Abstract
MCL-1, a pro-survival member of the BCL-2 family, was previously shown to have functions in ATR-dependent Chk1 phosphorylation following DNA damage. To further delineate these functions, we explored possible differences in DNA damage response caused by lack of MCL-1 in mouse embryo fibroblasts (MEFs). As expected, Mcl-1-/- MEFs had delayed Chk1 phosphorylation following etoposide treatment, compared to wild type MEFs. However, their response to hydroxyurea, which causes a G1/S checkpoint response, was not significantly different. In addition, appearance of g-H2AX was delayed in the Mcl-1-/- MEFs treated with etoposide. We next investigated whether MCL-1 is present, together with other DNA damage response proteins, at the sites of DNA damage. Immunoprecipitation of etoposide-treated extracts with anti-MCL-1 antibody showed association of MCL-1 with g-H2AX as well as NBS1. Immunofluorescent staining for MCL-1 further showed increased co-staining of MCL-1 and NBS1 following DNA damage. By using a system that creates DNA double strand breaks at specific sites in the genome, we demonstrated that MCL-1 is recruited directly adjacent to the sites of damage. Finally, in a direct demonstration of the importance of MCL-1 in allowing proper repair of DNA damage, we found that treatment for two brief exposures to etoposide over several days, which mimics the clinical situation of etoposide use, resulted in many more chromosomal abnormalities in the MEFs that lacked MCL-1. Together, these data indicate an important role for MCL-1 in coordinating DNA damage mediated checkpoint response, and have broad implications for the importance of MCL-1 in maintenance of genome integrity.
Introduction
Eukaryotic cells respond in a wide variety of ways to diverse genotoxic insults. These insults arise from both the environment, e.g., exposure to ultraviolet or ionizing radiation and various chemicals, as well as intracellularly from by-products of normal metabolic processes such as reactive oxygen species.Citation1 Since maintenance of genome integrity is one of the fundamental features of life, cells have developed a dynamic and coordinated response known as the DNA damage response (DDR). The DDR detects many types of DNA damage and signals its presence to numerous proteins that are involved in stopping the cell cycle at appropriate “checkpoints,” and subsequently to allow repair of the damage. Double stranded breaks (DSBs) by their very nature are the most dangerous of all lesions. If left unrepaired or mis-repaired these lesions can have deleterious effects on cells ranging from the propagation of mutations to chromosomal rearrangement or cell death. The signaling cascade generated as a result of DNA damage utilizes a multi-component protein machine, some members of which sense the actual damage while others are mediators and effectors.Citation2
In response to DNA damage, the PI 3-kinase-related kinase (PIKK) family of proteins such as Ataxia telangiectasia mutated (ATM) and Ataxia telangiectasia and RAD 3-related (ATR) are activated and initiate amplification of the damage response. The mediator proteins such as breast cancer 1, early onset (BRCA1), Mediator of DNA damage checkpoint 1 (MDC1) and p53 binding-protein 1 (53BP1) then acquire post-translational protein modifications, such as phosphorylation, which are generated by ATM or ATR. The modified mediator proteins are capable of further amplifying the damage response and relaying it to the downstream effector proteins Rad51, checkpoint kinase-1 (Chk1), Chk2 and p53.Citation3 One of the hallmarks of DNA damage is the formation of multi-protein foci in the nuclei of damaged cells. The composition of these foci depends not only on the type of damage but also the stage of damage, as the protein components are altered as the damage is recognized, processed and repaired.Citation4
The localized response to DSBs is best characterized by the phosphorylation of a minor histone, H2AX. Compared to other histones, H2AX is relatively rare and distributed throughout the mammalian chromatin. In response to DNA damage, the protruding tails of H2AX become phosphorylated on Ser 139 (to yield what is referred to as γ-H2AX) by the PIKK family of proteins. H2AX is one of the earliest proteins to be modified during the DDR, with the γ-H2AX achieving a plateau within 30 minutes.Citation5,Citation6 Staining of damage-induced foci has shown that γ-H2AX covers up to 2 Mbp of chromatin per DSB, making it a very useful marker for visualization of DSBs.Citation6
Generally, different subsets of proteins are recruited to the site of DSBs depending on the type of lesion, but there are some proteins that respond to multiple types of lesions. These proteins include ATM and ATR, the Mre11, Rad50 and NBS1 (MRN) complex, and Replication protein A (RPA).Citation7–Citation10 While each of the proteins in the MRN complex has specific functions, the complex as a whole has been proposed to be required for sensing of DNA damageCitation11,Citation12 and full activation of ATM and ATR kinases.Citation13,Citation14 The importance of this complex is highlighted by the fact that either an increase or decrease in MRN expression may result in disease, particularly an increased susceptibility to multiple types of cancers.Citation15
MCL-1 is a pro-survival member of the BCL-2 family. BCL-2 family members have a well-established role in the intrinsic apoptosis pathway, functioning through the interactions among several conserved BCL-2 homology (BH) domains. However, many studies have reported that these proteins have additional functions in cell cycle regulation and DNA damage pathways.Citation16–Citation20 In the case of MCL-1, it has an amino terminal domain unrelated to any other protein, and it has no homologues other than those in vertebrates. MCL-1 is an essential gene and its deficiency leads to peri-implantation lethality, but with no evidence of increased apoptosis in the embryos.Citation21 Intriguingly, the pro-survival activity of MCL-1 is required for the maintenance and development of T and B lymphocytes.Citation22,Citation23 Thus, it is not surprising that the increased expression of MCL-1 has been demonstrated in cancers of the hematopoietic lineage.Citation24,Citation25 Paradoxically, alongside the well established pro-survival, and hence oncogenic, effects there co-exists an anti-proliferative effect of MCL-1 which points towards a potential tumor suppressive role. Taken together these observations emphasize the pleiotropic functions of MCL-1, which is well known to be tightly regulated at the level of both mRNA and protein. For example, in a recent study, we demonstrated a function for MCL-1 in the checkpoint response following DNA damage, acting as a key regulator of the ATR-Chk1 pathway.Citation16
In attempting to further understand the regulatory role of MCL-1, we analyzed the etoposide responsiveness of cells lacking MCL-1, and found that they have a delayed and suppressed Chk1 and H2AX phosphorylation. We therefore investigated associations of MCL-1 with key proteins involved in the DDR and could demonstrate its association with γ-H2AX and NBS1 by several different means, as well as showing that MCL-1 was physically associated with chromatin adjacent to DSBs. Moreover, we have demonstrated that MCL-1 deficiency results in a suppressed molecular response to DNA damage and genetic instability in response to treatment with low concentrations of etoposide. Together, we can report for the first time, based on several different lines of evidence, that MCL-1 must be considered to have two independent functions—one as a BCL-2-like survival protein, and an alternate function as a component of the DDR machinery, localized immediately adjacent to DNA double strand breaks.
