Abstract
Wilms' tumour is a paediatric malignancy of the kidneys and is one of the most common solid childhood cancers. The Wilms' tumour 1 protein (WT1) is a transcription factor that can either activate or repress genes involved in growth, apoptosis and differentiation. It is frequently mutated or aberrantly expressed in Wilms' tumour, where the wild type protein would normally act as a tumour suppressor. Several studies, however, have found that wild type WT1 acts as an oncogene in adult tumours, primarily through the inhibition of apoptosis. The expression of WT1 correlates with the aggressiveness of several adult cancers, and its continued expression following treatment is indicative of a poor outcome.
We recently found that the treatment of tumour cell lines with cytotoxic drugs leads to the cleavage of WT1 by the serine protease HtrA2. HtrA2 binds to a specific region of WT1, the suppression domain, and then cleaves WT1 at multiple sites. The HtrA2-mediated proteolysis of WT1 leads to its removal from gene promoter regions and changes in gene expression. Cleavage of WT1 by HtrA2 enhances apoptosis. This event is advantageous to the treatment of adult tumours where WT1 acts as an oncogene. However, when WT1 is acting as a tumour suppressor in paediatric malignancies, proteolysis by HtrA2 would be antagonistic to therapy.
Introduction
Wilms tumor is a pediatric malignancy of the kidneys that affects 1 in 10,000 children.Citation1–Citation3 The tumor arises from abnormal development of the kidneys that leads to increased proliferation of the metanephric mesenchyme instead of differentiation to form the tubules and filtration system of the nephron. The Wilms tumor 1 protein WT1 was identified on the basis of its mutation in approximately 15% of Wilms tumors. Additional genes that have been implicated in Wilms tumor include β-cateninCitation4 and WTX.Citation5
The average age of diagnosis of Wilms tumor is five years and the vast majority of cases involve only one kidney. Most Wilms tumors are sporadic rather than arising from germline mutations.Citation1 Treatment is by a combination of surgery and chemotherapy, but radiotherapy is also used in particularly aggressive cases. Like other pediatric cancers, Wilms tumor has seen a significant increase in survival rate over the last thirty years. Tumors that are detected early have almost 90% survival rates, but those detected at a later stage are significantly lower. Furthermore, due to the toxic effects of treatment, the risk of secondary malignant neoplasms is high. In the treatment of children this can potentially have a significant affect on lifespan. The design of better-targeted therapies towards Wilms tumor would significantly reduce the secondary effects, and furthermore could provide a less traumatic treatment regime for children.
Although only 15% of Wilms tumors contain mutations in WT1, WT1 is overexpressed or shows an imbalance in isoforms in some Wilms tumors.Citation1,Citation6 Wild-type WT1 is also highly expressed in a significant proportion of other tumors including leukemia, lung cancer, melanoma and breast cancer.Citation6–Citation9 Moreover, in many cases, the expression of WT1 correlates with the aggressiveness of the cancer and its continued expression following treatment indicates a poor outcome. The role of WT1 in leukemia has attracted considerable attention over the last few years.Citation6,Citation10 74% of acute myeloid leukemia (AML) and 66% of acute lymphoblastic leukemia (ALL) samples show high levels of wild-type WT1 expression. Moreover, AML and ALL patients with elevated WT1 have a poor prognosis and significantly reduced 5-year survival rates. Mutations of the WT1 gene are frequently present in many pediatric and adult cancers.Citation1,Citation6
Taken together, several studies of a diverse range of adult tumors suggest that WT1 is likely to play a direct role in tumor formation and/or maintenance. Indeed, clinical trials using WT1 peptide vaccines have shown effects in reducing adult leukemia cell counts and also promoting shrinkage of solid tumors.Citation6,Citation9,Citation11–Citation13 Thus, WT1 is a significant target for therapy in both pediatric and adult tumors. However, because WT1 can act as both a tumor suppressor and an oncogene, effective therapies will need to enhance WT1 activity in the former and ablate it in the latter. Moreover, it is critical to gain an understanding of how existing cancer therapies affect cellular WT1, as this will have significant implications for the potential success of the treatment in tumors that are WT1-dependent.
The Destruction of WT1 by HtrA2
We recently reported that cytotoxic drug-treatment of cells derived from tumors that express WT1 results in the proteolysis of WT1 by HtrA2,Citation14,Citation15 (). This leads to the removal of WT1 from the promoters of the c-myc and JunB genes and loss of transcriptional repression. The altered regulation of WT1 target genes enhances apoptosis. We found that WT1 acts in an antiapoptotic manner and antagonizes the effects of cytotoxic drugs. We demonstrated that the downregulation of WT1 by siRNA significantly enhances the effects of cytotoxic drugs in promoting apoptosis. Our results suggest that WT1 is a critical barrier to apoptosis in cancer cells and epitomize the oncogenic actions of WT1.
