10.1016/j.tibs.2006.01.006; PMID: 16483782
- Santagata S, Hu R, Lin NU, Mendillo ML, Collins LC, Hankinson SE, Schnitt SJ, Whitesell L, Tamimi RM, Lindquist S, et al. High levels of nuclear heat-shock factor 1 (HSF1) are associated with poor prognosis in breast cancer. Proc Natl Acad Sci U S A 2011; 108:18378 - 83; http://dx.doi.org/10.1073/pnas.1115031108; PMID: 22042860
- Dai C, Whitesell L, Rogers AB, Lindquist S. Heat shock factor 1 is a powerful multifaceted modifier of carcinogenesis. Cell 2007; 130:1005 - 18; http://dx.doi.org/10.1016/j.cell.2007.07.020; PMID: 17889646
- Min J-N, Huang L, Zimonjic DB, Moskophidis D, Mivechi NF. Selective suppression of lymphomas by functional loss of Hsf1 in a p53-deficient mouse model for spontaneous tumors. Oncogene 2007; 26:5086 - 97; http://dx.doi.org/10.1038/sj.onc.1210317; PMID: 17310987
- Meng L, Gabai VL, Sherman MY. Heat-shock transcription factor HSF1 has a critical role in human epidermal growth factor receptor-2-induced cellular transformation and tumorigenesis. Oncogene 2010; 29:5204 - 13; http://dx.doi.org/10.1038/onc.2010.277; PMID: 20622894
- Gabai VL, Meng L, Kim G, Mills TA, Benjamin IJ, Sherman MY. Heat shock transcription factor Hsf1 is involved in tumor progression via regulation of hypoxia-inducible factor 1 and RNA-binding protein HuR. Mol Cell Biol 2012; 32:929 - 40; http://dx.doi.org/10.1128/MCB.05921-11; PMID: 22215620
- Dudeja V, Chugh RK, Sangwan V, Skube SJ, Mujumdar NR, Antonoff MB, Dawra RK, Vickers SM, Saluja AK. Prosurvival role of heat shock factor 1 in the pathogenesis of pancreatobiliary tumors. Am J Physiol Gastrointest Liver Physiol 2011; 300:G948 - 55; http://dx.doi.org/10.1152/ajpgi.00346.2010; PMID: 21330448
- Xi C, Hu Y, Buckhaults P, Moskophidis D, Mivechi NF. Heat shock factor Hsf1 cooperates with ErbB2 (Her2/Neu) protein to promote mammary tumorigenesis and metastasis. J Biol Chem 2012; 287:35646 - 57; http://dx.doi.org/10.1074/jbc.M112.377481; PMID: 22847003
- Meng L, Hunt C, Yaglom JA, Gabai VL, Sherman MY. Heat shock protein Hsp72 plays an essential role in Her2-induced mammary tumorigenesis. Oncogene 2011; 30:2836 - 45; http://dx.doi.org/10.1038/onc.2011.5; PMID: 21297664
- Gabai VL, Yaglom JA, Waldman T, Sherman MY. Heat shock protein Hsp72 controls oncogene-induced senescence pathways in cancer cells. Mol Cell Biol 2009; 29:559 - 69; http://dx.doi.org/10.1128/MCB.01041-08; PMID: 19001088
- Yaglom JA, Gabai VL, Sherman MY. High levels of heat shock protein Hsp72 in cancer cells suppress default senescence pathways. Cancer Res 2007; 67:2373 - 81; http://dx.doi.org/10.1158/0008-5472.CAN-06-3796; PMID: 17332370
- Luo J, Solimini NL, Elledge SJ. Principles of cancer therapy: oncogene and non-oncogene addiction. Cell 2009; 136:823 - 37; http://dx.doi.org/10.1016/j.cell.2009.02.024; PMID: 19269363
- Solimini NL, Luo J, Elledge SJ. Non-oncogene addiction and the stress phenotype of cancer cells. Cell 2007; 130:986 - 8; http://dx.doi.org/10.1016/j.cell.2007.09.007; PMID: 17889643
- Torres EM, Sokolsky T, Tucker CM, Chan LY, Boselli M, Dunham MJ, Amon A. Effects of aneuploidy on cellular physiology and cell division in haploid yeast. Science 2007; 317:916 - 24; http://dx.doi.org/10.1126/science.1142210; PMID: 17702937