Results
Lack of MCL-1 causes a delay in Chk1 phosphorylation on Ser 345.
Since our previous study showed that siRNA knockdown of MCL-1 in human cells resulted in reduced Chk1 activation,Citation16 we further investigated the kinetics of Chk1 phosphorylation in Mcl-1-/- MEFs treated with various concentrations of etoposide, which is a topoisomerase II inhibitor and a potent inducer of DSBs.Citation26 As shown in , in response to 1.5 µM etoposide treatment, WT MEFs showed phosphorylation of Chk 1 on Ser 345 within 15 minutes, with a robust response at 30 min and 1 h. Mcl-1-/- MEFs on the other hand showed phosphorylation, and hence activation, of Chk1 only at 3 h of treatment. A ten-fold higher concentration of etoposide resulted in more potent Chk1 phosphorylation within 15 and 30 min of treatment in WT MEFs, but the Mcl-1-/- cells showed only low levels of phosphorylation at 15 min, and still required 3 h to show a maximal response. However, when the cells were treated with 150 µM etoposide, while the WT MEFs showed a maximal response at an earlier time point than the Mcl-1-/- cells, there was less apparent difference in total levels of Chk1 phosphorylation. These results demonstrate that the absence of MCL-1 delays the onset of G2 checkpoint response most dramatically in response to low levels of DNA damage. This would imply that other components of the cellular DDR machinery are capable of responding to the more extensive DNA damage induced by the higher concentrations of etoposide independently of MCL-1.
MCL-1 does not have a role in G1 checkpoint response.
We next tested whether checkpoint response to hydroxyurea, which causes S-phase arrest, is altered in Mcl-1-/- MEFs. When WT or Mcl-1-/- MEFs were treated with hydroxyurea (), there was no apparent difference in the kinetics of Chk1 phosphorylation between the two cell types, although the extent of phosphorylation was lower in Mcl-1-/- MEFs. Similar results were observed following treatment with aphidicolin (data not shown). Together, these results suggest that MCL-1 does not play a role in G1/S checkpoint response.
Lack of MCL-1 causes a reduced H2AX phosphorylation in response to a low dose of etoposide.
Given these results, we next investigated the ability of H2AX to undergo phosphorylation in Mcl-1-/- and WT MEFs. Accumulation of γ-H2AX in nuclear foci is one of the earliest responses to DNA damage.Citation2 Treatment of cells with 1.5 µM of etoposide showed a much less of an increase in the level of γ-H2AX in Mcl-1-/- MEFs compared to WT MEFs (). H2AX was maximally phosphorylated within 1 h in WT cells, and the level of phosphorylation was maintained at 2 h. In contrast, the level of H2AX phosphorylation in Mcl-1-/- MEFs was much lower at 1 h, and increased at 2 h, but remained at a reduced level compared to the WT cells. These data imply that while MCL-1 is not the sole mediator of H2AX activation, it has a role in early events induced by low level of DNA damage caused by etoposide. It is also important to point out that, consistent with our previous findings in human cells,Citation16 an increase in the expression of murine MCL-1 was observed in the nuclei of WT MEF cells treated with a low concentration of etoposide. The human and murine MCL-1 proteins are detected by antibodies that do not cross-react between the two species.
MCL-1 is co-immunoprecipitated with modified histones following DNA damage.
Accumulation of γ-H2AX in nuclear foci is one of the earliest responses to DNA damage.Citation2 We began to explore the possible interaction of MCL-1 with some of the nuclear DDR proteins known to be present near sites of DNA damage. We first tested for MCL-1 association with γ-H2AX. HeLa cells were treated with etoposide for 3 h and were then fractionated into cytosolic, nuclear and chromatin fractions. Presence of endogenous MCL-1 was observed in each of these fractions following etoposide-induced damage. However, immunoprecipitation with anti-MCL-1 antibody showed association with endogenous phospho-Ser139 H2AX only in the chromatin fraction of etoposide-treated cells (). The purity of the fractions was ascertained by probing with vinculin, which is a cytoplasmic marker, or Oct1, which is a marker for nuclear and chromatin fractions.
We considered the possibility that the interaction between γ-H2AX and MCL-1 could be a consequence of independent binding of each protein to DNA. Treatment of extracts with DNaseI prior to co-immunoprecipitation showed that MCL-1 co-immunoprecipitated γ-H2AX from both the DNaseI soluble and insoluble extracts () suggesting that the observed results ( and B) were not due to non-specific association as a result of large protein-DNA complexes being bound to beads. Interestingly, MCL-1 antibody co-immunoprecipitated more γ-H2AX from the DNaseI released soluble chromatin fraction () than the insoluble fraction. DNaseI treatment was carried out for all the subsequent nuclear/chromatin extracts used in this study.