HtrA2 was identified as a WT1 interaction partner in a yeast two-hybrid screen, using the suppression domain (amino acids 71–101) as bait (see ). We found that HtrA2 cleaves WT1 at at least three sites. Based on the sizes of the WT1 fragments that are generated, and the reported cleavage site preference of HtrA2, the three potential cleavage sites are L94, V286 and L320 (). However, the proteolytic fragments were highly unstable in vivo and it is likely that other sites are also cleaved. In addition, the specific location of the three observed sites was not determined precisely. It will be important to directly analyze the number and location of the HtrA2 cleavage sites in WT1, for example by mass spectrometric analysis of the WT1 proteolytic products that are generated in vitro. This will reveal if any of the proteolytic fragments could potentially retain biological activity. It will also be interesting to determine if there are any cancer-relevant mutations within the proteolytic cleavage sites of WT1.
Antiapoptotic Functions of WT1
Several reports have revealed an antiapoptotic function for WT1, which is consistent with its role as an oncogene in some tumors.Citation12,Citation16–Citation19 How WT1 elicits its oncogenic effects is not clear, and is likely to show cell-type specificity. Several genes that regulate apoptosis have been reported as WT1 targets including Bak, Bcl2, Bcl2A1, c-myc and JunB.Citation16,Citation20–Citation26 WT1 null mice display increased apoptosis of the metanephirc mesenchyme, which leads to the absence of kidney development.Citation27 In addition, treatment of embryonic kidney explants with WT1 siRNA causes the metanephric mesenchyme to undergo apoptosis.Citation28
The +17 amino acid form of WT1, in particular, has been linked with antiapoptotic activity (see ). The ectopic expression of WT1 +17AA can confer resistance to apoptosis in cells treated with apoptosis inducing agents.Citation16,Citation17 Moreover, treatment of cells with siRNA that specifically target the WT1 +17AA isoforms results in the induction of apoptosis.Citation18,Citation22 A recent study noted that the predominant WT1 isoforms expressed in AML are those containing +17AA and that the levels of these isoforms were higher in relapse samples than in diagnostic samples.Citation29 This indicates that WT1 isoforms containing +17AA may be involved in the pathogenesis of relapse.
The resistance to apoptosis associated with WT1 +17AA correlates with increased Bcl-2 expression, and high levels of Bcl-2 have been reported in sporadic Wilms tumors that express elevated wild-type WT1 +17AA.Citation16 Stable expression of WT1 +17AA in cells treated with apoptotic stimuli also results in the downregulation of Bak and protection of cells from apoptosis.Citation12 Thus, the activation or repression of Bcl2 or Bak transcription by WT1 is both cell-type and WT1-isoform specific.
In our recent study of U2OS osteosarcoma cells, we found that WT1 was a repressor of the c-myc and JunB genes. The degradation of WT1 resulted in the removal of WT1 from its binding sites upstream of the c-myc and JunB promoters and their transcriptional upregulation. As stated above, WT1 shows cell context dependent effects on transcriptional regulation and therefore it will be important to analyze the effects of WT1 degradation by HtrA2 on WT1 target genes in multiple cell types. Analysis of U2OS cells, mouse embryonic fibroblasts and other cells lines revealed that WT1 generally acts as an antiapoptotic factor. Taken together with the several previous analyses mentioned above, it is evident that WT1 selectively regulates different genes in different cell types in different directions (activation or repression; see ). It is clear that the mechanism of cell type specificity elicited by WT1 in transcriptional regulation will require further studies. Analysis of the cell type-and context-specific expression of WT1 transcriptional cofactors will help in this regard. Moreover, recent genome-wide studies have uncovered a significant number of WT1 target genes that await further analysis.Citation30,Citation31
WT1 Interaction Partners
WT1 has been reported to interact with a variety of other proteins.Citation32 Indeed, WT1 is present in large complexes in cells.Citation26,Citation33 It is therefore likely that the HtrA2-mediated proteolysis of WT1 can be regulated by its association with other factors. It will be particularly interesting to determine if BASP1 can modulate the cleavage of WT1 by HtrA2. BASP1 binds to the suppression domain of WT1, the same region that also binds to HtrA2.Citation34,Citation35 BASP1 is temporally and spatially coexpressed with WT1 at many sites during development.Citation34 Interestingly, BASP1 has been found to be silenced by gene promoter methylation in hepatocellular carcinoma's and leukemia.Citation36–Citation38 BASP1 can also inhibit cell transformation by the oncogene v-myc.Citation39 The emerging tumor suppressor activities of BASP1 suggest that it is unlikely to inhibit the proteolysis of WT1, which could potentially be oncogenic. Alternatively, BASP1 might augment binding of HtrA2 to WT1 and enhance its cleavage.
Hsp70 also interacts with the N-terminus of WT1 within a region that contains the suppression domainCitation40 (). Although this study did not report an effect of Hsp70 on the transcriptional regulatory activity of WT1, a role for Hsp70 in regulating WT1 activity under apoptotic conditions remains to be tested. Indeed, Hsp70 has known antiapoptotic activities and it will therefore be interesting to determine if Hsp70 regulates the HtrA2-mediated proteolysis of WT1.Citation41
It is not obligatory that the same domain should be targeted in WT1 to elicit an effect on cleavage by HtrA2. For example, the conformation of WT1 could potentially be modified by protein-protein interactions and this could affect the way in which WT1 is presented to HtrA2. Another possibility is that specific cleavage sites in WT1 could be protected by interaction partners. The WT1 zinc finger region contains at least one HtrA2 cleavage site, and is also a major site of interaction with other factors.Citation32 For example, WT1 has been shown to interact with the tumor suppressor p53, an association that requires zinc fingers 1 and 2 of WT1.Citation42 WT1 enhances p53-dependent transcriptional activation by stabilizing p53, resulting in an increase in its half-life, and enhancing the binding of p53 to its consensus sequence. As the zinc finger region of WT1 is a site of HtrA2 proteolysis, this raises the possibility that p53-bound WT1 might regulate WT1 proteolysis by HtrA2.
