Abstract
Polycomb group (PcG) proteins are overexpressed in several human malignancies including breast cancer. In particular, aberrant expression of BMI1 and EZH2 has been linked to metastasis and poor prognosis in cancer patients. At present, very little is known about the pharmacological inhibitors of PcG proteins. Here we show that histone deacetylase inhibitors (HDACi) downregulate expression of BMI1. Treatment of MCF10A cells, which are immortal non-transformed breast epithelial cells, and breast cancer cells with HDACi led to decreased expression of BMI1. We further show that downregulation of BMI1 by HDACi results due to the transcriptional downregulation of BMI1 gene. Specifically, we show that primary transcription and promoter activity of BMI1 is suppressed upon treatment with HDACi. Furthermore, downregulation of BMI1 was accompanied by a decrease in histone 2A lysine 119 ubiquitination (H2AK119Ub), which is catalyzed by BMI1 containing polycomb repressive complex 1. HDACi treatment also led to derepression of growth inhibitory genes and putative tumor suppressors, which are known to be silenced by PcG proteins and polycomb repressive complexes (PRCs). In summary, our findings suggest that BMI1 is an important therapy target of HDACi, and that HDACi can be used alone or in combination with other therapies to inhibit growth of tumors that overexpress PcG proteins such as BMI1.
Introduction
Polycomb Group (PcG) proteins are evolutionarily conserved epigenetic regulators of development.Citation1–Citation3 These proteins also regulate proliferation and differentiation of cells via epigenetic silencing of important growth regulatory genes.Citation3,Citation4 There is increasing evidence that deregulated expression of PcG proteins contribute to cancer development. Aberrant expression of PcG proteins, in particular BMI1 and EZH2 is associated with several human malignancies. For example, BMI1 is overexpressed in a number of cancers, such as myeloid leukemia,Citation5 non-small cell lung cancer,Citation6 colorectal cancer,Citation7 breast and prostate cancers,Citation8,Citation9 and head and neck cancers.Citation10,Citation11 Importantly, overexpression of BMI1 and EZH2 correlates with therapy failure in cancer patients, and BMI1 expression is thought to promote stem-ness in tumor cells.Citation8,Citation12
PcG proteins assemble into polycomb repressive complexes (PRCs), which possess histone posttranslational modifications (PTMs) activities.Citation3 BMI1 is a rate-limiting constituent of PRC1, which ubiquitinates histone 2A at lysine 119 residue (H2AK119Ub modification).Citation13 On the other hand, PRC2 which initiates PRC-mediated gene silencing contain EZH2, SUZ12 and EED. Histone methyl transferase activity of EZH2 trimethylates lysine 27 residue of histone 3 (H3-K27me3 modification).Citation14,Citation15 Histone modifications catalyzed by PRCs lead to compaction of chromatin and silencing of important tumor suppressors, developmental regulators and differentiation-specific genes.Citation16 As BMI1 and EZH2, and PRC-mediated histone PTM activities are often upregulated in tumors, it is conceivable that inhibitors of BMI1 and EZH2 are likely to have high therapeutic value.
Recently, it was reported that histone deacetylase inhibitors (HDACi) could downregulate expression of PRC2 constituents EZH2, SUZ12 and EED in human acute leukemia cells.Citation17 HDACi are very promising epigenetic therapy reagents, which induce growth inhibition, cell cycle arrest, premature senescence and cell death in tumor cells.Citation18,Citation19 Although the primary targets of HDACi are histone deacetylases, the mechanisms of HDACiinduced phenotypes remain unclear.Citation18,Citation19 These mechanisms and phenotypes likely involve several molecular targets and multiple substrates of HDACs.Citation20 In this context, here we report that HDACi downregulate expression of the PcG protein BMI1 and PRC1-catalyzed H2AK119Ub activity in immortal breast epithelial and breast cancer cells. In agreement with previously reported results in human acute leukemia cells,Citation17 we also observed downregulation of EZH2 and EZH2-dependent H3K27me3 activity by HDACi in breast cancer cells. We further explored the mechanism of downregulation of BMI1 by HDACi. Our data suggest that HDACi suppress transcription of BMI1.
Results
HDACi downregulate BMI1 in breast epithelial cells.
We first determined the effects of three different HDACi on the expression of the BMI1 and EZH2 in a normal immortal breast epithelial cell line MCF10A, and a breast cancer cell line MCF7. Cells in culture were treated with different doses of sodium butyrate (NaB), and valproic acid (VPA) for 24 h. After treatment, cells were harvested and analyzed for the expression of BMI1 and EZH2 using western blot analysis. Our results indicated that HDACi treatment of all the cells led to dose-dependent decreased expression of BMI1 and EZH2 (). Similar results were obtained using Trichostatin A (TSA) (Suppl. Fig. S1). BMI1 and EZH2 were also downregulated by HDACi in other breast cancer cell lines such as MDA-MB-231, T47D and BT549 breast cancer cells (Suppl. Fig. S2). BMI1 is usually detected as two or more bands of different mobilities while EZH2 is always detected as a single band. Our unpublished data indicate that the multiple bands of BMI1 likely represent phosphorylated and unphosphorylated forms of BMI1. To confirm the biological activity of HDACi used in this study, we also determined expression of p21, and acetylated histone 3 (Ac-H3) in NaB- and VPA-treated cells. Consistent with previously reported data,Citation21–Citation24 HDACi treatment led to dose-dependent increase in p21 and Ac-H3. p21 was also upregulated by HDACi in MDA-MB-231 cells (Suppl. Fig. S2A), which express a mutant p53. These results are consistent with previous reports suggesting that HDACi generally induce p21 in a p53-independent manner.Citation21–Citation24
HDACi repress transcription of BMI1.
In order to examine the possible mechanism of BMI1 downregulation by HDACi, we determined whether the downregulation of BMI1 by HDACi is due to transcriptional repression of BMI1. First, we performed qRT-PCR analysis of BMI1 expression in mock and HDACi-treated MCF10A and MCF7 cells. qRT-PCR data suggested a dose-dependent decrease of BMI1 mRNA upon treatment with HDACi in both cell types (, Suppl. Fig. S3A). Using qRT-PCR assay, we also determined that EZH2 expression is similarly downregulated at the mRNA level by HDACi (. Suppl. Fig. S3A). Since downregulation of mRNA could reflect transcriptional or post-transcriptional regulation, we examined the effect of HDACi on primary transcription of BMI1 using primary transcript (PT) RT-PCR. Compared to standard RT-PCR assay, PT RT-PCR assay more accurately reflects transcriptional regulation of a gene in its native state.Citation25,Citation26 Using primers that can detect unspliced pre-mRNA, our result suggested that HDACi downregulate transcription of both BMI1 and EZH2 genes (, Suppl. Fig. S3B).