We next examined the intracellular localization of MCL-1 and γ-H2AX in untreated and etoposide treated HeLa cells by immunofluorescence. As expected, in untreated cells no γ-H2AX was detected, while MCl-1 showed diffuse staining in both the cytoplasm and the nucleus. Following 3 h etoposide treatment, both MCL-1 and γ-H2AX showed an increase in their nuclear staining, and some overlap of the nuclear staining was consistently detected (). These results suggest that MCL-1 and γ-H2AX share the same location in the nucleus of cells following DNA damage.
Verification of MCL-1/γ-H2AX co-immunoprecipitation and antibody specificity.
The results suggesting that MCL-1 and γ-H2AX co-localize following DNA damage are based on both co-immunoprecipitation and immunofluorescence studies. To further emphasize the specificity of these interactions, and to rule out the possibility that the interaction may be due to non-specific binding of the antibodies, we tested an additional antibody to MCL-1. We used an antibody (MCL-1 RC13) which recognizes an alternative epitope in MCL-1. As shown in MCL-1 (RC13) successfully co-immunoprecipitated γ-H2AX in extracts from etoposide treated cells while the untreated cells showed only a very faint association. No non-specific interaction was observed between the control IgG and γ-H2AX in etoposide treated total nuclear extracts. We further confirmed the specificity of antibody in Mcl-1-/- MEFs that were transiently transfected with human MCL-1. As apparent from , the antibody reacted only with human MCL-1 with no cross reactivity with any other protein in the MCL-1-deficient cells.
It is worth mentioning here that our immunoprecipitation studies yield an MCL-1 band at the expected molecular weight and that we have previously established the specificity of the MCL-1 band by mass spectrometry.Citation17 Moreover, further confirmation of antibody specificity has been demonstrated in MCL-1 knock down experiments where siRNA targeted to MCl-1 showed diminished expression of the band detected by the antibody.Citation16
MCL-1/γ-H2AX association requires functional ATR.
We next tested the ability of MCL-1 to associate with γ-H2AX in response to exposure to UVB. The mechanism by which UV induces damage is by collision or stalling of replication forks at the site of UV induced base adducts.Citation27 Our previous study showed an increase in the expression of MCL-1 in response to a low level of UVB damage,Citation16 although high levels of UV irradiation are known to cause degradation of MCL-1.Citation28 Since our previous study showed that MCL-1 functions downstream of ATR, we sought to determine whether association with γ-H2AX requires functional ATR. Seckel syndrome cells, heterozygous for a mutation in ATR,Citation29 were treated with either etoposide for 3 h or exposed to UVB followed by recovery for 3 h. Total nuclear extracts were immunoprecipitated with anti-MCL-1 antibody. Interestingly, Seckel cells showed diminished association between MCL-1 and γ-H2AX in response to UVB but not etoposide treatment (). Analysis of proteins remaining in the extracts after immunoprecipitation (post-IP) showed that the lack of association was not due to the inability of UVB-treated Seckel cells to phosphorylate H2AX on the serine 139 residue. By contrast, in normal human fibroblasts (Hs68 cells), γ-H2AX was co-immunoprecipitated with MCL-1 following etoposide as well as UVB exposure. It is noteworthy that Seckel cells have a greater abundance of MCL-1 compared to the normal fibroblasts. Further analysis of MCL-1 and γ-H2AX association in etoposide or UVB treated ATM-deficient cells showed co-immunoprecipitation of these proteins following treatment with either etoposide or UVB (). Since ATR functions predominantly in the UV-induced response, these results suggest that MCL-1/γ-H2AX association requires the presence of functional ATR during DDR.
MCL-1 associates with the DNA damage response protein NBS1.
The above observations suggest that MCL-1 accumulates together with the γ-H2AX within chromatin following DNA damage. These results however, are not indicative of the presence of MCL-1 at the site of damage as studies of Berkovich et al.Citation30 clearly revealed that γ-H2AX is not situated at the site of damage, but lies adjacent to it. In the context of DSBs, NBS1 plays a more prominent role than γ-H2AX as it associates directly with the DNA breaks.Citation30 In order to gain greater insight into the role of MCL-1 in the DNA damage response we next investigated the potential interaction between MCL-1 and NBS1. Results of immunoprecipitation studies showed no association between MCL-1 and NBS1 in the total nuclear extracts in normally proliferating, non-DNA damage induced HeLa cells, although the basal levels of MCL-1 in the nuclear extracts were low compared to those detected after DNA damage (). On the other hand, when HeLa cells were treated with 15 µM of etoposide for 3 h an association was observed between MCL-1 and NBS1 in total nuclear extracts that were immunoprecipitated with anti- MCL-1 antibody. This result was further supported by immunoprecipitation with anti-NBS1 antibody, in which case MCL-1 was co-immunoprecipitated. In cells that were exposed to a low dose of UVB and then allowed to recover for 3 h, anti-NBS1 co-immunoprecipitated MCL-1 although the reciprocal immunoprecipitation was not as effective. This could be due to a difference in immunoprecipitation efficiency of the two antibodies. Both anti-MCL-1 and anti-NBS1 were able to effectively co-immunoprecipitate γ-H2AX upon induction of DNA damage, as might be expected from the above results and other published studies.Citation31
Next, we investigated the possible co-localization of MCL-1 and NBS1 by immunostaining of cells and visualization by confocal microscopy. Interestingly, some overlap in the staining of these two proteins was observed in untreated cells, but a dramatic increase was observed in the co-staining of the two proteins in the nuclei of the etoposide treated cells (). These results, together with those of the immunoprecipitation experiments, suggest that in response to genotoxic stress, MCL-1 is able to translocate to the vicinity of DNA damage and interact with checkpoint response proteins.