Prostate apoptosis response-4 (Par-4) engages in interaction with two distinct domains in WT1, one of which we showed to be WT1 splice isoform-specificCitation17,Citation43 (). Through these interactions, Par-4 can act as either a coactivator or corepressor of WT1. Par-4 is rapidly induced at the translation level by proapoptotic stimuli and interacts with WT1. It is likely that Par-4 modifies the regulation of WT1 target genes that are involved in apoptosis, and indeed, the Bcl2 promoter shows both WT1 and Par-4 occupancy.Citation44 As mentioned above, the WT1 +17AA form of WT1 in particular has been demonstrated to be antiapoptotic and is over-represented in several tumor types. The association of Par-4 with the WT1 +17AA region suggests that Par-4 may play a role in the oncogenic activities of WT1. Our recent study found that the +17AA form of WT1 is cleaved by HtrA2, but it remains to be tested if Par-4 can regulate this event.
There are several other WT1 interaction partners of WT1,Citation32 but those mentioned above have established roles in apoptosis and are thus likely modulators of WT1-mediated proteolysis by HtrA2. Considering the cell-context action of WT1 in transcriptional regulation, it will be important to gain a better understanding of the dynamic between WT1 and its cofactors. For example, regulation through cell type specificity of the WT1 cofactors, subcellular localization or post-translational modification.
WT1 Isoforms and Post-Translational Modifications
Our recent report found that the four major isoforms of WT1 (variant in +17AA and KTS) are all cleaved by HtrA2. Two forms of WT1 that lack the N-terminal suppression domain, but contain the transcriptional activation domain, remain to be tested. One form arises from internal translation and lacks the first 126 amino acids of the major forms of WT1.Citation45 The second form, AWT1, is produced from an alternative promoter and lacks the N-terminal 125 amino acids, but contains an extra four amino acids at the N-terminus that are not present in the major forms of WT1.Citation46 The AWT1 promoter is paternally imprinted, which is frequently lost in some Wilms tumors, leading to an increase in the expression of AWT1. Because these two forms of WT1 do not contain the suppression domain, they lack the major HtrA2 binding site that we identified. It will therefore be interesting to determine the effects of HtrA2 on these isoforms of WT1. If they escape regulation by HtrA2, it is possible that they are particularly oncogenic when compared to the more common isoforms.
Ubiquitin-conjugating enzyme 9 (UBC9) has been shown to interact with the N-terminus of WT1, which is likely related to WT1 sumoylationCitation47,Citation48 (see ). WT1 is sumoylated at two sites, K73 and K177, the former of which lies within the HtrA2-binding site. This raises the possibility that sumoylation of WT1 might regulate cleavage by HtrA2. Previous studies failed to determine a function for WT1 sumoylation.Citation48 However, these studies were not performed under apoptotic conditions, which are required for the activation of HtrA2. It will therefore be interesting to determine if the sumoylation of WT1 plays a role in its proteolysis by HtrA2 and if this affects the antiapoptotic activity of WT1.
Functions of HtrA2
The serine protease HtrA2/Omi is homologous to the bacterial high-temperature requirement (HtrA) stress responsive genes, DegP and DegS.Citation49 It encodes a trypsin-like protease with an N-terminal mitochondrial targeting sequence, followed by a transmembrane segment, an inhibitor of apoptosis (IAP)-binding motif (IBM), the catalytic domain, and a C-terminal PDZ interaction domain. HtrA2/Omi is expressed as a pro-enzyme and is targeted to the mitochondrial inter membrane space (IMS) where it is attached to the inner mitochondrial membraneCitation50,Citation51 and undergoes proteolytic maturation. Though HtrA2/Omi mainly localizes to the IMS, a fraction of endogenous HtrA2/Omi resides in the nucleus.Citation14,Citation51–Citation54
Mice with a targeted deletion of the HtrA2/Omi gene display reduced body weight, a significant reduction in the size of organs such as heart, thymus and spleen and progressive loss of neurons in the striatum of the basal ganglia. This results in a progressive neurodegenerative disorder with a parkinsonian phenotype that leads to death of the mice around 30 days after birth.Citation55 An almost identical phenotype was earlier reported in Mnd2 (motor neuron degeneration 2) mice, which are homozygous for a spontaneous occurring HtrA2 Ser276Cys mutation that greatly reduces its catalytic activity,Citation56 hence confirming the importance of the catalytic domain for the maintenance of healthy mitochondria. Though a link between mitochondrial dysfunction and neurodegenerative disorders is well established, the underlying mechanisms are poorly understood.Citation57 It has been speculated that HtrA2/Omi fulfils its protective role in neuronal cells by functioning as a mitochondrial chaperone, similar to its homolog DegP in the periplasmic space in bacteria.Citation49 In agreement with this, loss of HtrA2/Omi has been shown to result in the accumulation of unfolded proteins in the mitochondria and transcriptional upregulation of nuclear genes including the transcription factor CHOP in the brain, which contributes to neuronal cell death.Citation58
