
Parkinson disease (PD) is a progressive neurodegenerative disorder affecting approximately 1% of the population over age 65. It is characterized by loss of dopaminergic neurons in the substantia nigra along with protein aggregates known as Lewy bodies in surviving neurons. The disease leads to debilitating motor symptoms, as well as some nonmotor signs. PD does not have a single known cause, and is thought to result from a combination of genetic and environmental risk factors. To date, about a dozen genes have been implicated in the disease. Among these risk factors, multiple groups have identified that an aspartate to asparagine substitution (D620N) in VPS35 results in autosomal dominant familial PD.Citation1,Citation2
VPS35 is a component of the highly conserved membrane-associated protein complex known as retromer. It was initially discovered to function in the endosome-to-Golgi retrieval of membrane proteins, such as the cation-independent mannose 6-phosphate receptor, which sorts hydrolases to the endosomal pathway and is then recycled back to the trans-Golgi network (TGN) for subsequent rounds of transport. Retromer consists of a cargo-selective complex (CSC) that includes VPS35, VPS26, and VPS29, as well as a sorting nexin dimer that is thought to play a role in membrane deformation. More recently, retromer has also been implicated in endosome-to-cell surface sorting due to its role in recruitment of the WASH complex.Citation3 The latter complex mediates actin patch formation on endosomes by regulating the actin nucleating activity of Arp2/3, and is critical for the proper routing of various membrane proteins to the cell surface.Citation4
Thus far, information about the consequences of the D620N mutation and its mechanism of action has been limited. We found that the D620N mutation in VPS35 does not affect assembly of the CSC, nor does it appear to influence the canonical retromer function of endosome-to-Golgi cargo retrieval, which is in agreement with other recent findings.Citation5 The disease-causing mutation does, however, impair the ability of retromer to bind and recruit the WASH complex to endosomes. Consequently, processes that rely on WASH complex-mediated trafficking to the plasma membrane are disturbed in VPS35 D620N-expressing cells. For example, levels of the glucose transporter GLUT1 are decreased at the cell surface. PD mutant cells are also slower to spread out after trypsinisation. This process relies on integrins, the transport of which is known to be affected by WASH disruption.
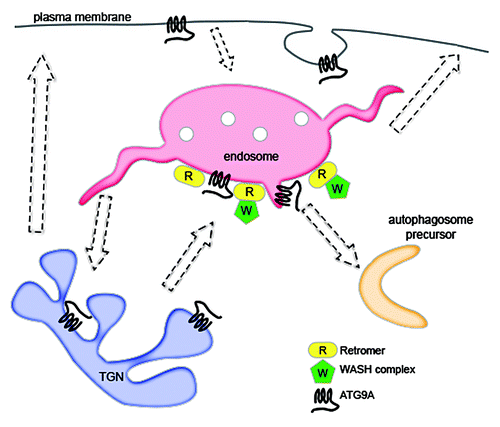
Genes that play pivotal roles in membrane trafficking have a tendency to affect the bulk degradation pathway known as macroautophagy (hereafter, autophagy), in which portions of the cytoplasm are engulfed by a double membraned autophagosome and targeted to lysosomes for degradation. Because autophagosome formation involves many compartments, including endosomes, it was logical to explore the effect of the VPS35 D620N mutation on autophagy. We found that the PD mutation impaired the formation of autophagosomes and the clearance of autophagy substrates. This was due, at least in part, to the mistrafficking of the multipass transmembrane autophagy protein ATG9A, which is necessary for proper autophagy induction. Normally, this protein moves between the TGN, plasma membrane, as well as early and recycling endosomes, and interacts with autophagic precursor proteins in the latter compartmentCitation6 (). However, in mutant VPS35-expressing cells, ATG9A was trapped in a perinuclear location and was unable to traffic to autophagic structures. Interestingly, both the defective ATG9A trafficking and the resultant inhibition of autophagosome formation were recapitulated by artificial displacement of the WASH complex or by depletion of the WASH1 component, which is the factor responsible for the activation of Arp2/3. It is well established that autophagy is central to neurodegenerative diseases, as it is able to clear a number of disease-causing intracytoplasmic proteins, including α-synuclein, tau, and proteins with polyglutamine expansions. Conversely, inhibition of autophagy results in a neurodegenerative phenotype, underpinned by reduced clearance of defective mitochondria and aggregate-prone proteins, and ultimately decreased cell viability. The D620N mutation has been shown to impair protection of dopaminergic neurons against a mitochondrial poison,Citation7 and we further found that WASH1 depletion in neuroblastoma cells decreased their survival. Thus, the autophagy inhibition observed with the VPS35 D620N mutation and concomitant WASH dysfunction is a disease-relevant phenotype, and could contribute to PD etiology.
Figure 1. Model of retromer and WASH complex in autophagy. Retromer directly interacts with the WASH complex to recruit it to endosomes. Proper WASH complex localization is necessary for normal trafficking of ATG9A, which passes through various organelles prior to contributing to nascent autophagosomes. A PD mutation in a retromer component impairs WASH complex recruitment and the ability of ATG9A to traffic to autophagic compartments.
Outlook
A critical priority in the future will be to form a deeper mechanistic understanding of the role of the WASH complex in autophagy. We expect that its reduced endosomal recruitment is accompanied by a decrease in actin patch formation at this compartment. It will be necessary to target the actin nucleation promoting activity of WASH1 to investigate whether this particular function affects autophagosome formation. By driving actin patch formation, the WASH complex regulates endosomal tubulation and fission,Citation4 and this process is likely to be of great importance to early events in the autophagic pathway. As implied by our data, it might regulate the trafficking of necessary autophagy proteins such as ATG9A to the appropriate compartments. Moreover, considering that the recycling endosome is a membrane source for autophagosomes and appears to be the nucleation site for their formation,Citation6 the integrity of tubulation and scission mechanisms—such as those facilitated by the WASH complex—may be essential for the biogenesis of a unique compartment.
Related Research Data
References
- Vilariño-Güell C, et al. Am J Hum Genet 2011; 89:162 - 7; http://dx.doi.org/10.1016/j.ajhg.2011.06.001; PMID: 21763482
- Zimprich A, et al. Am J Hum Genet 2011; 89:168 - 75; http://dx.doi.org/10.1016/j.ajhg.2011.06.008; PMID: 21763483
- Harbour ME, et al. J Cell Sci 2010; 123:3703 - 17; http://dx.doi.org/10.1242/jcs.071472; PMID: 20923837
- Derivery E, et al. Dev Cell 2009; 17:712 - 23; http://dx.doi.org/10.1016/j.devcel.2009.09.010; PMID: 19922875
- Tsika E, et al. Hum Mol Genet 2014; http://dx.doi.org/10.1093/hmg/ddu178; PMID: 24740878
- Puri C, et al. Cell 2013; 154:1285 - 99; http://dx.doi.org/10.1016/j.cell.2013.08.044; PMID: 24034251
- Bi F, et al. Int J Biol Sci 2013; 9:149 - 55; http://dx.doi.org/10.7150/ijbs.5617; PMID: 23411763