
Abstract
In recent years a growing recognition that molecularly-targeted therapies face formidable obstacles has revived interest in more generic tumor cell phenotypes that could be exploited for therapy. Two recent reports demonstrate that cancer cell survival is critically dependent on the activity of MTH1, a nucleotide pyrophosphatase that converts the oxidized nucleotides 8-oxo-dGTP and 2-OH-dATP to the corresponding monophosphates, thus preventing their incorporation into genomic DNA. Tumor cells frequently overexpress MTH1, probably because malignant transformation creates oxidative stress that renders the nucleotide pool highly vulnerable to oxidation. As a result, MTH1 inhibition in cancer cells results in accumulation and incorporation of 8-oxo-dGTP and 2-OH-dATP into DNA, leading to DNA damage and cell death. This toxic effect is highly cancer cell-specific, as MTH1 is generally dispensable for the survival of normal, untransformed cells. Importantly, MTH1 proves to be a “druggable” enzyme that can be inhibited both by an existing protein kinase inhibitor drug, crizotinib, and by novel compounds identified through screening. Inhibition of MTH1 leading to toxic accumulation of oxidized nucleotides specifically in tumor cells therefore represents an example of a “non-personalised” approach to cancer therapy.
In recent years a dominant paradigm guiding the development of novel cancer therapies has been to target tumor-specific molecular alterations, typically affecting oncogenes. Although amazingly successful in chronic myeloid leukemias that express the Philadelphia chromosome, where imatinib and related Bcr-Abl tyrosine kinase inhibitors have revolutionised treatment, this strategy has been much less productive in the case of the more prevalent solid tumor types.Citation1 Problems encountered include a paucity of recurrently mutated oncogenes, substantial inter- and intra-tumor heterogeneity, and rampant acquired resistance mechanisms as exemplified by the remarkable, but short-lived, response to the B-RAF-inhibitor vemurafenib in B-RAF mutant melanoma.Citation2 As a result there is a resurgence of interest in identifying more generic features of the tumor cell phenotype that could be targeted for therapy. One recent example is familial breast and ovarian cancers lacking functional BRCA1/BRCA2 due to mutations, where a deficiency in DNA repair via homologous recombination confers sensitivity to inhibitors of poly (ADP) ribose polymerase (PARP).Citation3 Such tumors constitute only a small proportion of the total burden of breast and ovarian cancer, however there are indications that defects in homologous recombination (sometimes referred to as a “BRCAness” phenotype) may occur more widely in multiple sporadic tumor types.Citation4 Similarly, p53 tumor suppressor function is thought to be compromised by diverse mechanisms in a significant proportion of human cancers, leading to defects in cell cycle arrest in G1 and G2 under conditions of genotoxic stress. Because conventional chemotherapies are generally specific for proliferating cells it has been proposed that a “pre-emptive” induction of p53-mediated cell cycle arrest in normal tissues using either MDM2 inhibitors, such as nutlin,Citation5 or low doses of certain genotoxic agents,Citation6 could open a therapeutic window that would allow selective killing of proliferating, p53-deficient tumor cells in a subsequent round of lethal chemotherapy (a concept dubbed “Cyclotherapy”Citation5). Finally, there is evidence that malignant cells exhibit elevated basal levels of autophagy, raising the possibility that inhibiting this process could lead to cancer-specific toxicity.Citation7
Two recent reportsCitation8,Citation9 highlight another potential generic Achilles heel in cancer; namely the possibility of converting endogenous oxidized nucleotide precursors, generally more abundant in tumor cells, into toxic DNA damage lesions. A study by Gad et al..Citation8 extends from earlier observations that overexpression of the nucleotide hydrolase MTH1 protected primary fibroblasts against Ras oncogene-induced premature senescence by suppressing reactive oxygen species (ROS)-induced DNA damage.Citation10 MTH1 (also known as NUDT1) is a nudex family pyrophosphatase that converts the oxidized nucleotide triphosphates of 8-oxo-deoxy-guanine (8-oxo-dG) and 2-OH-deoxy-adenosine (2-OH-dA) into the corresponding monophosphates, thereby preventing their utilization by DNA polymerases and thus incorporation into genomic DNA.Citation11 Incorporation of 8-oxo-dG and 2-OH-dA is potentially dangerous to cells for two reasons, first, repair by base excision repair (BER) and mismatch repair (MMR) involves the formation of single strand DNA breaks and gaps, structures which normally exist only transiently but which can nevertheless be converted to cytotoxic double strand breaks through DNA replication and other mechanisms. Second, incorporated 8-oxo-dG can mispair with thymine and is thus mutagenic when replicated.