Although the phenotype of mice with loss of HtrA2/Omi activity shows no resistance to apoptosis, earlier studies have clearly demonstrated that HtrA2/Omi has pro-apoptotic functions. Apoptotic stimuli induce HtrA2/Omi translocation from the mitochondria into the cytosol where it contributes to apoptosis through caspase dependent and independent mechanisms.Citation49 Cytosolic HtrA2/Omi facilitates caspase activation by degrading their natural inhibitors (IAP), and this mechanism was shown to be evolutionary conserved in Drosophila.Citation59
A Nuclear Function for HtrA2
As stated above, a pool of HtrA2 is located in the nucleus. Interestingly the tumor suppressor WARTS has been shown to be degraded by HtrA2 primarily through direct interaction in the nucleus.Citation53 Moreover, Marabese and coworkers were recently able to demonstrate that after apoptotic stimuli HtrA2 accumulates in the nucleus and cleaves the transcription factor p73α.Citation54 Similar to our observation with WT1, they also observed accumulation of endogenous p73 in HtrA2 knockout mouse embryo fibroblasts confirming that loss of HtrA2 activity leads to accumulation of target proteins in the nucleus. In summary these data strongly support a nuclear function for HtrA2.
WT1 has been shown to shuttle between the cytoplasm and nucleus and contains both nuclear localization sequences and a region required for nuclear export.Citation3 It is not clear if the HtrA2 mediated cleavage of WT1 occurs exclusively within the nucleus or cytoplasm. Following the cleavage of WT1 by HtrA2, the C-terminal fragments that contain the NLS' are primarily located in the nucleus, while the N-terminal fragments are located in the cytoplasm.Citation14 Thus, we were unable to conclude if the HtrA2-mediated proteolysis of WT1 is restricted to a specific cellular compartment. Because HtrA2 is present throughout the cell, we favour the scenario that WT1 is targeted in both compartments. However, it will be important to shed more light on this, especially as WT1 has been found in the polysomes and perhaps plays a role in mRNA function in the cytoplasm.Citation60
The regulation of HtrA2 by other signalling pathways is not well studied. However, the serine/threonine kinases Akt1 and Akt2 have recently been shown to phosphorylate HtrA2 and attenuate its serine protease activity and pro-apoptotic function.Citation61 It remains to be seen whether the nuclear HtrA2 pool can be inactivated by Akt and whether this HtrA2 phosphorylation event is primarily responsible for the high levels of WT1 in tumor cells. Our experiments using HtrA2 siRNAs in U2OS and H1299 cells suggested that WT1 is continually susceptible to HtrA2 under “resting conditions”. Even so, treatment of cells with cytotoxic drugs dramatically enhances WT1 cleavage by HtrA2. Further experiments will be required to determine if either, nuclear HtrA2 is activated, cytoplasmic HtrA2 enters the nucleus or nucleocytoplasmic shuttling of WT1 leads to cytoplasmic degradation of WT1 and its titration from the nucleus. Whichever mechanism is at play, a better understanding of HtrA2 activity and regulation is required, particularly of the nuclear pool. Related to this point, it is notable that the amino acid sequence preference of HtrA2 does not show high selectivity.Citation62 Nevertheless, HtrA2 shows very high specificity in the substrates that it cleaves. A large proteome-wide screen identified only 15 substrates among over 1,000 proteins,Citation63 and in addition 14 others have been independently identified and verified in vivo (see ). It is possible that HtrA2 binding proteins might regulate its activity and substrate specificity, perhaps mediated by the HtrA2 PDZ region. It will therefore be interesting to identify potential modulators of WT1 processing by HtrA2 or indeed if any of the known WT1 interaction partners can fulfil this function.
Therapeutic Implications of HtrA2-Mediated Proteolysis of WT1
As mentioned above, WT1 acts as a tumor suppressor in Wilms tumors and in some childhood leukemia's, but in adult tumors acts as an oncogene. Our finding that chemotherapeutic agents can induce the proteolysis of WT1 by HtrA2 leads to two very important questions regarding the current therapies for WT1-dependent cancers. Firstly, if WT1 is acting as a tumor suppressor, then treatments that induce HtrA2 activity toward WT1 will potentially be detrimental. Secondly, in adult tumors, the effectiveness of chemotherapeutic agents to induce the HtrA2-dependent cleavage of WT1 will likely contribute to the success of treatment. It will therefore be important to gain a better understanding of the dynamics by which different types of chemotherapeutic agents induce WT1 proteolysis by HtrA2. Such studies may lead to a more informed selection of the appropriate chemotherapeutic regime for WT1 dependent cancers.