Downregulation of BMI1 by HDACi is indirect.
Next to determine whether the HDACi-mediated transcriptional repression of BMI1 is a direct or an indirect effect, we performed a time-course experiment. MCF10A and MCF7 cells were treated with 4 mM NaB for 0, 3, 6, 12 and 24 h, and total cell lysates were analyzed for the expression of BMI1, Ac-H3 and p21. Our results indicated that HDCAi repression of BMI1 is time dependent but relatively a late event when compared to upregulation of histone acetylation (a direct effect of HDACi) and p21 induction (). Significant upregulation of Ac-H3 (acetylated Histone 3) is detected within 3 h, while >2 fold induction of p21 was detected by this time point. H3-acetylation showed no further increase and p21 upregulation peaked around 12 h time point. In contrast to H3-acetylation and p21 upregulation, about two fold downregulation of BMI1 occurred only by 12 h and a significant downregulation was noticed only at the 24 h time point (). We also confirmed our results using RT-PCR assays, which showed that the significant transcriptional downregulation of BMI1 occurs at/after 12 h time point (). Our data also indicated that significant downregulation of EZH2 occurs after 12 h of treatment in MCF10A and MCF7 cells (not shown).
In case of HDACi targets that are upregulated by HDACi-treatment, it has been shown that increase in histone acetylation correlates with increased binding of acetylated histones to genes encoding a particular target such as p21.Citation23 To explain downregulation of BMI1, we hypothesized that it is possible that despite globally increased histone acetylation, the actual binding of acetylated histone to the promoter regions of BMI1 may be decreased resulting into closed chromatin conformation and hence repression of it. To probe this hypothesis, we performed a chromatin immunoprecipitation linked PCR (ChIP) assay to determine the binding of Ac-H3 to the promoter regions of BMI1 and p21. Our results indicated that in case of BMI1 promoter there is no significant change in binding of Ac-H3 with or without HDACi treatment (). As expected, the binding of Ac-H3 increased to p21 promoter after HDACi treatment (). Thus the downregulation of BMI1 does not result due to decreased binding of Ac-H3 to its promoter region. These results are not surprising as it is known that changes in binding of acetylated histones in response to HDACi treatment is gene specific.Citation23 Collectively, our data indicate that HDACi indirectly regulate the transcription of BMI1.
Downregulation of BMI1 by HDACi is independent of c-Myc.
Next, we hypothesized that HDACi may downregulate a positive regulator of BMI1, which may indirectly result in repression of BMI1. Recently, we have reported that BMI1 is positively regulated by the transcription factor c-Myc via an E-box binding site present in its promoter.Citation25 Furthermore, c-Myc has been identified as one of the targets of HDACi in certain cell types.Citation27–Citation30 Hence, we examined whether the transcriptional downregulation of BMI1 is mediated by repression of c-Myc by HDACi in MCF10A and MCF7 cells. Our data indicated that consistent with previous observations in other cell types,Citation27–Citation30 c-Myc is downregulated by HDACi in both MCF10A and MCF7 cells (). Next, we transfected MCF7 cells with BMI1 promoter-reporters that contain an intact E-box (WT), a mutant E-box (Mut), or have a deletion that spans E-box (ΔMyc), and treated cells with NaB. Quantification of these promoter-reportersdriven luciferase activity showed that HDACi treatment led to a dose-dependent decrease in BMI1 promoter activity independent of the presence or absence of a functional E-box (). However, consistent with our published data,Citation25 E-box mutated and deleted promoter-reporter constructs exhibited lower activity (). Hence, we conclude that the transcriptional repression of BMI1 by HDACi is independent of c-Myc downregulation by HDACi in MCF7 cells, and that the residual c-Myc is sufficient to transactivate BMI1 promoter in these cells.
HDACi treatment lead to downregulation of H2AK119Ub and H3K27me3.
Next, we determined whether downregulation of BMI1 results in decrease in BMI1-dependent PRC1 activity, and EZH2-dependent PRC2 activity. Using antibodies against BMI1, H2AK119Ub, EZH2 and H3K27me3, we performed western blot analysis of control and HDACi treated MCF10A and MCF7 cells. The results indicated that HDACi treatment led to decreased levels of both H2AK119Ub and H3K27me3 (). In order to correlate the downregulation of BMI1 and EZH2 with decreased levels of H2AK119Ub and H3K27me3 respectively, we performed co-immunostaining of control and HDACi treated MCF10A cells using respective antibodies. Our results indicated that on a single cell basis, HDACi treatment led to downregulation of BMI1, which resulted in corresponding decrease in H2AK119Ub (). Similarly, treatment with HDACi led to downregulation of nuclear EZH2 and corresponding decrease in levels of H3K27me3. HDACi also downregulated H2AK119Ub and H3K27me3 in MCF7 cells (not shown).
HDACi treatment lead to premature senescence or apoptosis.
HDACi-treatment is known to exert anti-oncogenic effects via an array of phenotypes such as cell death, senescence and differentiation.Citation18,Citation19 Hence, we determined HDACi-induced phenotypes of MCF10A and MCF7 cells. We observed visual morphological differences after HDACi-treatment in these two cell lines. MCF10A cells appeared to have growth arrested but there was little or no visual cell death; on the other hand HDACitreatment of MCF7 cells led to induction of apoptotic morphology. Using trypan blue exclusion assay, we quantified live and dead cells in control and HDACi-treated MCF10A and MCF7 culture. Our data indicated that NaB treatment of MCF7 but not MCF10A cells led to significant cell death particularly at a higher drug dose (4 mM NaB) ().
Induction of a cell death phenotype by HDACi-treatment of MCF7 cells but not MCF10A cells was further confirmed by analysis of PARP, and cell death mediators such as BAX and BCL2. Our data indicate that NaB treatment led to downregulation of BCL2 and generation of cleaved PARP product in MCF7 but not in MCF10A cells (). Interestingly BCL2 levels increased in MCF10A cells (). Results of trypan blue staining were further confirmed by Annexin V assay using FACS analysis of NaB-treated MCF10A and MCF7 cells. As determined by Annexin V positive fraction of cells, our data indicated that after HDACi treatment, there was no increase in apoptosis in MCF10A cells; however, the percentage of apoptotic cells increased from 1% in untreated control cells to 9% in 2 mM NaB- and 16% in 4 mM NaB-treated cells (). There was a minor increase in necrotic cell population (PI and Annexin V double positive) in both MCF10A and MCF7 cultures, which may reflect a non-specific effect of HDACi ().