MCL-1 is recruited to the site of DNA strand breaks.
The above results using co-immunoprecipitation and immunofluorescence staining showed that MCL-1 is present in the nuclear complexes that include γ-H2AX and NBS1. However, as this is an unexpected finding based on the normal ‘BCL-2-like’ functions of MCL-1, we sought to use yet another approach to determine whether MCL-1 is closely associated with DNA double strand breaks. It is known that the protein aggregates detected by light microscopy do not necessarily directly reflect proteins that are recruited immediately adjacent to the sites of damage.Citation30,Citation32 We therefore analyzed the recruitment of proteins to DSBs using a system that generates site-specific DSBs at endogenous sites in the genome, which can be probed using a ChIP assay.Citation30,Citation32 The system requires a HA-ER-I-Ppo1 retroviral plasmid, where I-Ppo1 is a restriction endonuclease which recognizes and cleaves a 15 bp target sequence in endogenous mammalian 28S ribosomal DNA.Citation33,Citation34 In this system, nuclear translocation of HA-ER-I-Ppo1 is achieved by treatment of cells with 4-hydroxytamoxifen (4-OHT). This induction of I-Ppo1 results in the generation of DSBs at the selected sites in the genome.
We first used this system to examine the change in expression of MCL-1 following the defined DSBs in HeLa cells expressing HA-ER-I-Ppo1. Immunoblot analysis of 4-OHT-treated cells showed an increase in the cytosolic expression as well as nuclear abundance of MCL-1 (), as we previously observed following low level DNA damage with either etoposide or UV.Citation16 To further assess whether MCL-1 is recruited to the sites of damage, HeLa cells infected with the HA-ER-I-Ppo1 retrovirus were either left untreated or I-Ppo1 was induced by treatment with 4-OHT for 2 h or 16 h. ChIP was performed using antibodies to MCL-1 or to NBS1, to determine which proteins were associated with chromatin immediately adjacent to the I-Ppo1-digested sites. Following immunoprecipitation with either anti-NBS1 or anti-MCL-1, PCR amplification of DNA adjacent to the I-Ppo1 restriction site, using primers specific to 28S ribosomal DNA, yielded a specific product in I-Ppo1-induced extracts, while no such product was observed in the DNA from uninduced cells. As shown in and C, binding of both MCL-1 and NBS1 was detected following 2 h of I-Ppo-1 induction, and increased binding was observed following induction for 16 h. The use of this I-Ppo1 system enabled us to confirm previous findingsCitation30 with regards to the binding of NBS1 to DSBs, as well as to unequivocally demonstrate for the first time that MCL-1 is also recruited to the sites immediately adjacent to discrete double strand breaks in response to DNA damage.
Accumulation of chromosomal aberrations in Mcl-1-/- MEFs following DNA damage.
The initial results in this study showed a difference in checkpoint response between WT and Mcl-1-/- MEFs specific to etoposide treatment. Etoposide is a strong inducer of DSBs and thus mammalian cells treated with etoposide must mount an effective checkpoint response involving cell cycle arrest and DNA repair or, if repair is unsuccessful, cell death. The absence of an effective checkpoint response can lead to chromosomal abnormalities, which result in genomic instability and are a hallmark of cancer. We therefore investigated the potential DNA damage in WT vs. Mcl-1-/- MEF cells following treatment with a low concentration of etoposide. We used a strategy similar to that in a recent study, which mimics the clinical scenario where an increase in the initial drug concentration is followed by a steady decline.Citation35 We expected that, if lack of MCL-1 does not allow a sufficiently robust checkpoint response, cells may divide with damaged DNA, which could be detected by conventional cytogenetics. As shown in , in two separate experiments, metaphase analyses of cells that were exposed to two treatments with etoposide followed by a recovery period, demonstrated that WT MEFs were able to efficiently repair the damage caused by etoposide. In two separate analyses, only three chromosomal aberrations were seen in untreated cells, and a total of seven aberrations after etoposide treatment. In contrast, similarly treated Mcl-1-/- MEFs accumulated one or more chromosomal aberrations in almost all of the metaphase spreads after etoposide treatment. While untreated Mcl-1-/- cells had only five chromosomal aberrations, the same cells treated with etoposide accumulated over 50 aberrations, suggesting that these cells were deficient in their ability to respond to DNA damage and/or repair the damage caused by etoposide treatment.
Discussion
It is well established that in response to DSBs, one of the key phenomena is the aggregation of a large number of proteins at the site of damage.Citation36 Analysis of damage-induced foci has provided valuable information on the kinetics and co-localization of proteins that form the DDR machinery. We investigated the potential role of MCL-1 in DDR events based on our recent observations showing that MCL-1 plays an essential role in ATR-dependent activation of Chk1 following DNA damage.Citation16 Our initial characterization revealed associations between MCL-1 and key DDR proteins, γ-H2AX and NBS1. Interestingly, MCL-1 has been shown to be associated with a number of nuclear proteins, either by co-immunoprecipitation or by yeast two-hybrid analyses.Citation17,Citation37,Citation38 Thus, it is clear from these multiple studies that MCL-1 must have a role in the nucleus, and this is likely to be independent of its role as a BCL-2 family protein, which is largely dependent upon its localization at mitochondrial membranes.