Furthermore, mutations in WT1 occur in approximately 10% of AML patients at diagnosis, cluster mainly to exons 7 and 9, and are associated with a higher rate of chemo-resistant leukemia, conferring to a negative prognostic outcome.Citation64 It will be important to analyse the functional properties of the mutated WT1 proteins in cancer and determine if they are resistant to HtrA2-mediated proteolysis and thereby confer chemoresistance.
Conclusions
Accumulating evidence has demonstrated that WT1 plays a role in several adult malignancies and that, unlike in some pediatric cancers, WT1 can act as an oncogene. The finding that HtrA2 can degrade WT1 opens up new therapeutic possibilities for the treatment of adult tumors. In addition, these findings suggest that the treatment of pediatric tumors, in which WT1 acts as a tumor suppressor might be optimized to reduce potential harmful effects resulting from HtrA2 mediated proteolysis of WT1.
Figures and Tables
Figure 1 Cytotoxic drugs stimulate the processing of WT1 by HtrA2. HtrA2 (H) is present in the mitochondria and nucleus. Upon exposure to apoptotic agents, HtrA2 cleaves WT1 (W), resulting in its loss from gene promoters, with concomitant changes in the expression of these genes. Those genes that were subject to repression or activation by WT1 would be upregulated and downregulated respectively. The HtrA2-dependent proteolysis of WT1 acts to drive apoptosis.
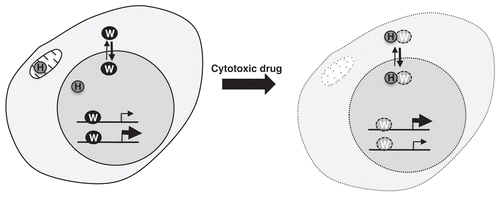
Figure 2 Functional motifs and interactions of WT1. A linear schematic of WT1 is shown with numbering indicating amino acids. Zn is zinc finger, A is the activation domain, R is the repression domain, SD is the suppression domain. The alternative splice sites (17AA and KTS) are indicated. The HtrA2 cleavage sites, the self-association, nuclear export and nucleic acid-binding domains are indicated below and post-translational modifications (sumoylation and phosphorylation) are shown above. The binding sites in WT1 for BASP1, HtrA2, Hsp70, Par-4 and p53 are indicated.
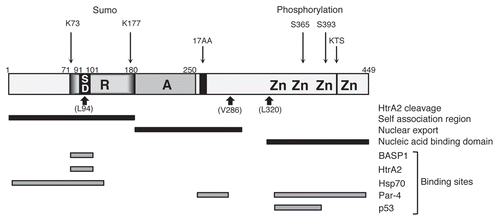
Table 1 List of HtrA2 substrates, with proposed cellular compartment in which cleavage occurs and potential function of substrate cleavage
Acknowledgements
Our work on WT1 has been funded by the AICR, Cancer Research UK, and the Wellcome Trust.
References
- Rivera MN, Haber DA. Wilms' tumour: connecting tumorigenesis and organ development in the kidney. Nat Rev Cancer 2005; 5:1 - 3
- Scholz H, Kirschner KM. A role for the Wilms' tumor protein WT1 in organ development. Physiology 2005; 20:54 - 59
- Hohenstein P, Hastie ND. The many facets of the Wilms' tumour gene, WT1. Hum Mol Genet 2006; 15:196 - 201
- Li CM, Kim CE, Margolin AA, Guo M, Zhu J, Mason JM, et al. CTNNB1 mutations and overexpression of Wnt/beta-catenin target genes in WT1-mutant Wilms' tumors. Am J Pathol 2004; 165:1943 - 1953
- Rivera MN, Kim WJ, Wells J, Driscoll DR, Brannigan BW, Han M, et al. An X chromosome gene, WTX, is commonly inactivated in Wilms tumor. Science 2007; 315:642 - 645
- Yang L, Han Y, Suarez Saiz F, Minden MD. A tumor suppressor and oncogene: the WT1 story. Leukemia 2007; 21:868 - 876
- Pritchard-Jones K, King-Underwood L. The Wilms tumour gene WT1 in leukaemia. Leuk Lymphoma 1997; 27:207 - 220
- Silberstein GB, Van Horn K, Strickland P, Roberts CT Jr, Daniel CW. Altered expression of the WT1 wilms tumor suppressor gene in human breast cancer. Proc Natl Acad Sci USA 1997; 94:8132 - 8137
- Oji Y, Kitamura Y, Kamino E, Kitano A, Sawabata N, Inoue M, et al. WT1 IgG antibody for early detection of nonsmall cell lung cancer and as its prognostic factor. Int J Cancer 2009; 125:381 - 387
- Ariyaratana S, Loeb DM. The role of the Wilms tumour gene (WT1) in normal and malignant haematopoiesis. Expert Rev Mol Med 2007; 9:1 - 17
- Oka Y, Tsuboi A, Oji Y, Kawase I, Sugiyama H. WT1 peptide vaccine for the treatment of cancer. Curr Opin Immunol 2008; 20:211 - 220
- Tatsumi N, Oji Y, Tsuji N, Tsuda A, Higashio M, Aoyagi S, et al. Wilms' tumor gene WT1-shRNA as a potent apoptosis-inducing agent for solid tumors. Int J Oncol 2008; 32:701 - 711
