
DNA double-strand breaks (DSBs) can be lethal to a cell. However, most sexually reproducing organisms deliberately induce a substantial amount of developmentally programmed DSBs that are subsequently repaired via homologous recombination in meiosis. The goal of this self-damage and self-repair process is to establish physical connections between homologous chromosomes, thereby ensuring accurate chromosome segregation and producing haploid germ cells (sperm and eggs) (). Recombination also disrupts the linkage of polymorphisms on the same chromosome and thus promotes genome diversity and evolution. Alterations in normal recombination patterns cause human aneuploidy, and these errors are a major cause of spontaneous abortion and congenital birth defects.Citation1
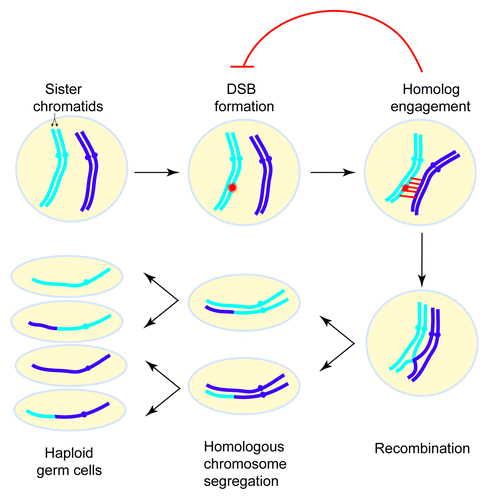
Figure 1. Overview of the events of meiosis. In meiosis I, homologous chromosomes exchange genetic information via recombination and are then segregated. In meiosis II, sister chromatids separate. Meiotic recombination is initiated by DNA double-strand breaks (DSBs). Homolog engagement triggers structural changes in certain chromosomal subdomains that suppress further DSB formation nearby. For simplicity only one pair of homologous chromosomes is depicted.
Meiotic recombination does not occur randomly. It is more likely to happen in some genomic regions than others, largely due to nonrandom DSB distribution. There are large DSB-hot and -cold domains (tens of kilobases [kb]), within which are short regions called hotspots (typically several hundred base pairs wide), where DSBs preferentially form. This DSB landscape is shaped by a hierarchical combination of many factors including whole chromosome variation, large subchromosomal domains, cohesins and other chromosome structure proteins, chromatin structure, and local nucleotide composition.Citation2 Notably, these factors act at different size scales and many of the molecular mechanisms connecting them to DSB formation are still not well understood.
As potentially hazardous events, meiotic DSBs are tightly controlled in their timing, amount, and location. Emerging evidence in several organisms implies that the first step in recombination (DSB formation) is regulated by subsequent steps such as DSB repair. Recently, we explored such a feedback circuit in which homolog engagement shapes meiotic DSB number and spatial patterning in Saccharomyces cerevisiae ().Citation3 For this purpose, we loosely define homolog engagement such that it could include the progression of recombination intermediates and/or the formation of synaptonemal complex (SC). (The SC is a meiosis-specific structure comprising the proteinaceous axes of a pair of homologous chromosomes held together by transverse filaments; it serves as a scaffold stabilizing the juxtaposition of homologous chromosomes and promotes the completion of recombination). Evidence in mice, flies and worms has suggested that negative feedback induced by engaging homologous chromosomes controls DSB formation.Citation4-Citation6
A test of this hypothesis arose from studies of the ZMM group of proteins (Zip1–4, Msh4–5, Mer3, Spo16, and Pph3). Mutants lacking ZMM proteins display defects in SC and recombination.Citation7 We found that ZMM mutants formed a substantially greater number of DSBs, as judged by a variety of molecular and genetic assays. This was a surprise because ZMM proteins have traditionally been viewed as acting strictly downstream of DSB formation, but the new findings show that ZMMs are genetically both upstream and downstream. A simple way to explain these results is to propose that chromosomes stop making DSBs once they successfully engage their homologous partners. The homolog engagement defects in ZMM mutants allow chromosomes to continue making DSBs when normally they would have stopped. A plausible mechanism is that formation of SC leads to structural changes in chromosomes that inactivate or remove the DSB-forming machinery.
Importantly, mapping of DSBs in zip3 mutants demonstrated that this feedback loop helps shape the genome-wide DSB distribution. Interestingly, different chromosomal subdomains responded differently to the DSB increase in zip3 mutants, with domains of greater or lesser change alternating along chromosomes. One possible explanation is that defective homolog engagement in zip3 mutants relieves the DSB suppression that would normally occur near sites of recombination. If so, this further implies that ZMM-dependent DSB suppression in wild-type cells spreads along chromosomes from sites of homolog engagement, with the magnitude of suppression decreasing with the distance from the engagement site.
Besides homolog engagement, there are other regulatory elements shaping DSB distributions on different size scales.Citation2 For example, few DSBs form in ~20-kb zones from telomeres, centromeres and the rDNA in wild-type. Despite a 1.8-fold increase of total DSBs in zip3 mutants, DSB frequencies in subtelomeric, and pericentromeric regions were elevated less than genome average, and were unchanged or reduced near the rDNA.Citation3 Accordingly, ZMM-dependent DSB suppression is different from and subordinate to the DSB suppression mechanisms acting in these subdomains.
Since DSB feedback circuits have been reported in other species as well, it will be interesting to map DSBs in mutants with feedback defects in other organisms, continuing the journey of exploring previously unknown regulators of the recombination landscape.
References
- Hassold T, et al. Hum Mol Genet 2007; 16 Spec No. 2:R203 - 8; http://dx.doi.org/10.1093/hmg/ddm243; PMID: 17911163
- Pan J, et al. Cell 2011; 144:719 - 31; http://dx.doi.org/10.1016/j.cell.2011.02.009; PMID: 21376234
- Thacker D, et al. Nature 2014; 510:241 - 6; http://dx.doi.org/10.1038/nature13120; PMID: 24717437
- Bhagat R, et al. Cytogenet Genome Res 2004; 107:160 - 71; http://dx.doi.org/10.1159/000080594; PMID: 15467361
- Henzel JV, et al. Genetics 2011; 187:685 - 99; http://dx.doi.org/10.1534/genetics.110.124958; PMID: 21212235
- Kauppi L, et al. Genes Dev 2013; 27:873 - 86; http://dx.doi.org/10.1101/gad.213652.113; PMID: 23599345
- Lynn A, et al. Chromosome Res 2007; 15:591 - 605; http://dx.doi.org/10.1007/s10577-007-1150-1; PMID: 17674148