A large number of checkpoint proteins are phosphorylated in response to damage and these phosphorylations modulate not only the protein function but also protein-protein interactions in checkpoint pathways.Citation39–Citation41 Our results () showed a diminished association between MCL-1 and γ-H2AX in UVB treated Seckel cells. This inability of MCL-1 to efficiently interact with γ-H2AX in UVB treated ATR defective cells, suggests a possible upstream role for ATR in modulating MCL-1 functions at the chromatin level. In contrast, the ability of MCL-1 to co-immunoprecipitate with γ-H2AX in etoposide treated Seckel cells could be due to residual ATR function due to a relatively high level of damage induced by sustained treatment with etoposide, compared to short exposure to UVB. Alternatively, the latter association could be due to ATM effects. However, the results shown in where MCL-1 was able to efficiently interact with γ-H2AX in both etoposide or UVB treated ATM deficient cells appear to rule out an essential role for ATM activity in MCL-1/γ-H2AX interaction. The etoposide-induced checkpoint, due to its sensitivity to caffeine, is generally believed to be ATM and/or ATR dependent. The studies of Costanzo et al.Citation42,Citation43 showed a requirement for ATR in etoposide-induced checkpoint by demonstrating that ATR depletion restored replication in etoposide treated cells. These studies established ATR as the kinase that mediates the etoposide-induced checkpoint. Together, these results implicate the ATR signaling pathway in mediating MCL-1/γ-H2AX interactions and are supportive of our previous studies in which MCL-1 was demonstrated to play an important role in the ATR-induced activation of Chk1.Citation16
NBS1 functions in both cell cycle checkpoint regulation and DNA repair in response to damage.Citation44,Citation45 It is noteworthy that in etoposide treated cells, the MRN complex functions downstream of the ATR pathway, while in response to other forms of damage such as ionizing radiation, it functions as a part of the ATM pathway (reviewed in ref. Citation46). The association between MCL-1 and NBS1 is significant due to the important role that NBS1 plays in the localization, signal transduction and catalytic activation of the MRN complex.Citation47,Citation48 Moreover, the MRN complex is required for maintaining replication fork integrity, DSB repair, activation of G2/M checkpoint and maintenance of telomere length.Citation49,Citation50 Therefore, the presence of MCL-1 in the same complex as NBS1 following damage could suggest a role for MCL-1 in one or more of these events, particularly a role in the G2/M checkpoint. The G2 checkpoint pathway is regulated by Chk1 and initiated by inhibition of cyclin dependent protein kinase 1 (CDK1) activity (reviewed in ref. Citation51). Once recruited to the site of damage, MCL-1 can contribute to the inhibition of the activity of CDK1, which is essential for timely progression of the cell cycle in two ways. Firstly, MCL-1 by virtue of its ability to bind to Tyr15-phosphorylated (inactive) CDK1Citation17 can arrest the cells at G2 and thus delay the cell cycle progression.Citation52 Secondly, MCL-1 can exert an inhibitory effect indirectly by preventing the dephosphorylation of Tyr15 of CDK1 through its effects on Chk1.Citation16 Following DNA damage, CDC25 phosphatase, which is a positive regulator of cell cycle progression, is inhibited by Chk1 mediated phosphorylation which subsequently prevents the dephosphorylation and hence the activation of CDK1.Citation53
Although the analysis of γ-H2AX provides a very useful means of indirectly visualizing the DNA damage in eukaryotic cells, Berkovich et al. have clearly shown that γ-H2AX is found not at DSBs, but adjacent to them.Citation30 By using a recently developed strategy of creating DSBs at specific endogenous sites in the human genome we have shown that MCL-1 could be detected at the sites of damage, just as NBS1, a key component of the MRN complex was shown at these sites. These results suggest that MCL-1 is likely present both immediately adjacent to sites of damage, as well as distal to these sites.
Use of a low dose of etoposide in our studies allowed us to investigate a role of MCL-1 in the DDR, as opposed to the use of high concentrations of etoposide, which can lead to the activation of redundant pathways and may confound interpretation about the role of individual proteins. The delay in Chk1 phosphorylation and reduced γ-H2AX levels in cells lacking MCL-1, as well as the inability of Mcl-1-/- MEFs to efficiently repair DNA damage in response to low concentrations of etoposide, suggest that the loss of MCL-1 affects both the initiation as well as the maintenance of the G2 checkpoint. Chk1 activity has been shown to be an important determinant for proper initiation as well as the maintenance of the checkpoint.Citation54 Hence these results further confirm a critical role for MCL-1 in the DNA damage checkpoint pathway.
The observation that the loss of MCL-1 delays but does not abrogate Chk1 phosphorylation following DNA damage (), suggests a redundancy in checkpoint pathways. It is now well established that ATR dependent Chk1 activation critically depends on the adaptor protein claspin.Citation55,Citation56 The results from the current report, together with those from our previous study showing a role for MCL-1 in Chk1 activation,Citation16 suggest a very similar function for MCL-1 in response to DNA damage. However, unlike claspin, MCL-1 does not require the induction of DNA damage for its association with Chk1.Citation16 Hence, one interpretation for our results could be that a high concentration (150 µM) of etoposide induces extensive damage, which leads to the activation of claspin in both the WT and Mcl-1-/- MEFs, while a low concentration of etoposide can activate claspin only if the damage is sustained in a time dependent manner.
We observed a differential response to treatment with etoposide of Mcl-1-/- MEFs compared to WT cells. In contrast, when cells were treated with hydroxyurea the kinetics of Chk1 phosphorylation were essentially the same in both cell types. Hydroxyurea causes replication stress leading to replication fork stress and generation of single stranded DNA which activates the ATR-Chk1 pathway, with a more pronounced effect in early S-phase.Citation57 In contrast, etoposide causes both replication stress and DSBs and causes S as well as G2 checkpoint activation.Citation46 The current study demonstrated a role for MCL-1 at late S and G2 checkpoints, which are predominantly regulated by the ATR-Chk1 pathway.Citation58 Mouse embryonic cells have been shown to have short G1 checkpoints and do not undergo arrest at G1 in response to DNA damage.Citation59 This observation demonstrates the importance of S and G2 checkpoints for maintaining the genomic integrity in embryonic stem cells. Intriguingly, MCL-1 has been shown to be important at several stages in the development of hematopoietic cells (reviewed in ref. Citation60), and has an essential role in early vertebrate development. While MCL-1's anti-apoptotic effect likely explains its role in hematopoiesis, it is tempting to speculate that the DNA damage checkpoint function of MCL-1 is responsible for its role in implantation and development. The exact molecular mechanism by which MCL-1 functions in DDR is unknown and will be the subject of future studies. However, given the fact that MCL-1 is present at sites of DNA damage, associated with key DDR proteins, and its deficiency impairs the ability to efficiently repair the damage, it is tempting to suggest that MCL-1 may act as a key adaptor protein involved in coordinating effective assembly of DDR complexes, thus facilitating cell cycle arrest following DNA damage.