- Zamora-Avila DE, Zapata-Benavides P, Franco-Molina MA, Saavedra-Alonso S, Trejo-Avila LM, Reséndez-Pérez D, et al. WT1 gene silencing by aerosol delivery of PEI-RNAi complexes inhibits B16-F10 lung metastases growth. Cancer Gene Ther 2009; 16:892 - 899
- Hartkamp J, Carpenter B, Roberts SGE. The Wilms' tumor suppressor protein WT1 is processed by the serine protease HtrA2/Omi. Mol Cell 2010; 37:159 - 171
- Essafi A, Hastie ND. WT1 the oncogene: a tale of death and HtrA. Mol Cell 2010; 37:153 - 155
- Mayo MW, Wang CY, Drouin SS, Madrid LV, Marshall AF, Reed JC, et al. WT1 modulates apoptosis by transcriptionally upregulating the bcl-2 protooncogene. EMBO J 1999; 18:3990 - 4003
- Richard DJ, Schumacher V, Royer-Pokora B, Roberts SG. Par4 is a coactivator for a splice isoform-specific transcriptional activation domain in WT1. Genes Dev 2001; 15:328 - 339
- Ito K, Oji Y, Tatsumi N, Shimizu S, Kanai Y, Nakazawa T, et al. Antiapoptotic function of 17AA(+) WT1 (Wilms' tumor gene) isoforms on the intrinsic apoptosis pathway. Oncogene 2006; 25:4217 - 4229
- Hartkamp J, Roberts SGE. The role of the Wilms' tumour-suppressor protein WT1 in apoptosis. Biochem Soc Trans 2008; 36:629 - 631
- Udtha M, Lee SJ, Alam R, Coombes K, Huff V. Upregulation of c-MYC in WT1-mutant tumors: assessment of WT1 putative transcriptional targets using cDNA microarray expression profiling of genetically defined Wilms' tumors. Oncogene 2003; 22:3821 - 3826
- Han Y, San-Marina S, Liu J, Minden MD. Transcriptional activation of c-myc proto-oncogene by WT1 protein. Oncogene 2004; 23:6933 - 6941
- Renshaw J, Orr RM, Walton MI, Te Poele R, Williams RD, Wancewicz EV, et al. Disruption of WT1 gene expression and exon 5 splicing following cytotoxic drug treatment: antisense downregulation of exon 5 alters target gene expression and inhibits cell survival. Mol Cancer Ther 2004; 3:1467 - 1484
- Morrison DJ, English MA, Licht JD. WT1 induces apoptosis through transcriptional regulation of the proapoptotic Bcl-2 family member Bak. Cancer Res 2005; 65:8174 - 8182
- Simpson LA, Burwell EA, Thompson KA, Shahnaz S, Chen AR, Loeb DM. The antiapoptotic gene A1/BFL1 is a WT1 target gene that mediates granulocytic differentiation and resistance to chemotherapy. Blood 2006; 107:4695 - 4702
- Kim HS, Kim MS, Hancock AL, Harper JC, Park JY, Poy G, et al. Identification of novel Wilms' tumor suppressor gene target genes implicated in kidney development. J Biol Chem 2007; 282:16278 - 16287
- Green LM, Wagner KJ, Campbell HA, Addison K, Roberts SGE. Dynamic interaction between WT1 and BASP1 in transcriptional regulation during differentiation. Nucleic Acids Res 2009; 37:431 - 440
- Kreidberg JA, Sariola H, Loring JM, Maeda M, Pelletier J, Housman D, Jaenisch R. WT-1 is required for early kidney development. Cell 1993; 74:679 - 691
- Davies JA, Ladomery M, Hohenstein P, Michael L, Shafe A, Spraggon L, Hastie N. Development of an siRNA-based method for repressing specific genes in renal organ culture and its use to show that the Wt1 tumour suppressor is required for nephron differentiation. Hum Mol Genet 2004; 13:235 - 246
- Gu W, Hu S, Chen Z, Qiu G, Cen J, He B, et al. High expression of WT1 gene in acute myeloid leukemias with more predominant WT1 +17AA isoforms at relapse. Leuk Res 2010; 34:46 - 49
- Hartwig S, Ho J, Pandey P, Macisaac K, Taglienti M, Xiang M, et al. Genomic characterization of Wilms' tumor suppressor 1 targets in nephron progenitor cells during kidney development. Development 2010; 137:1189 - 1203
- Kim MK, McGarry TJ, O'Broin P, Flatow JM, Golden AA, Licht JD. An integrated genome screen identifies the Wnt signaling pathway as a major target of WT1. Proc Natl Acad Sci USA 2009; 106:11154 - 11159
- Roberts SGE. Transcriptional regulation by WT1 in development. Curr Opin Genet Dev 2005; 15:542 - 547
- Iben S, Royer-Pokora B. Analysis of native WT1 protein from frozen human kidney and Wilms' tumors. Oncogene 1999; 18:2533 - 2536
- McKay LM, Carpenter B, Roberts SGE. Regulation of the Wilms' tumour suppressor protein transcriptional activation domain. Oncogene 1999; 18:6546 - 6554