Because MCF10A cells did not exhibit significant cell death; we reasoned that these cells may get growth arrested via induction of premature senescence, which is known to be induced by HDACi in certain cell types.Citation20,Citation31,Citation32 To examine this possibility, we performed Senescence Associated-beta-galactosidase (SA-β-gal) stainingCitation33 of control and NaB-treated (96 h) cells and quantified stained cells under a light microscope. We also quantified number of SA-β-gal positive cells in cells that were treated with NaB for 48 h and then grown in drug-free medium for additional 48 h. Our data indicated that HDACi-treatment led to induction of senescence-like state upon HDACi-treatment in MCF10A cells ().
HDACi treatment lead to derepression of PcG targets.
To determine the functional significance of HDACi-induced downregulation of BMI1 and EZH2, we determined the expression of potential targets of PcG proteins and PRCs. Specifically, tumor suppressor p57 and IGFBP2 have been shown to be repressed by EZH2 and EZH2-containing PRC2.Citation34,Citation35 Using RT-PCR analysis, our data suggested a modest upregulation of p57 by downregulation of BMI1 using an RNAi approach in MCF10A cells (Suppl. Fig. S4A and B). Moreover, using ChIP analysis we also detected binding of BMI1 to p57 promoter (Suppl. Fig. S4C). Thus, our data suggest that in addition to EZH2, BMI1 is also a bona fide repressor of p57. Next, we treated MCF10A and MCF7 cells with different concentrations of NaB and examined the expression of p57, IGFBP2 and IGFBP3 by qRT-PCR analysis. Our results indicated that HDACi-treatment led to dosedependent upregulation of p57, IGFBP2 and IGFBP3 in both MCF10A and MCF7 cells (, Suppl. Fig. S5). VPA and TSA also upregulated p57, IGFBP2 and IGFBP3 in MCF10A and MCF7 cells (Suppl. Fig. S5). However, compared to MCF7 cells, upregulation of IGFBP2 and IGFBP3 was much more pronounced in MCF10A cells, which may be related to senescence induction rather than cell death phenotype in MCF10A cells. As expected, MCF10A and MCF7 cells did not express p16INK4a and there was no increase in its expression after HDACi-treatment (Suppl. Fig. S5).
Discussion
Epigenetic alterations are common to many cancers including breast cancer. Recently, PcG proteins have emerged as an important class of proteins regulating epigenetic alterations via histone PTM activities.Citation1–Citation3 PcG proteins, in particular BMI1 and EZH2 are often overexpressed in many types of cancers, where they are associated with epigenetic silencing of important tumor suppressors. Hence, PcG proteins and PcG proteins mediated histone PTM activities are attractive cancer therapy targets. At present very little is known about therapy reagents that can target PcG proteins to reverse epigenetic silencing of tumor suppressors and differentiation-related genes. In this report, we provide evidence for negative regulation of expression of PcG proteins BMI1 and EZH2 by HDACi in immortal breast epithelial cells and breast cancer cells.
HDACi of diverse classes such as NaB, VPA, TSA, suberoylanalide hydroxamic acid (SAHA), LBH589 and LAQ824 etc., are thought to modulate gene expression and induce complex anti-oncogenic phenotypes by directly modulating chromatin structure via increased acetylation of core histones.Citation18,Citation19 Activation of growth inhibitory genes such as p21 has been shown to coincide with increased acetylation of H3 and H4 by HDACi.Citation19,Citation20 HDACi can also downregulate growth promoting genes such as c-Jun,Citation36 CyclinsCitation37,Citation38 and c-Myc.Citation27–Citation30 At present, very little is known about the mechanisms of downregulation of prooncogenic or growth promoting genes by HDACi. It may involve transcriptional regulation of a particular gene or post-translational regulation of a protein that is HDACi target. For example, HDACi repress ER-α-dependent transcription,Citation17,Citation37 and promotes proteasomal degradation of Cyclin D1.Citation37 HDACi has also been reported to promote ubiquitin-dependent proteasomal degradation of DNMT1.Citation39
With respect to the mechanism of downregulation of BMI1, our studies suggest that it is primarily downregulated at the transcription level by HDACi. BMI1 is positively regulated by c-Myc and E2F transcription factors.Citation25,Citation40 c-Myc is also a target of HDACiCitation27–Citation30 and our data also suggest that HDACi can downregulate c-Myc in breast cancer cells. Hence it is possible that HDACi-induced repression of BMI1 involves regulation of c-Myc and E2F by HDACi. However, promoter reporter assays suggest that the promoter region of BMI1 that does not contain either E2F- or c-Myc-binding site is also responsive to HDACi, thus suggesting that HDACi-mediated repression of BMI1 is independent of these transcription factors. Our detailed time course experiment suggests that the repression of BMI1 is an indirect effect of HDACitreatment. Furthermore our ChIP experiment suggests that the transcriptional regulation of BMI1 is independent of increased histone acetylation by HDACi. Based on these data, we speculate that the repression of BMI1 by HDACi is mediated by a negative regulator(s) of BMI1, which is induced upon HDACi treatment in normal immortal and transformed breast epithelial cells.
Induction of cell death in breast cancer cells by HDACi is consistent with similar reports in the literature.Citation18,Citation19 Importantly, data presented in the present study also suggest that in contrast to fully transformed cancer cells, HDACi-treatment leads to induction of premature senescence in immortal cells such as MCF10A. Immortalization is a prerequisite for cell transformation. 41 Senescence on the other hand, is an anti-oncogenic phenotype and its induction contributes to tumor suppressive effect of various chemotherapy drugs.Citation41 Hence, the induction of premature senescence in immortal cells by HDACi can prevent further transformation of immortal cells that will eventually give rise to fully transformed phenotype in vitro and possibly tumor formation in vivo. Induction of apoptosis and modulation of various genes associated with it by HDACi has been extensively studied in various cell types.Citation18,Citation19 Downregulation of BCL2 by HDACi has been reported in various cancer cell lines.Citation42–Citation44 This study does not particularly address the induction of cell death pathways by HDACi in breast cancer cells; however, we did notice downregulation of BCL2 by HDACi in MCF7 cells but not in MCF10A cells. Although, MCF10A cells expressed relatively low levels of BCL2, HDACi-treatment led to upregulation of BCL2 in these cells. Induction of a premature senescent phenotype in MCF10A cells is consistent with published data, which suggests that BCL2 is upregulated in senescent fibroblastsCitation45 and that BCL2 determines therapy outcome with respect to senescence and apoptosis.Citation46 Based on our data, we speculate that the differential regulation of BCL2 by HDACi may determine the phenotype of HDACi-treated cells. Apart from BCL2, it is also possible that other regulators of apoptosis are differentially expressed in MCF10A and MCF7 cells in response to HDACi treatment, which remain to be determined.