Postulating that oxidized nucleotides might be more abundant in cancer cells compared with untransformed cells owing to abnormalities in redox regulation due to malignant transformation, Gad et al. assessed the effects of inhibiting MTH1 expression in a panel of tumor cell lines.Citation8 They show that siRNA-mediated knock down of MTH1 severely impaired cancer cell viability, and that cell death was associated with spontaneous DNA damage and markedly increased incorporation of 8-oxo-dG and 2-OH-dA into genomic DNA. In comparison, depletion of MTH1 was much less toxic in normal cells. Importantly, cell survival could be rescued through overexpression of siRNA-resistant wild-type MTH1 but not a catalytically inactive mutant, indicating a requirement for MTH1 catalytic activity to sustain cancer cell viability. Rescue experiments using additional mutants with substrate-selective hydrolase deficiency provided evidence that fraudulent incorporation of both 8-oxo-dG and 2-OH-dA contributed to toxicity. Remarkably, these effects were not confined to cells in culture, as xenografts formed using cancer cells bearing a tetracycline-inducible MTH1 siRNA were effectively sterilised when tetracycline was added to the drinking water of tumor-bearing mice. Thus, MTH1 was also essential for tumor formation in vivo.
Gad et al. followed up these proof-of-principle studies with an in vitro screen for chemical inhibitors of MTH1 catalytic activity as potential anti-cancer drug leads.Citation8 Two potent and specific MTH1 inhibitors, TH287 and TH588, were identified and shown to kill cancer cells by inducing DNA damage and enhancing incorporation of 8-oxo-dG and 2-OH-dA into genomic DNA in much the same way as MTH1 depletion by siRNA. Crucially, overexpression of the bacterial homolog of MTH1, MutT, which can also hydrolyse 8-oxo-dGTP but is not sensitive to TH588, rendered cells partially resistant to the inhibitor, providing strong evidence that MTH1 catalytic activity was a key, if perhaps not the sole, target conferring tumor cell toxicity. Importantly, the growth of xenografts formed using metastatic melanoma explants derived from a heavily treated patient was inhibited by TH588, indicative of significant anti-cancer activity against bona fide human tumor cells in vivo.Citation8
The accompanying article from Huber et al.Citation9 takes a very different approach, although again the starting point is a previous study of Ras transformation.Citation12 SCH51344 was discovered in a phenotypic screen for compounds capable of suppressing the anchorage-independent growth of Ras-transformed cells. Perplexingly, although SCH51344 efficiently suppressed cell transformation as judged by this criterion, it did not inhibit ERK MAPK signaling, a key downstream effector of Ras oncogenic activity.Citation12 Thus, although clearly anti-oncogenic, the mechanism of action of SCH51344 remained unclear.
To unravel this mystery Huber et al. adopted a chemical-proteomic strategy, generating an SCH51344 affinity matrix and identifying proteins that bound directly to the drug.Citation9 Remarkably, this revealed MTH1 and adenosine kinase (ADK) as the primary cellular targets of SCH51344. Subsequent experiments demonstrated that SCH51344 potently inhibits MTH1 catalytic activity, leading to severely impaired viability together with spontaneous DNA damage and increased incorporation of 8-oxodG and 2-OH-dA into genomic DNA of cancer cells. In contrast, depletion or inhibition of ADK using a specific inhibitor failed to recapitulate any of the effects of SCH51344. As in the study of Gad et al., overexpression of wild-type MTH1, but not a catalytically inactive mutant, partially protected cancer cells against killing, arguing strongly that MTH1 was indeed the biological target through which SCH51344 suppressed Ras transformation.