- Carpenter B, Hill KJ, Charalambous M, Wagner KJ, Lahiri D, James DI, et al. BASP1 is a transcriptional cosuppressor for the Wilms' tumour suppressor protein WT1. Mol Cell Biol 2004; 24:537 - 549
- Moribe T, Iizuka N, Miura T, Stark M, Tamatsukuri S, Ishitsuka H, et al. Identification of novel aberrant methylation of BASP1 and SRD5A2 for early diagnosis of hepatocellular carcinoma by genome-wide search. Int J Oncol 2008; 33:949 - 958
- Ross ME, Zhou X, Song G, Shurtleff SA, Girtman K, Williams WK, et al. Classification of pediatric acute lymphoblastic leukemia by gene expression profiling. Blood 2003; 102:2951 - 2959
- Wang J, Coombes KR, Highsmith WE, Keating MJ, Abruzzo LV. Differences in gene expression between B-cell chronic lymphocytic leukemia and normal B cells: a meta-analysis of three microarray studies. Bioinformatics 2004; 20:3166 - 3178
- Hartl M, Nist A, Khan MI, Valovka T, Bister K. Inhibition of Myc-induced cell transformation by brain acid-soluble protein 1 (BASP1). Proc Natl Acad Sci USA 2009; 106:5604 - 5609
- Maheswaran S, Englert C, Zheng G, Lee SB, Wong J, Harkin DP, et al. Inhibition of cellular proliferation by the Wilms tumor suppressor WT1 requires association with the inducible chaperone Hsp70. Genes Dev 1998; 12:1108 - 1120
- Galluzzi L, Giordanetto F, Kroemer G. Targeting HSP70 for cancer therapy. Mol Cell 2009; 36:176 - 177
- Maheswaran S, Englert C, Bennett P, Heinrich G, Haber DA. The WT1 gene product stabilizes p53 and inhibits p53-mediated apoptosis. Genes Dev 1995; 9:2143 - 2156
- Johnstone RW, See RH, Sells SF, Wang J, Muthukkumar S, Englert C, et al. A novel repressor, par-4, modulates transcription and growth suppression functions of the Wilms' tumor suppressor WT1. Mol Cell Biol 1996; 16:6945 - 6956
- Cheema SK, Mishra SK, Rangnekar VM, Tari AM, Kumar R, Lopez-Berestein G. Par-4 transcriptionally regulates Bcl-2 through a WT1-binding site on the bcl-2 promoter. J Biol Chem 2003; 278:19995 - 20005
- Scharnhorst V, Dekker P, van der Eb AJ, Jochemsen AG. Internal translation initiation generates novel WT1 protein isoforms with distinct biological properties. J Biol Chem 1999; 274:23456 - 23462
- Dallosso AR, Hancock AL, Brown KW, Williams AC, Jackson S, Malik K. Genomic imprinting at the WT1 gene involves a novel coding transcript (AWT1) that shows deregulation in Wilms' tumours. Hum Mol Genet 2004; 13:405 - 415
- Wang ZY, Qiu QQ, Seufert W, Taguchi T, Testa JR, Whitmore SA, et al. Molecular cloning of the cDNA and chromosome localization of the gene for human ubiquitin-conjugating enzyme 9. J Biol Chem 1996; 271:24811 - 24816
- Smolen GA, Vassileva MT, Wells J, Matunis MJ, Haber DA. SUMO-1 modification of the Wilms' tumor suppressor WT1. Cancer Res 2004; 64:7846 - 7851
- Vande Walle L, Lamkanfi M, Vandenabeele P. The mitochondrial serine protease HtrA2/Omi: an overview. Cell Death Differ 2008; 15:453 - 460
- Hegde R, Srinivasula SM, Zhang Z, Wassell R, Mukattash R, Cilenti L, et al. Identification of Omi/HtrA2 as a mitochondrial apoptotic serine protease that disrupts inhibitor of apoptosis protein-caspase interaction. J Biol Chem 2002; 277:432 - 438
- Martins LM, Iaccarino I, Tenev T, Gschmeissner S, Totty NF, Lemoine NR, et al. The serine protease Omi/HtrA2 regulates apoptosis by binding XIAP through a reaper-like motif. J Biol Chem 2002; 277:439 - 444
- Gray CW, Ward RV, Karran E, Turconi S, Rowles A, Viglienghi D, et al. Characterization of human HtrA2, a novel serine protease involved in the mammalian cellular stress response. Eur J Biochem 2000; 267:5699 - 5710
- Kuninaka S, Iida SI, Hara T, Nomura M, Naoe H, Morisaki T, et al. Serine protease Omi/HtrA2 targets WARTS kinase to control cell proliferation. Oncogene 2007; 26:2395 - 2406
- Marabese M, Mazzoletti M, Vikhanskaya F, Broggini M. HtrA2 enhances the apoptotic functions of p73 on bax. Cell Death Differ 2008; 15:849 - 858
- Martins LM, Morrison A, Klupsch K, Fedele V, Moisoi N, Teismann P, et al. Neuroprotective role of the Reaper-related serine protease HtrA2/Omi revealed by targeted deletion in mice. Mol Cell Biol 2004; 24:9848 - 9862