Although the individual PcG proteins could regulate gene expression, they are known to act as silencers via PRCs. PRC1 and PRC2 are the two major repressive complexes implicated in the development,Citation1–Citation3 and their deregulation is thought to contribute to tumorigenesis.Citation1–Citation3 The widely accepted model of PcG mediated gene silencing proposes that silencing of PcG targets requires both PRC1 and PRC2, and that these complexes act in hierarchical fashion.Citation1–Citation3 The silencing is initiated by PRC2 complex (initiation complex), specifically, its HMT activity trimethylates H3K27 and H3K9. The trimethylation of H3 creates an epigenetic signature or docking site for PRC1 (maintenance complex), which binds to chromatin with methylated H3 and ubiquitinates H2A. The binding of PRC1 and ubiquitination of H2A is thought to cause further chromatin compaction and also directly or indirectly interfere with the recruitment of Pol II basal transcriptional machinery, ultimately resulting in gene silencing. By downregulating the expression of BMI1 and EZH2, which are the major components of PRC1 and PRC2 respectively, HDACi can disrupt the assembly of PRCs, and downregulate histone PTM activities of both PRC1 and PRC2. Indeed, our data indicate that HDACi-treatment lead to substantial reduction in both H3K27me3 and H2AK119Ub levels in MCF10A and MCF7 cells.
The most well studied target of PcG proteins and PRCs is p16INK4A locus,Citation47 which is deleted and/or methylated in most breast cancer cells including MCF7 and MCF10A cells. Hence, PRCs most likely target other tumor suppressors in breast cancer cells. Identification of tumor suppressors that are silenced by PRCs in breast cancer cells and breast tumors is important for the development of breast cancer therapies. Studies present here predict that HDACi should activate such tumor suppressors resulting in inhibition of tumor growth. One such potential tumor suppressor is p57 (also known as CDKN1C), which was recently shown to be silenced in breast tumors.Citation35 Other studies have also implicated p57 in the development of breast cancer.Citation48,Citation49 Importantly, expression of p57 was shown to be reactivated by inhibition of PcG protein EZH2 and PRC2-mediated H3K27me3 activity.Citation35 Our data suggest that BMI1 and possibly PRC1-mediated H2AK119Ub activity also regulate expression of p57. Hence, p57 expression can be used as a functional output of the inhibition of PRC1 and PRC2 activities by HDACi in breast cancer cells. Indeed our studies show strong induction of p57 by HDACi in both MCF7 and MCF10A cells. In addition to p57, we also observed upregulation of IGFBP2 and IGBP3, the two other related growth inhibitors by HDACi in breast cancer cells, which may play a role in HDACi-mediated phenotypes such as cell death and induction of premature senescence.
In summary, we have presented evidence for BMI1 as being an important transcriptional repression target of HDACi in breast cancer cells and immortal breast epithelial cells. Our studies also highlight the functional relevance of inhibition of PcG proteins by HDACi. BMI1 has been proposed to promote stem-ness in tumors and its overexpresion has been correlated with a death from cancer signature in cancer patients.Citation8,Citation12 Recently a micro RNA cluster miR-200c was shown to regulate BMI1 expression in mammary epithelial stem cells, and its downregulation was shown to result in BMI1 upregulation and expansion of breast cancer stem cells.Citation50 Thus BMI1 is an attractive therapy target to treat breast cancer, and eradicate breast cancer stem cells from the primary tumor mass, which can otherwise give rise to tumor recurrence. In summary, our studies have important implications for the development of breast cancer therapy targeted towards inhibition of PcG proteins and reactivation of tumor suppressors that are silenced by PRCs in breast cancer cells, and eradication of breast cancer stem cells.
Materials and Methods
Cells, cell culture methods and HDACi treatment of cells.
MCF10A, MCF7 and other breast cancer cell lines were obtained from ATCC (Manassas, VA, USA), and cultured as described.Citation51 Freshly plated cells were allowed to grow for 16 to 20 h and were then treated with various HDACi at indicated concentrations for 0–24 h. NaB, VPA and TSA were obtained from Sigma-Aldrich (St. Louis, MO). Stock solutions of NaB and VPA were prepared in water, while TSA was dissolved in dimethyl sulfoxide (DMSO). Stock solutions of HDACi were diluted in culture medium as required.
PCR primers, quantitative real-time RT-PCR, primary transcript RT-PCR and ChIP assays.
The PCR primers used for RT-PCR (RT), primary transcript RT PCR (PT-PCR) and chromatin immunoprecipitation (ChIP) assays are listed in . Quantitative reverse-transcription polymerase chain reaction (qRT-PCR) and primary transcript RT-PCR (PT RT-PCR) assays were performed as described.Citation25 Briefly, total RNA was isolated from cell lines treated with different HDACi (NaB, VPA or TSA) using Trizol (Invitrogen, Carlsbad, CA, USA). qRT-PCR and PT RT-PCR assays were performed using SensiMix Plus SYBR kit (Quantace Inc.,), on a Rotor-Gene 6000 series PCR system (Corbett Life Sciences-Qiagen Inc., Valencia, CA). For RT-PCR, 2 µg of total RNA was reverse transcribed into cDNA using M-MLV reverse transcriptase (USB Corporation, Cleveland, OH, USA) and Oligo dT primer (Operon Biotechnologies, Huntsville, AL).
For PT RT-PCR, 1 µg of total RNA was subjected to DNase (Invitrogen, Carlsbad, CA) treatment as per manufacturer's instructions to remove any contaminating DNA, and cDNA was generated using Random Hexamer Primers (Fermentas Inc., Glen Burnie, MD). PCR primers were designed to specifically amplify approximately 200 bp of reverse transcribed unspliced RNAs (pre mRNAs). cDNAs were amplified for different number of cycles and data were analyzed using Rotor-Gene Q software package (Corbett Life Sciences-Qiagen Inc., Valencia, CA) to determine Ct values and fold changes in expression. qRT-PCR and PT RT-PCR experiments were performed in triplicates, and data were plotted as the mean ± S.D. of three separate experiments. ChIP assays were performed as describedCitation23,Citation52 with few modifications. Briefly, cells were treated with 1% formaldehyde for 20 min at room temperature. The cross-linked chromatin was isolated and sonicated to yield 200–500 bp fragments. IPs were carried out using a rabbit polyclonal against Ac-H3 and a control IgG. The Ac-H3- and IgG-bound chromatins were amplified using primers specific for BMI1 and p21 promoters () by PCR as described above. The PCR products were run on an agarose gel and visualized by ethidium bromide staining. The intensity of each band was quantified by densitometry using Image J software (NIH, Bethesda, MD).