Because MTH1 hydrolyses the triphosphates of 8-oxodG and 2-OH-dA Huber et al. hypothesized that its active site might also be vulnerable to protein kinase inhibitors, since protein kinases also utilize ATP (and sometimes GTP). Accordingly, a library of protein kinase inhibitor drugs was screened for molecules capable of inhibiting MTH1 catalytic activity. This search revealed crizotinib, a licensed MET-ALK tyrosine kinase inhibitor drug, to be a potent and relatively selective inhibitor of MTH1. Crizotinib is a chiral molecule, and surprisingly, it turns out that whereas the R-enantiomer is active against (and licensed for use against tumors bearing) activated MET-ALK kinases, it is the S-enantiomer that inhibits MTH1. Armed with this knowedge Huber et al. then showed that S-crizotinib kills cancer cells in vitro, inducing spontaneous DNA damage and increased incorporation of 8-oxodG and 2-OH-dA in much the same way as SCH51344. Puzzlingly, although overexpression of MTH1 alleviated DNA damage by S-crizotinib, it did not confer increased cell survival as with SCH51344. The explanation for this is currently unclear; one possibility is that the effect of S-crizotinib on cell survival is mediated through multiple targets including MTH1.
Drugs that act by inhibiting nucleotide biosynthesis, such as methotrexate and hydroxyurea, have of course been long-standing components of the anti-cancer drug armamentarium. It is important to note however that because such agents induce DNA damage and cytotoxicity through nucleotide pool depletion and imbalances, their primary mode of action does not in principle discriminate between normal and cancer cells. By contrast, Gad et al. and Huber et al. propose a very different concept; namely that cancer cells are generically distinct from normal cells in generating a potentially lethal burden of 8-oxodG and 2-OH-dA that is only prevented from entering genomic DNA through the sanitising action of MTH1 (). Thus, when MTH1 is inhibited in cancer cells these aberrant nucleotides are incorporated into genomic DNA at highly elevated levels, leading to an abundance of repair-associated single-strand breaks and gaps that are likely then converted to cytotoxic DSBs through replication or other mechanisms. In fact, the scale of misincorporation seems sufficient to overwhelm the capacity of the BER system, since in both studies alkaline comet assays using purified BER glycosylases OGG1 and MYH1 indicate that substantial amounts of unexcised 8-oxo-G and 2-OH-A accumulate in the genomic DNA of MTH1-inhibited cells. This raises the possibility that inhibition of MTH1 may be mutagenic as well as clastogenic in cancer cells. Whether this contributes to toxicity is currently unclear, however it could potentially complicate MTH1 inhibition as a therapeutic strategy if mutation hastened tumor adaptation.
Figure 1. MTH1 inhibition is selectively toxic to cancer cells. Tumor cells frequently exhibit elevated levels of ROS compared with normal, untransformed cells. ROS can damage many cellular macromolecules including the pool of deoxynucleotide triphosphates (dNTPs) that serve as precursors for DNA synthesis (gray dots). Oxidized nucleotides formed through the action of ROS include 8-oxo-dGTP (red dots) and 2-OH-dATP (green dots), both of which can be utilized by DNA polymerases and thus incorporated into genomic DNA. MTH1 hydrolyses 8-oxo-dGTP and 2-OH-dATP to the corresponding monophophosphate forms, thus preventing incorporation. MTH1 is frequently overexpressed in cancer, possibly as an adaptation to redox stress during tumor progression, and acts to prevent excessive accumulation of 8-oxo-dGTP and 2-OH-dATP in the nucleotide pool. When MTH1 is inhibited in cancer cells by siRNA depletion or through pharmacological inhibition, oxidized nucleotides accumulate, leading to incorporation of 8-oxo-dGTP and 2-OH-dATP into genomic DNA. Inc. 8-oxo-dG and 2-OH-dA is recognized and excised by BER and MMR, resulting in the formation of single-strand breaks and gaps that are subsequently likely converted to DSBs through replication and potentially other mechanisms. The resulting DNA damage is sufficiently severe to induce tumor cell death through a mechanism that is independent of the p53 tumor suppressor. Because normal cells display much lower levels of ROS MTH1 inhibition does not result in excessive accumulation or incorporation of 8-oxo-dGTP and 2-OH-dATP into genomic DNA, thus limiting toxicity. Please refer to the text for further details.