- Jones JM, Datta P, Srinivasula SM, Ji W, Gupta S, Zhang Z, et al. Loss of Omi mitochondrial protease activity causes the neuromuscular disorder of mnd2 mutant mice. Nature 2003; 425:721 - 727
- Lin MT, Beal MF. Mitochondrial dysfunction and oxidative stress in neurodegenerative diseases. Nature 2006; 443:787 - 795
- Moisoi N, Klupsch K, Fedele V, East P, Sharma S, Renton A. Mitochondrial dysfunction triggered by loss of HtrA2 results in the activation of a brain-specific transcriptional stress response. Cell Death Differ 2009; 16:449 - 464
- Challa M, Malladi S, Pellock BJ, Dresnek D, Varadarajan S, Yin YW, et al. Drosophila Omi, a mitochondrial-localized IAP antagonist and proapoptotic serine protease. EMBO J 2007; 26:3144 - 3156
- Niksic M, Slight J, Sanford JR, Caceres JF, Hastie ND. The Wilms' tumour protein (WT1) shuttles between nucleus and cytoplasm and is present in functional polysomes. Hum Mol Genet 2004; 13:463 - 471
- Yang L, Sun M, Sun XM, Cheng GZ, Nicosia SV, Cheng JQ. Akt attenuation of the serine protease activity of HtrA2/Omi through phosphorylation of serine 212. J Biol Chem 2007; 282:10981 - 10987
- Martins LM, Turk BE, Cowling V, Borg A, Jarrell ET, Cantley LC, Downward J. Binding specificity and regulation of the serine protease and PDZ domains of HtrA2/Omi. J Biol Chem 2003; 278:49417 - 49427
- Vande Walle L, Van Damme P, Lamkanfi M, Saelens X, Vandekerckhove J, Gevaert K, Vandenabeele P. Proteome-wide Identification of HtrA2/Omi Substrates. J Proteome Res 2007; 6:1006 - 1015
- Owen C, Fitzgibbon J, Paschka P. The clinical relevance of Wilms Tumour 1 (WT1) gene mutations in acute leukaemia. Hematol Oncol 2010; 28:13 - 19
- Savopoulos JW, Carter PS, Turconi S, Pettman GR, Karran EH, Gray CW, et al. Expression, purification and functional analysis of the human serine protease HtrA2. Protein Expr Purif 2000; 19:227 - 234
- Seong YM, Choi JY, Park HJ, Kim KJ, Ahn SG, Seong GH, et al. Autocatalytic processing of HtrA2/Omi is essential for induction of caspase-dependent cell death through antagonizing XIAP. J Biol Chem 2004; 279:37588 - 37596
- Park HJ, Kim SS, Seong YM, Kim KH, Goo HG, Yoon EJ, et al. Beta-amyloid precursor protein is a direct cleavage target of HtrA2 serine protease: implications for the physiological function of HtrA2 in the mitochondria. J Biol Chem 2006; 281:34277 - 34287
- Yang QH, Church-Hajduk R, Ren J, Newton ML, Du C. Omi/HtrA2 catalytic cleavage of inhibitor of apoptosis (IAP) irreversibly inactivates IAPs and facilitates caspase activity in apoptosis. Genes Dev 2003; 17:1487 - 1496
- Srinivasula SM, Gupta S, Datta P, Zhang Z, Hegde R, Cheong N, et al. Inhibitor of apoptosis proteins are substrates for the mitochondrial serine protease Omi/HtrA2. J Biol Chem 2003; 278:31469 - 31472
- Trencia A, Fiory F, Maitan MA, Vito P, Barbagallo AP, Perfetti A, et al. Omi/HtrA2 promotes cell death by binding and degrading the anti-apoptotic protein ped/pea-15. J Biol Chem 2004; 279:46566 - 46572
- Cilenti L, Soundarapandian MM, Kyriazis GA, Stratico V, Singh S, Gupta S, et al. Regulation of HAX-1 anti-apoptotic protein by Omi/HtrA2 protease during cell death. J Biol Chem 2004; 279:50295 - 50301
- Vande Walle L, Wirawan E, Lamkanfi M, Festjens N, Verspurten J, Saelens X, et al. The mitochondrial serine protease HtrA2/Omi cleaves RIP1 during apoptosis of Ba/F3 cells induced by growth factor withdrawal. Cell Res 2010; 20:421 - 433
- Wang CY, Lin YS, Su WC, Chen CL, Lin CF. Glycogen synthase kinase-3 and Omi/HtrA2 induce annexin A2 cleavage followed by cell cycle inhibition and apoptosis. Mol Biol Cell 2009; 20:4153 - 4161
- Park HM, Kim GY, Nam MK, Seong GH, Han C, Chung KC, et al. The serine protease HtrA2/Omi cleaves Parkin and irreversibly inactivates its E3 ubiquitin ligase activity. Biochem Biophys Res Commun 2009; 387:537 - 542
- Balakrishnan MP, Cilenti L, Mashak Z, Popat P, Alnemri ES, Zervos AS. THAP5 is a human cardiac-specific inhibitor of cell cycle that is cleaved by the proapoptotic Omi/HtrA2 protease during cell death. Am J Physiol Heart Circ Physiol 2009; 297:643 - 653