Promoter-reporter assays.
The BMI promoter-reporter constructs (pGL3-Bmi PrWT, pGL3-Bmi PrMut and pGL3-Bmi Pr.Myc) have been described previously.Citation25 Luciferase activity of these reporters was determined in control and HDACitreated MCF7 cells as described.Citation25 Briefly, MCF7 cells were transfected with 1.8 µg of the reporter construct together with 0.20 µg pRL-TK (Promega, Madison, WI), using FuGENE HD (Roche Applied Science, Indianapolis, IN) in 100 µl Opti-MEM medium. Twenty-four hours after transfection, the cells were treated with NaB at indicated concentrations for 24 h. At the end of treatment, cell lysates were prepared and analyzed for Firefly and Renilla luciferase activities using the Dual-Luciferase Reporter Assay System (Promega, Madison, WI). All transfection experiments were performed in triplicates, and data were plotted as the mean ± S.D. of three separate experiments.
Histone isolation.
For the analysis of histone PTMS, total histones were prepared using acid extraction method as described.Citation53 Briefly, the cell pellet from control and HDACi-treated cells was lysed in cold lysis buffer (0.5% Triton X-100, 50 mM Tris (pH 7.5), 150 mM sodium chloride, 1 mM EDTA, 5 mM sodium butyrate, 1 mM phenylmethylsulfonyl fluoride, 1 µg/mL each of leupeptin and pepstatin, 1 mM sodium orthovanadate, and 10 mM sodium fluoride) for 30 min, centrifuged at 8,000 rpm for 10 min at 4°C to spin down the nuclei. The pellet was resuspended in 0.2N HCl overnight at 4°C for acid extraction. The samples were centrifuged, and supernatant containing the histones was transferred to a clean tube. The protein was quantified and 2.5 µg of the protein containing total histone was loaded on 15% SDS-PAGE gel for western blot analysis using antibodies specific to total and modified histones as required.
Immunological reagents, western blot analysis and immunofluorescence assay.
Various antibodies were obtained from commercial sources. p21 monoclonal antibody (mAb), c-Myc rabbit polyclonal (pAb), PARP-1 mAb, p53 mAb and Cyclin D1 mAb were from Santa Cruz Biotechnology (Santa Cruz, CA). BMI1 mAb was obtained from Invitrogen (Carlsbad, CA). Acetylated levels of histone H3 were detected using anti-acetyl histone H3 (Lys 9) mAb from Cell Signaling Technology (Danvers, MA). EZH2 mAb and E-Cadherin mAb were obtained from BD Biosciences (San Jose, CA). pAbs against H3K27me3 and H2AK119Ub were obtained from Millipore (Billerica, MA). β-actin mAb used to assess β-actin levels as a loading control was obtained from Sigma-Aldrich (St. Louis, MO). For ChIP analysis, Ac-H3 pAb was obtained from Active Motif (Carlsbad, CA). Western blot analyses of total cell extracts from control and HDACi-treated cells using various antibodies were performed as described.Citation25
Immunofluorescence (IF) assays were performed as described.Citation51,Citation54 Briefly, cells were plated in chamber slides and treated with HDACi as described above. Cells were fixed in 4% formaldehyde, permeabilized with 0.5% Triton X-100 for 5 min, and co-immunostained with anti-BMI1 and anti-H2AK119Ub, and anti-EZH2 and H3K27me3 antibodies followed by staining with Alexa Fluor 488- and Alexa Fluor 594-conjugated secondary antibodies (Molecular Probes) as required. The slides were mounted with Vectashield mounting medium containing 4′,6-diamidino-2-phenylindole (DAPI) (Vector Laboratories). After staining, cells were photographed (40X) using a Nikon Eclipse 80i confocal microscope.
Senescence and apoptosis assays.
Induction of senescence by HDACi was studied using staining for the SA-β-gal marker as described.Citation33 Cell death was determined using trypan blue staining and a FITC Annexin V apoptosis detection kit (BD Pharmingen, San Diego, CA). Trypan blue-stained cells were counted under a light microscope (10X). For Annexin V staining, after the drug treatment of cells, floating and adherent cells were collected by centrifugation and washed twice with cold PBS. Cells were then resuspended in 100 µL staining solution containing Annexin V-FITC and propidium iodide (PI, 50 µg/ml) (BD Pharmingen, San Diego, CA) in a HEPES-Calcium Chloride binding buffer. Following incubation at room temperature for 15 min, cells were analyzed by flow cytometry for Annexin V and PI stained fractions on a FACSCalibur with CellQuest software (BD Biosciences-Immunocytometry Systems, San Jose, CA).
Figures and Tables
Figure 1 HDACi downregulate BMI1. (A) MCF10A and MCF7 cells were treated with different concentrations of NaB for 24 h, cells were harvested and the expression of EZH2, BMI1, p21, Ac-H3 and β-actin (loading control) was determined by western blot analysis. The relative expression of BMI1 and EZH2 was determined by densitometric analysis of BMI1 and EZH2 signals normalized for the corresponding β-actin signal. The densitometric analysis was performed using Image J software (NIH, Bethesda, MD). (B) expression of EZH2, BMI1, p21, Ac-H3 and β-actin was determined by western blot analysis of control and VPA-treated MCF10A and MCF7 cells as indicated. The relative expression of BMI1 and EZH2 was determined as described in the preceding Figure legend (A).
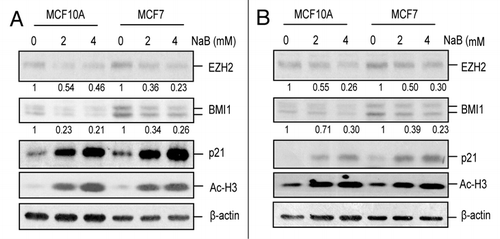
Figure 2 HDACi transcriptionally downregulate BMI1. (A) the relative mRNA levels of BMI1 and EZH2 were determined in control (0 mM) and NaB (2 mM and 4 mM)-treated MCF10A and MCF7 cells by qRT-PCR using primers specific for BMI1 and EZH2 as described in Materials and Methods. Results represent the mean ± S.D. of three separate experiments in each case. (B) the relative primary transcript (PT) levels of BMI1 and EZH2 were determined in control and NaB-treated MCF10A and MCF7 cells (as indicated) by qRT-PCR analysis using primer sets that can quantify primary transcripts (PT RT-PCR) as described in Materials and Methods. Results represent the mean ± S.D. of three separate experiments in each case.