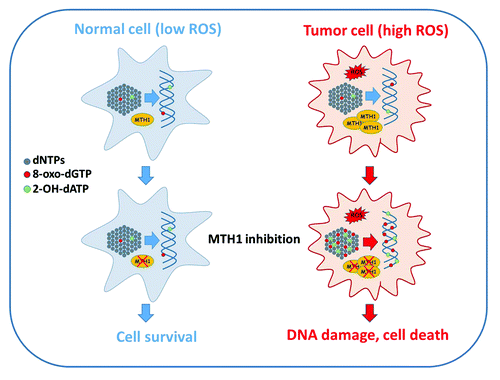
Several important questions emerge from these studies. First, how specific are the cytotoxic effects of MTH1 inhibition for cancer cells? Both studies present evidence that genetically normal, untransformed cells are less sensitive to MTH1 inhibition than the panel of cancer cells tested, a finding consistent with the relatively benign phenotype of MTH1 knockout mice,Citation13 which are developmentally normal and fertile (although they show a very slight predisposition to spontaneous tumorsCitation13) This indicates that MTH1 is dispensable in normal healthy cells and tissues, at least in mice. In addition, both studies show that transformation of genetically normal cells using Ras or SV40 LT oncogenes confers a degree of sensitization to MTH1 inhibition, although clearly such manipulations do not recapitulate the full range of genetic and phenotypic diversity of human cancers. On the other hand, MTH1 is overexpressed in some cancers at least,Citation14-Citation16 possibly as a result of adaptation during tumor development, while tumor cell killing through MTH1 inhibition does not require p53, a function commonly lost in cancer. Given that the excess of oxidized nucleotides in the cancer cells studied most probably arise from ROS generated through redox imbalances it will be important now to establish just how “generic” such aberrations are in human cancer. Finally, if MTH1 inhibition does indeed prove to be a viable anti-cancer strategy, it will be critical to determine what, if any, resistance mechanisms might arise to counter this novel approach. Answers to these questions will be eagerly awaited.
| Abbreviations: | ||
| BER | = | base excision repair |
| MMR | = | mismatch repair |
| PARP | = | poly (ADP) ribose polymerase |
| ROS | = | reactive oxygen species |
| 8-oxo-dG | = | 8-oxo-deoxyguanosine |
| 2-OH-dA | = | 2-hydroxy-deoxyadenosine |
Disclosure of Potential Conflicts of Interest
No potential conflicts of interest were disclosed.
Acknowledgments
V.A.J.S. is a Ramón y Cajal fellow. Her work is supported by a grant from the Spanish MINECO (SAF2010-22126). D.A.G. is an IMBRAIN Investigator at the Universidad de La Laguna, Tenerife, and a Visiting Group Leader at the Beatson Institute for Cancer Research, Glasgow, UK. IMBRAIN is supported by the EU FP7 program and the Gobierno de Canarias (FP7-REGPOT-2012-CT2012-31637-IMBRAIN).