- Pavelka N, Rancati G, Zhu J, Bradford WD, Saraf A, Florens L, Sanderson BW, Hattem GL, Li R. Aneuploidy confers quantitative proteome changes and phenotypic variation in budding yeast. Nature 2010; 468:321 - 5; http://dx.doi.org/10.1038/nature09529; PMID: 20962780
- Torres EM, Dephoure N, Panneerselvam A, Tucker CM, Whittaker CA, Gygi SP, Dunham MJ, Amon A. Identification of aneuploidy-tolerating mutations. Cell 2010; 143:71 - 83; http://dx.doi.org/10.1016/j.cell.2010.08.038; PMID: 20850176
- Tang YC, Williams BR, Siegel JJ, Amon A. Identification of aneuploidy-selective antiproliferation compounds. Cell 2011; 144:499 - 512; http://dx.doi.org/10.1016/j.cell.2011.01.017; PMID: 21315436
- Demidenko ZN, An WG, Lee JT, Romanova LY, McCubrey JA, Blagosklonny MV. Kinase-addiction and bi-phasic sensitivity-resistance of Bcr-Abl- and Raf-1-expressing cells to imatinib and geldanamycin. Cancer Biol Ther 2005; 4:484 - 90; http://dx.doi.org/10.4161/cbt.4.4.1702; PMID: 15846067
- O’Callaghan-Sunol C, Gabai VL, Sherman MY. Hsp27 modulates p53 signaling and suppresses cellular senescence. Cancer Res 2007; 67:11779 - 88; http://dx.doi.org/10.1158/0008-5472.CAN-07-2441; PMID: 18089808
- Yaglom JA, Gabai VL, Meriin AB, Mosser DD, Sherman MY. The function of HSP72 in suppression of c-Jun N-terminal kinase activation can be dissociated from its role in prevention of protein damage. J Biol Chem 1999; 274:20223 - 8; http://dx.doi.org/10.1074/jbc.274.29.20223; PMID: 10400639
- Yaglom J, O’Callaghan-Sunol C, Gabai V, Sherman MY. Inactivation of dual-specificity phosphatases is involved in the regulation of extracellular signal-regulated kinases by heat shock and hsp72. Mol Cell Biol 2003; 23:3813 - 24; http://dx.doi.org/10.1128/MCB.23.11.3813-3824.2003; PMID: 12748284
- Rérole AL, Gobbo J, De Thonel A, Schmitt E, Pais de Barros JP, Hammann A, Lanneau D, Fourmaux E, Deminov O, Micheau O, et al. Peptides and aptamers targeting HSP70: a novel approach for anticancer chemotherapy. Cancer Res 2011; 71:484 - 95; http://dx.doi.org/10.1158/0008-5472.CAN-10-1443; PMID: 21224349
- Neznanov N, Komarov AP, Neznanova L, Stanhope-Baker P, Gudkov AV. Proteotoxic stress targeted therapy (PSTT): induction of protein misfolding enhances the antitumor effect of the proteasome inhibitor bortezomib. Oncotarget 2011; 2:209 - 21; PMID: 21444945
- Demidenko ZN, Vivo C, Halicka HD, Li CJ, Bhalla K, Broude EV, Blagosklonny MV. Pharmacological induction of Hsp70 protects apoptosis-prone cells from doxorubicin: comparison with caspase-inhibitor- and cycle-arrest-mediated cytoprotection. Cell Death Differ 2006; 13:1434 - 41; http://dx.doi.org/10.1038/sj.cdd.4401812; PMID: 16311509
- Yerlikaya A, Okur E, Eker S, Erin N. Combined effects of the proteasome inhibitor bortezomib and Hsp70 inhibitors on the B16F10 melanoma cell line. Mol Med Rep 2010; 3:333 - 9; http://dx.doi.org/10.3892/mmr_000000262; PMID: 21472244
- Kim G, Meriin AB, Gabai VL, Christians E, Benjamin I, Wilson A, Wolozin B, Sherman MY. The heat shock transcription factor Hsf1 is downregulated in DNA damage-associated senescence, contributing to the maintenance of senescence phenotype. Aging Cell 2012; 11:617 - 27; http://dx.doi.org/10.1111/j.1474-9726.2012.00827.x; PMID: 22510478
- Zaarur N, Gabai VL, Porco JA Jr., Calderwood S, Sherman MY. Targeting heat shock response to sensitize cancer cells to proteasome and Hsp90 inhibitors. Cancer Res 2006; 66:1783 - 91; http://dx.doi.org/10.1158/0008-5472.CAN-05-3692; PMID: 16452239