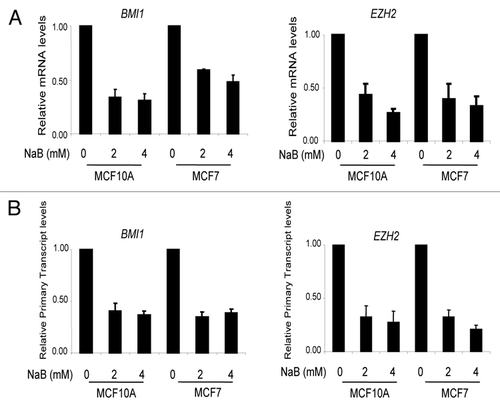
Figure 3 Downregulation of BMI1 is indirect and a late event. (A) MCF10A and MCF7 cells were untreated (0 h) or treated with 4 mM NaB for different time points (3–24 h), and relative expression of BMI1, p21, Ac-H3 was determined by western blot and densitometric analyses as described in legend. (B) Total RNA was isolated from MCF10A and MCF7 cells treated with 4 mM NaB for different time points, and expression of BMI1 was determined by RT-PCR analysis to confirm their transcriptional downregulation. Relative transcript levels of BMI1 were determined by densitometric analyses of corresponding signals normalized to β-actin in each case. (C) Binding of Ac-H3 to BMI1 and p21 promoters was determined by ChIP analysis, which was performed using primers specific for each gene () as described in Materials and Methods. The end products of PCR were run on an agarose gel and visualized by ethidium bromide staining. The relative binding of Ac-H3 to individual promoters was determined by densitometric analysis of immunoprecipitated and PCR amplified DNA normalized to PCR amplified input DNA in each case.
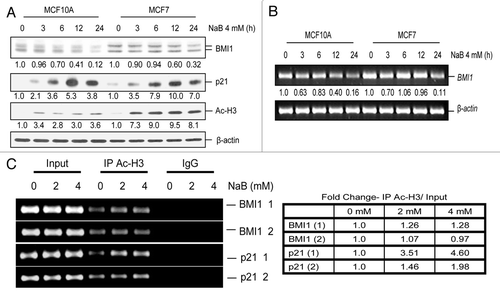
Figure 4 HDACi transcriptionally downregulate BMI1 independent of c-Myc. (A) MCF10A and MCF7 cells were treated with different concentrations of NaB (0, 2 mM and 4 mM) for 24 h, and the expression of c-Myc and β-actin was determined by western blot analysis as described in Materials and Methods. (B) MCF7 cells were transiently transfected with wild type BMI1 (pGL3-BmiPr WT), mut BMI1 (pGL3-Bmipr Mut) and the Myc site deleted minimal (pGL3 Bmipr ΔMyc) promoter reporters together with pRL-TK plasmid, and 48 h post transfection cells were either treated (2 mM and 4 mM) or untreated (0 mM) with NaB. The normalized luciferase activity of each promoter-reporter construct was determined as described in Materials and Methods.
Figure 5 HDACi downregulate histone PTMs mediated by BMI1 and EZH2 containing PRCs. (A) Total histones were prepared by acid extraction from NaB-treated (2 mM and 4 mM), and untreated (0 mM) MCF10A and MCF7 cells as indicated. H3K27me3 and H2AK119Ub levels were determined by western blot analysis (IB) using antibodies specific for the indicated histone PTMs. The membrane with transferred histones was stained with Ponceau to visualize histones and to determine the extent of equal loading. (B) MCF10A cells were left untreated (0 mM) or treated with 2 mM NaB in chamber slides, fixed and co-immunostained using antibodies specific for EZH2 and H3 K27me3 (left), and BMI and H2AK119Ub (right), followed by staining with different Alexa Fluor conjugated secondary antibodies as described in Materials and Methods. After staining, the cells were photographed (40X) using a Nikon Eclipse 80i confocal microscope.
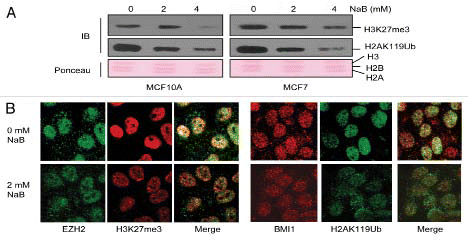
Figure 6 HDACi-induce cell death in MCF7 and premature senescence in MCF10A cells. (A) MCF10A and MCF7 cells were treated with different concentrations (as indicated) of NaB, and number of living and dead cells were quantified using trypan blue exclusion assays as described in Experimental Procedures. (B) The expression of total PARP, cleaved PARP (an indication of apoptosis), BAX, BCL2 and β-actin (loading control) was determined in NaB-treated (2 mM and 4 mM) and untreated (0 mM) MCF10A and MCF7 cells (as indicated) by western blot analysis as described in Materials and Methods. (C) Fluorescence-activated cell sorting (FACS) profile of Annexin V/PI stained untreated (0 mM) and NaB-treated (2 mM and 4 mM) MCF10A cells (upper) and MCF7 cells (lower) was performed as described in Materials and Methods. The percentage of Annexin positive cells as an indication of apoptotic cell death is indicated in lower right quadrant. The percentage of double positive (Annexin V and PI) cells indicating the % necrotic cell death is indicated in upper right quadrant of FACS profile of the respective cells. (D) Induction of premature senescence in NaB-treated (2 mM and 4 mM), and untreated (0 mM) was determined by counting the number of SA-β-gal positive cells. SA-β-gal assay was done as described in Materials and Methods. Cells were either treated for 96 h with drug or first treated for 48 h and then cultured in drug free medium for additional 48 h before staining for the SA-β-gal marker.
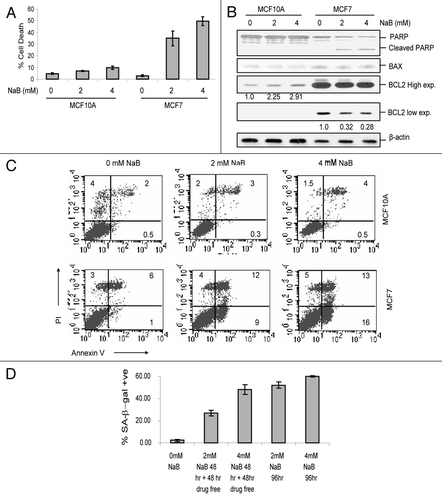
Figure 7 HDACi treatment lead to derepression of p57, IGFBP2 and IGFBP3. (A) Downregulation of BMI1 by NaB was confirmed using qRT-PCR analysis before examining the expression of PcG targets. qRT-PCR assay for BMI1 in untreated (0 mM) and NaB-treated (2 mM and 4 mM) MCF10A and MCF7 cells was done as described in and Materials and Methods. (B–D) the expression of p57/CDKN1C (B), IGFBP2 (C) and IGFBP3 (D) in untreated (0 mM) and NaB-treated (2 mM and 4 mM) MCF10A and MCF7 cells was determined by qRT-PCR assay as described in and Materials and Methods. (E) Upregulation of p21 by NaB was confirmed in NaB-treated MCF10A and MCF7 cells by qRT-PCR analysis as described above.