Materials and Methods
Cell lines.
HeLa cells were obtained from ATCC (Manassas, VA). ATM-deficient HT-144 cells were a kind gift from Dr. P. Olive (British Columbia Cancer Agency, BC). ATR-defective fibroblasts, F02-98, were a kind gift from Dr. P. Jeggo (University of Sussex, UK). Hs68, which are primary human foreskin fibroblastsCitation61 maintained within 30–60 mean population doublings, were kindly provided by Dr. K. Riabowol (University of Calgary, AB). HeLa, Hs68 and F02-98 cells were grown in DMEM supplemented with 10% fetal bovine serum. Mcl-1-/- MEFs were a kind gift from Dr. J. Opferman (St. Jude Children's. Research Hospital, Memphis, TN) and were obtained from Mcl-1flox/null embryos harvested on E10.5 (mixed 129/B6 background). The primary P3 cells were immortalized by SV40 and cloned. Single cell clones were cultured and treated with TAT-Cre fusion protein to delete the conditional allele and then were single cell cloned again. Individual clones were expanded, genotyped and tested for protein expression. They are neomycin resistant due to the presence of the Null allele which is a beta-galactosidase/Neomycin fusion which acts as a reporter. Both Mcl-1-/- and WT MEF cells were grown in DMEM supplemented with 10% fetal bovine serum.
Antibodies and reagents.
Anti-MCL-1 (Sc-19), Oct I (C-21) and normal rabbit and mouse IgG were purchased from Santa Cruz Biotechnology (Santa Cruz, CA). Antibodies recognizing human MCL-1 (RC-13, mouse monoclonal ab3184), NBS1 (mouse monoclonal ab49958 and rabbit polyclonal ab23996), anti-phospho-histone H2AX (Ser 139) (mouse monoclonal ab18311and rabbit polyclonal ab2893) were purchased from Abcam (Cambridge, MA). Phospho-Chk1 (ser 345) was from Cell Signaling Technology (Beverly, MA). Monoclonal antihuman vinculin was purchased from Sigma-Aldrich (Oakville, ON). Anti-Mouse MCL-1 was from Rockland Immunochemicals for Research (Gilbertsville, PA). Etoposide and hydroxyurea were purchased from Calbiochem (La Jolla, CA). Fetal bovine serum and Protein-G Agarose beads were purchased from Invitrogen (Carlsbad, CA).
Cell treatments.
Optimal concentrations of etoposide were determined for each cell line by treating the cells with a concentration range for 24 h. The lowest concentration that caused G2 arrest with the least amount of apoptosis was selected as the optimal concentration. For HeLa, Hs68, F02-98 and HT 144 cells 15 µM etoposide was found to be optimal. For the MEF cells used in this study, 1.5 µM etoposide was found to be optimal. For UV treatments, cells were irradiated using a UVB source (5 J/m2/sec) for 20 seconds for a total of 100 J/m2, after which cells were washed twice with phosphate-buffered saline (PBS) and then allowed to incubate under normal conditions for three hours prior to protein extraction. For hydroxyurea, 0.2 mM was found to be optimal for MEFs.
Subcellular fractionation.
Subcellular fractionation was carried out as described.Citation62 Briefly, cells were resuspended in Buffer A (10 mM HEPES pH 7.9, 10 mM KCl, 1.5 mM MgCl2, 0.34 M sucrose and 10% glycerol), containing protease inhibitor cocktail (Roche, Laval, Quebec) and 200 µM sodium vanadate and 1 mM DTT. To each extract, 0.1% Triton-X100 was added and samples were incubated on ice for 7 min. The samples were centrifuged for 5 minutes at 1,300 × g. Supernatant containing cytoplasmic proteins was centrifuged further for 10 min at 20,000 × g. The pellets containing the nuclei were washed with Buffer A and resuspended in Buffer B (0.2 mM EGTA pH 8.0, 3 mM EDTA pH 8.0) and incubated on ice for 30 min. The extracts were centrifuged at 1,700 × g for 5 min. The pellets were washed and centrifuged for 5 min at 1,700 × g. The pellets were resuspended in solubilization buffer (20 mM Tris HCl pH 8.0, 1% NP40, 10% glycerol, 137 mM NaCl, 10 mM NaF, supplemented with protease inhibitor cocktail, 200 µM sodium vanadate and 5 µg/ml each of DNase I and RNase A) and sonicated for 5 sec. In experiments where nuclear proteins were extracted together with chromatin, 5 µg/ml each of DNase I and RNase A were added to the Buffer B and the nuclear preparations were sonicated for 5 s. These preparations were designated as the total nuclear extracts.
Protein concentrations were determined by BCA protein assay. For immunoprecipitation, cytoplasmic, nuclear or chromatin extracts were pre-cleared with 20 µl of Protein G agarose beads for 30 minutes. One µg/ml anti-MCL-1 or anti-NBS1 or normal rabbit or mouse IgG antibody was added and after an overnight incubation and the immunoprecipitates were collected by adding 50 µl of Protein G agarose beads. Beads were washed four times with solubilization buffer.