References
- Horne SD, Stevens JB, Abdallah BY, Liu G, Bremer SW, Ye CJ, Heng HH. Why imatinib remains an exception of cancer research. J Cell Physiol 2013; 228:665 - 70; http://dx.doi.org/10.1002/jcp.24233; PMID: 23018746
- Bucheit AD, Davies MA. Emerging insights into resistance to BRAF inhibitors in melanoma. Biochem Pharmacol 2014; 87:381 - 9; http://dx.doi.org/10.1016/j.bcp.2013.11.013; PMID: 24291778
- Farmer H, McCabe N, Lord CJ, Tutt AN, Johnson DA, Richardson TB, Santarosa M, Dillon KJ, Hickson I, Knights C, et al. Targeting the DNA repair defect in BRCA mutant cells as a therapeutic strategy. Nature 2005; 434:917 - 21; http://dx.doi.org/10.1038/nature03445; PMID: 15829967
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 2004; 4:814 - 9; http://dx.doi.org/10.1038/nrc1457; PMID: 15510162
- van Leeuwen IM. Cyclotherapy: opening a therapeutic window in cancer treatment. Oncotarget 2012; 3:596 - 600; PMID: 22711025
- Blagosklonny MV. “Targeting the absence” and therapeutic engineering for cancer therapy. Cell Cycle 2008; 7:1307 - 12; http://dx.doi.org/10.4161/cc.7.10.6250; PMID: 18487952
- Mancias JD, Kimmelman AC. Targeting autophagy addiction in cancer. Oncotarget 2011; 2:1302 - 6; PMID: 22185891
- Gad H, Koolmeister T, Jemth AS, Eshtad S, Jacques SA, Ström CE, Svensson LM, Schultz N, Lundbäck T, Einarsdottir BO, et al. MTH1 inhibition eradicates cancer by preventing sanitation of the dNTP pool. Nature 2014; 508:215 - 21; http://dx.doi.org/10.1038/nature13181; PMID: 24695224
- Huber KV, Salah E, Radic B, Gridling M, Elkins JM, Stukalov A, Jemth AS, Göktürk C, Sanjiv K, Strömberg K, et al. Stereospecific targeting of MTH1 by (S)-crizotinib as an anticancer strategy. Nature 2014; 508:222 - 7; http://dx.doi.org/10.1038/nature13194; PMID: 24695225
- Rai P, Young JJ, Burton DG, Giribaldi MG, Onder TT, Weinberg RA. Enhanced elimination of oxidized guanine nucleotides inhibits oncogenic RAS-induced DNA damage and premature senescence. Oncogene 2011; 30:1489 - 96; http://dx.doi.org/10.1038/onc.2010.520; PMID: 21076467
- Sakumi K, Furuichi M, Tsuzuki T, Kakuma T, Kawabata S, Maki H, Sekiguchi M. Cloning and expression of cDNA for a human enzyme that hydrolyzes 8-oxo-dGTP, a mutagenic substrate for DNA synthesis. J Biol Chem 1993; 268:23524 - 30; PMID: 8226881
- Walsh AB, Dhanasekaran M, Bar-Sagi D, Kumar CC. SCH 51344-induced reversal of RAS-transformation is accompanied by the specific inhibition of the RAS and RAC-dependent cell morphology pathway. Oncogene 1997; 15:2553 - 60; http://dx.doi.org/10.1038/sj.onc.1201424; PMID: 9399643
- Tsuzuki T, Egashira A, Kura S. Analysis of MTH1 gene function in mice with targeted mutagenesis. Mutat Res 2001; 477:71 - 8; http://dx.doi.org/10.1016/S0027-5107(01)00108-7; PMID: 11376688
- Speina E, Arczewska KD, Gackowski D, Zielińska M, Siomek A, Kowalewski J, Oliński R, Tudek B, Kuśmierek JT. Contribution of hMTH1 to the maintenance of 8-oxoguanine levels in lung DNA of non-small-cell lung cancer patients. J Natl Cancer Inst 2005; 97:384 - 95; http://dx.doi.org/10.1093/jnci/dji058; PMID: 15741575
- Kennedy CH, Cueto R, Belinsky SA, Lechner JF, Pryor WA. Overexpression of hMTH1 mRNA: a molecular marker of oxidative stress in lung cancer cells. FEBS Lett 1998; 429:17 - 20; http://dx.doi.org/10.1016/S0014-5793(98)00505-5; PMID: 9657375
- Okamoto K, Toyokuni S, Kim WJ, Ogawa O, Kakehi Y, Arao S, Hiai H, Yoshida O. Overexpression of human mutT homologue gene messenger RNA in renal-cell carcinoma: evidence of persistent oxidative stress in cancer. Int J Cancer 1996; 65:437 - 41; http://dx.doi.org/10.1002/(SICI)1097-0215(19960208)65:4<437::AID-IJC7>3.0.CO;2-Y; PMID: 8621223