Table 1 List of the Primers
Additional material
Download Zip (457.2 KB)Acknowledgements
We thank Adam Gluskin for performing preliminary experiments related to this project. We thank Sunil Metkar for help with FACS analysis. This work was supported in part by grant RO1 CA094150 from the National Cancer Institute, NIH to G.P.D. M.D. gratefully acknowledges support from Breast and Ovarian Research Program of the NorthShore University HealthSystem, Evanston, IL.
References
- Schwartz YB, Pirrotta V. Polycomb silencing mechanisms and the management of genomic programmes. Nat Rev Genet 2007; 8:9 - 22
- Simon JA, Kingston RE. Mechanisms of polycomb gene silencing: knowns and unknowns. Nat Rev Mol Cell Biol 2009; 10:697 - 708
- Sparmann A, van Lohuizen M. Polycomb silencers control cell fate, development and cancer. Nat Rev 2006; 6:846 - 856
- Martinez AM, Cavalli G. The role of polycomb group proteins in cell cycle regulation during development. Cell Cycle 2006; 5:1189 - 1197
- Sawa M, Yamamoto K, Yokozawa T, Kiyoi H, Hishida A, Kajiguchi T, et al. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int J Hematol 2005; 82:42 - 47
- Vonlanthen S, Heighway J, Altermatt HJ, Gugger M, Kappeler A, Borner MM, et al. The bmi-1 oncoprotein is differentially expressed in non-small cell lung cancer and correlates with INK4A-ARF locus expression. Br J Cancer 2001; 84:1372 - 1376
- Kim JH, Yoon SY, Kim CN, Joo JH, Moon SK, Choe IS, et al. The Bmi-1 oncoprotein is overexpressed in human colorectal cancer and correlates with the reduced p16INK4a/p14ARF proteins. Cancer Lett 2004; 203:217 - 224
- Glinsky GV, Berezovska O, Glinskii AB. Microarray analysis identifies a death-from-cancer signature predicting therapy failure in patients with multiple types of cancer. J Clin Invest 2005; 115:1503 - 1521
- Kim JH, Yoon SY, Jeong SH, Kim SY, Moon SK, Joo JH, et al. Overexpression of Bmi-1 oncoprotein correlates with axillary lymph node metastases in invasive ductal breast cancer. Breast 2004; 13:383 - 388
- Kang MK, Kim RH, Kim SJ, Yip FK, Shin KH, Dimri GP, et al. Elevated Bmi-1 expression is associated with dysplastic cell transformation during oral carcinogenesis and is required for cancer cell replication and survival. Br J Cancer 2007; 96:126 - 133
- Song LB, Zeng MS, Liao WT, Zhang L, Mo HY, Liu WL, et al. Bmi-1 is a novel molecular marker of nasopharyngeal carcinoma progression and immortalizes primary human nasopharyngeal epithelial cells. Cancer Res 2006; 66:6225 - 6232
- Glinsky GV. Death-from-cancer signatures and stem cell contribution to metastatic cancer. Cell Cycle 2005; 4:1171 - 1175
- Cao R, Tsukada Y, Zhang Y. Role of Bmi-1 and Ring1A in H2A ubiquitylation and Hox gene silencing. Mol Cell 2005; 20:845 - 854
- Cao R, Wang L, Wang H, Xia L, Erdjument-Bromage H, Tempst P, et al. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002; 298:1039 - 1043
- Cao R, Zhang Y. SUZ12 is required for both the histone methyltransferase activity and the silencing function of the EED-EZH2 complex. Mol Cell 2004; 15:57 - 67
- Ringrose L. Polycomb comes of age: genome-wide profiling of target sites. Curr Opin Cell Biol 2007; 19:290 - 297
- Fiskus W, Pranpat M, Balasis M, Herger B, Rao R, Chinnaiyan A, et al. Histone deacetylase inhibitors deplete enhancer of zeste 2 and associated polycomb repressive complex 2 proteins in human acute leukemia cells. Mol Cancer Ther 2006; 5:3096 - 3104
- Minucci S, Pelicci PG. Histone deacetylase inhibitors and the promise of epigenetic (and more) treatments for cancer. Nature Rev 2006; 6:38 - 51
- Glozak MA, Seto E. Histone deacetylases and cancer. Oncogene 2007; 26:5420 - 5432
- Xu WS, Parmigiani RB, Marks PA. Histone deacetylase inhibitors: molecular mechanisms of action. Oncogene 2007; 26:5541 - 5552
- Gui CY, Ngo L, Xu WS, Richon VM, Marks PA. Histone deacetylase (HDAC) inhibitor activation of p21WAF1 involves changes in promoter-associated proteins, including HDAC1. Proc Natl Acad Sci USA 2004; 101:1241 - 1246
- Ocker M, Schneider-Stock R. Histone deacetylase inhibitors: signalling towards p21cip1/waf1. Int J Biochem Cell Biol 2007; 39:1367 - 1374
- Richon VM, Sandhoff TW, Rifkind RA, Marks PA. Histone deacetylase inhibitor selectively induces p21WAF1 expression and gene-associated histone acetylation. Proc Natl Acad Sci USA 2000; 97:10014 - 10019
- Rosato RR, Almenara JA, Grant S. The histone deacetylase inhibitor MS-275 promotes differentiation or apoptosis in human leukemia cells through a process regulated by generation of reactive oxygen species and induction of p21CIP1/WAF1 1. Cancer Res 2003; 63:3637 - 3645
- Guo WJ, Datta S, Band V, Dimri GP. Mel-18, a polycomb group protein, regulates cell proliferation and senescence via transcriptional repression of Bmi-1 and c-Myc oncoproteins. Mol Biol Cell 2007; 18:536 - 546
- Murray PJ. The primary mechanism of the IL-10-regulated antiinflammatory response is to selectively inhibit transcription. Proc Natl Acad Sci USA 2005; 102:8686 - 8691
- Jiang G, Espeseth A, Hazuda DJ, Margolis DM. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J Virol 2007; 81:10914 - 10923