Immunoblotting.
For samples separated on SDS polyacrylamide gels, transfer to nitrocellulose was performed using a semidry transfer system as described previously.Citation17 Representative blots are shown in figures, but in all cases, similar results were obtained in a minimum of three replicates. In most cases, four or more replicates were performed.
Confocal microscopy.
Immunofluorescence staining was performed as described previously.Citation16 Briefly, HeLa cells were seeded on chamber slides (Lab-Tek, Hatfield, PA). Cells were treated or not with 15 µM etoposide for 3 h and fixed with fresh 4% paraformaldehyde. Cells were permeabilized with 100% methanol and probed with 1:200 anti-MCL-1 or 1:500 γ-H2AX or 1:300 of the anti-NBS1 antibodies. Bound antibody was detected by goat anti-rabbit antibody conjugated to Alexa Fluor 488 or goat anti-mouse antibody conjugated to Alexa Fluor 594 (Molecular Probes, Carlsbad, CA). The cell nuclei were stained with 1:20,000 dilution of either Hoechst 33342 or Draq5. Confocal images of fluorescently labeled cells were acquired using a Leica AOBS SP2 laser scanning confocal microscope (Leica, Heidelberg, Germany) with Leica confocal TCS SP2 software. All image processing operations were performed using Volocity software (Improvision, Lexington, MA).
Chromatin immunoprecipitation (CHiP).
For the ChIP assay, a protocol described by Berkovich et al. was followed.Citation32 The plasmid pBABE-HA-ER-I-Ppo1 was kindly provided by Dr. Michael Kastan (St. Jude Children's Research Hospital, Memphis, TN). HeLa cells were infected with the HA-ER-I-Ppo1 retrovirus. I-Ppo1 is a restriction endonuclease which recognizes and cleaves a 15 bp target sequence in endogenous mammalian 28S ribosomal DNA.Citation33,Citation34 Nuclear translocation of HA-ER-I-Ppo1 was induced by treatment of cells with 1 µM of 4-OH-tamoxifen (4-OHT). The cells were harvested at 2 and 16 h after the treatment. For each condition, HeLa cells were crosslinked by directly adding formaldehyde to reach a final concentration of 1%. After 10 min incubation at room temperature (RT) with continuous agitation the crosslinking was stopped by the addition of 0.125 M glycine. This was followed by incubation at RT for 5 min with continuous agitation. The crosslinked cells were washed twice with cold PBS. The cells were scraped in 1 ml PBS supplemented with protease inhibitor cocktail and 200 µM sodium vanadate, centrifuged at 750 × g for 5 min at 4°C and resuspended in 1 ml of buffer I (10 mM HEPES at pH 6.5, 1 mM EDTA, 0.5 mM EGTA, 0.25% TritonX-100 supplemented with protease inhibitor cocktail and 200 µM sodium vanadate). Cells were incubated on ice for 10 min and pelleted by centrifugation at 1,700 × g for 5 min at 4°C. The supernatant was aspirated and the nuclei were resuspended in 1 ml of lysis buffer II (10 mM HEPES at pH 6.5, 1 mM EDTA, 0.5 mM EGTA and 200 mM NaCl supplemented with protease inhibitor cocktail and 200 µM sodium vanadate). After centrifugation the nuclei were resuspended in 0.3 ml nuclei lysis buffer (50 mM Tris HCl, pH 8.1, 10 mM EDTA and 0.5% SDS supplemented with protease inhibitor cocktail and 200 µM sodium vanadate). The chromatin was sonicated 5 × 10 s at level 5 output in a 550 Sonic Dismembrator (Fisher Scientific). The fragmented chromatin was centrifuged at 20,000 × g for 10 min at 4°C and diluted 1:5 in IP dilution buffer (1% Triton X-100, 2 mM EDTA, 20 mM Tris HCl (pH 8.1) and 150 mM NaCl supplemented with protease inhibitor cocktail and 200 µM sodium vanadate). The extracts were pre-cleared by adding 30 µl of Protein G beads for 1 h. The precipitates were centrifuged at 20,000 × g and pre-clearing repeated for an additional 2 h. Anti-MCL-1 or anti-NBS1 were added at 1 µg/ml overnight. The precipitates were recovered by adding 30 µl of Protein G beads for 3 h.
The antibody/protein/DNA complexes were eluted in elution buffer (0.1 M NaHCO3 and 1% SDS). This was followed by incubation with 1 µl of 20 µg/ml Proteinase K (Fermentas, Burlington, ON) at 55°C for 1 h. The samples were incubated at 65°C to reverse formaldehyde crosslinking. The resultant DNA was cleaned up using Qiaquick PCR purification kit (Qiagen, Mississauga, ON) according to the manufacturer's recommendations. The eluted DNA was analyzed by PCR using primers that recognize sites 489 bp 3′ to the I-Ppo-I cut site. The primer sequences were 5′-TGG AGC AGA AGG GCA AAA GC-3′ and 5′-TAG GAA GAG CCG ACA TCG AAG G-3′, which yield a 200 bp product. A sample of the input DNA was taken from each preparation prior to immunoprecipitation and used to quantify the amount of DNA in each sample to be used for subsequent immunoprecipitation. These samples were also used as positive controls for the PCR assays. The PCR parameters were the same as described previously.Citation30,Citation32
Metaphase spreads.