- Skov S, Rieneck K, Bovin LF, Skak K, Tomra S, Michelsen BK, et al. Histone deacetylase inhibitors: a new class of immunosuppressors targeting a novel signal pathway essential for CD154 expression. Blood 2003; 101:1430 - 1438
- Wang LG, Liu XM, Fang Y, Dai W, Chiao FB, Puccio GM, et al. De-repression of the p21 promoter in prostate cancer cells by an isothiocyanate via inhibition of HDACs and c-Myc. Int J Oncol 2008; 33:375 - 380
- Xu Y, Voelter-Mahlknecht S, Mahlknecht U. The histone deacetylase inhibitor suberoylanilide hydroxamic acid downregulates expression levels of Bcr-abl, c-Myc and HDAC3 in chronic myeloid leukemia cell lines. Int J Mol Med 2005; 15:169 - 172
- Munro J, Barr NI, Ireland H, Morrison V, Parkinson EK. Histone deacetylase inhibitors induce a senescencelike state in human cells by a p16-dependent mechanism that is independent of a mitotic clock. Exp Cell Res 2004; 295:525 - 538
- Ogryzko VV, Hirai TH, Russanova VR, Barbie DA, Howard BH. Human fibroblast commitment to a senescence-like state in response to histone deacetylase inhibitors is cell cycle dependent. Mol Cell Biol 1996; 16:5210 - 5218
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92:9363 - 9367
- Tan J, Yang X, Zhuang L, Jiang X, Chen W, Lee PL, et al. Pharmacologic disruption of Polycomb-repressive complex 2-mediated gene repression selectively induces apoptosis in cancer cells. Gene Dev 2007; 21:1050 - 1063
- Yang X, Karuturi RK, Sun F, Aau M, Yu K, Shao R, et al. CDKN1C (p57) is a direct target of EZH2 and suppressed by multiple epigenetic mechanisms in breast cancer cells. PLoS One 2009; 4:5011 - 5011
- Yamaguchi K, Lantowski A, Dannenberg AJ, Subbaramaiah K. Histone deacetylase inhibitors suppress the induction of c-Jun and its target genes including COX-2. J Biol Chem 2005; 280:32569 - 32577
- Alao JP, Stavropoulou AV, Lam EW, Coombes RC, Vigushin DM. Histone deacetylase inhibitor, trichostatin A induces ubiquitin-dependent cyclin D1 degradation in MCF-7 breast cancer cells. Mol Cancer 2006; 5:8 - 8
- Hu J, Colburn NH. Histone deacetylase inhibition downregulates cyclin D1 transcription by inhibiting nuclear factor-kappaB/p65 DNA binding. Mol Cancer Res 2005; 3:100 - 109
- Zhou Q, Agoston AT, Atadja P, Nelson WG, Davidson NE. Inhibition of histone deacetylases promotes ubiquitin-dependent proteasomal degradation of DNA methyltransferase 1 in human breast cancer cells. Mol Cancer Res 2008; 6:873 - 883
- Nowak K, Kerl K, Fehr D, Kramps C, Gessner C, Killmer K, et al. BMI1 is a target gene of E2F-1 and is strongly expressed in primary neuroblastomas. Nucleic Acids Res 2006; 34:1745 - 1754
- Dimri GP. What has senescence got to do with cancer?. Cancer Cell 2005; 7:505 - 512
- Bali P, Pranpat M, Swaby R, Fiskus W, Yamaguchi H, Balasis M, et al. Activity of suberoylanilide hydroxamic Acid against human breast cancer cells with amplification of her-2. Clin Cancer Res 2005; 11:6382 - 6389
- Duan H, Heckman CA, Boxer LM. Histone deacetylase inhibitors downregulate bcl-2 expression and induce apoptosis in t(14;18) lymphomas. Mol Cellular Biol 2005; 25:1608 - 1619
- Singh TR, Shankar S, Srivastava RK. HDAC inhibitors enhance the apoptosis-inducing potential of TRAIL in breast carcinoma. Oncogene 2005; 24:4609 - 4623
- Wang E. Regulation of apoptosis resistance and ontogeny of age-dependent diseases. Exp Geront 1997; 32:471 - 484
- Schmitt CA, Fridman JS, Yang M, Lee S, Baranov E, Hoffman RM, et al. A senescence program controlled by p53 and p16INK4a contributes to the outcome of cancer therapy. Cell 2002; 109:335 - 346
- Bracken AP, Kleine-Kohlbrecher D, Dietrich N, Pasini D, Gargiulo G, Beekman C, et al. The Polycomb group proteins bind throughout the INK4A-ARF locus and are disassociated in senescent cells. Gene Dev 2007; 21:525 - 530
- Larson PS, Schlechter BL, King CL, Yang Q, Glass CN, Mack C, et al. CDKN1C/p57kip2 is a candidate tumor suppressor gene in human breast cancer. BMC Cancer 2008; 8:68 - 68
- Nijjar T, Wigington D, Garbe JC, Waha A, Stampfer MR, Yaswen P. p57KIP2 expression and loss of heterozygosity during immortal conversion of cultured human mammary epithelial cells. Cancer Res 1999; 59:5112 - 5118
- Shimono Y, Zabala M, Cho RW, Lobo N, Dalerba P, Qian D, et al. Downregulation of miRNA-200c links breast cancer stem cells with normal stem cells. Cell 2009; 138:592 - 603
- Dimri M, Naramura M, Duan L, Chen J, Ortega-Cava C, Chen G, et al. Modeling breast cancer-associated c-Src and EGFR overexpression in human MECs: c-Src and EGFR cooperatively promote aberrant three-dimensional acinar structure and invasive behavior. Cancer Res 2007; 67:4164 - 4172
- Kia SK, Gorski MM, Giannakopoulos S, Verrijzer CP. SWI/SNF mediates polycomb eviction and epigenetic reprogramming of the INK4b-ARF-INK4a locus. Mol Cell Biol 2008; 28:3457 - 3464
- Dimri M, Bommi P, Sahasrabuddhe AA, Khandekar JD, Dimri GP. Dietary omega-3 polyunsaturated fatty acids suppress expression of EZH2 in breast cancer cells. Carcinogenesis 2009;
- Datta S, Hoenerhoff MJ, Bommi P, Sainger R, Guo WJ, Dimri M, et al. Bmi-1 cooperates with H-Ras to transform human mammary epithelial cells via dysregulation of multiple growth-regulatory pathways. Cancer Res 2007; 67:10286 - 10295