WT and Mcl-1-/- MEFs were treated with 1.5 µM etoposide for 3 h with an intervening recovery period of 72 h followed by a second round of treatment and recovery. WT and Mcl-1-/- MEFs were treated with colcemid (KaryoMax-Invitrogen), which was added directly to the culture dish at 1:1,000 dilution, followed by an incubation for 1.5 h. Cells were trypsinized and 1.5 ml of warm 0.075 M KCl (37°C) was added to the trypsinized cells. Cells were incubated at RT for 7–10 min. Cells were centrifuged at 10,000 rpm for 5 min and supernatant was removed. This was followed by the addition of 2 ml of freshly made methanol/acetic acid (3:1). Cells were incubated at RT for 15 min. To obtain metaphase spreads, cells were spread on wet microscope slides. Slides were stained with DAPI (1:20,000). Ten metaphases each from WT or Mcl-1-/- MEFs were scored for chromosomal damage.
Figures and Tables
Figure 1 Mcl-1-/- MEFs display a delay in Chk1 phosphorylation on Ser 345. (A) WT and Mcl-1-/- MEFs were either left untreated (0) or treated with 1.5 µM etoposide (top part) for 15, 30 min, 1 h or 3 h and cytoplasmic extracts immunoblotted for phospho-Ser345 Chk1, unphosphorylated Chk1, MCL-1 or vinculin as a loading control. Middle and bottom parts: WT and Mcl-1-/- MEFs were treated with 15 and 150 µM of etoposide, respectively, for the same time periods as indicated for the top part. (B) WT and Mcl-1-/-MEFs were either left untreated (0) or treated with 0.2 mM hydroxyurea (HU) for 1, 2 or 3 h and cytoplasmic extracts were prepared. The blot was probed for phospho-Ser345 Chk1, MCL-1 or vinculin as a loading control.
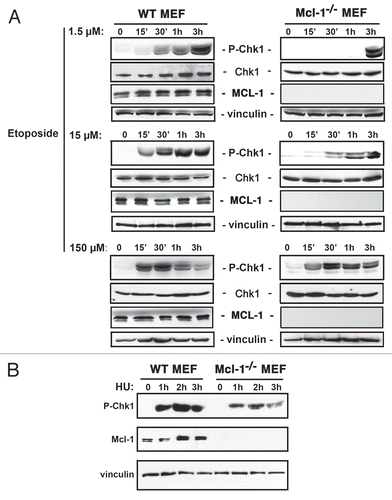
Figure 2 Mcl-1-/- MEFs display reduced H2AX phosphorylation in response to a low dose of etoposide. WT and Mcl-1-/- MEFs were either left untreated (0) or treated with 1.5 µM etoposide (Etop) for 1 or 2 h. Total nuclear extracts were prepared. The blot was probed with anti-γH2AX, anti-MCL-1 or anti-Histone 4 as a loading control. Samples were run on the same gel, with intervening lanes deleted, as indicated by vertical bar.
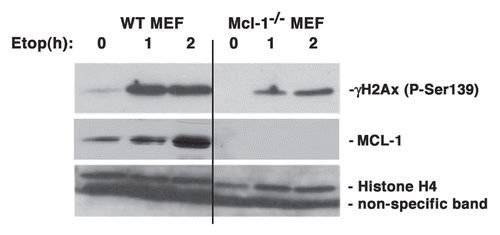
Figure 3 MCL-1 co-immunoprecipitates with -H2AX following DNA damage. (A) HeLa cells were untreated (C) or treated with etoposide (E) for 3 h. Immunoprecipitation of endogenous MCL-1 was performed from cytoplasmic, nuclear or chromatin extracts (or antibody (Ab) alone with no extract was used as a control). Immunoprecipitates were probed with anti-γ-H2AX and anti-MCL-1. The unbound proteins in the post IP extracts were probed with anti-vinculin antibody to indicate cytosol purity and anti-Oct1 antibody to indicate purity of nuclear and chromatin fractions. (B) HeLa cells were untreated or treated with etoposide (as indicated) for 3 h. Immunoprecipitation of endogenous MCL-1 was performed from total nuclear extracts (or antibody (Ab) alone where no extract was used as a control). Extracts in lanes 2 and 3 were prepared using the standard protocol described in Materials and Methods. Extract in lane 4 was prepared using the standard protocol with the exception that no DNaseI and RNase A were added prior to the co-immunoprecipitation. Immunoprecipitates were probed with anti-γ-H2AX and anti-MCL-1. (C) HeLa cells were untreated or treated with etoposide for 3 h. Cells were fixed and stained with anti-MCL-1 or anti-γ-H2AX antibodies and visualized by confocal microscopy using goat anti-rabbit Alexa Fluor 488 (green; for MCL-1) or goat anti-mouse Alexa Fluor 594 (red; for γ-H2AX). Nuclei were stained with Draq5.
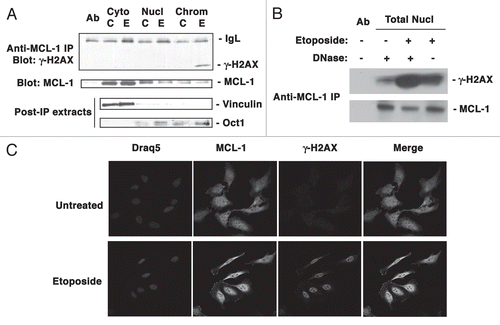
Figure 4 Verification of anti-MCL-1 antibody specificity. (A) HeLa cells were either untreated or treated with Etoposide (as indicated) for 3 h. Total nuclear extracts were immunoprecipitated with anti-MCL-1 (RC13) antibody. Immunoprecipitation of etoposide treated total nuclear extracts with mouse IgG was used as a control. Immunoprecipitates were probed for γ-H2AX or MCL-1. (B) Mcl-1-/- MEFs were either transiently transfected with empty vector or vector containing human MCL-1. Cells were fixed and stained with anti-MCL-1 antibody and visualized by confocal microscopy using goat anti-rabbit Alexa Fluor 594 (red; for MCL-1). Nuclei were stained with Hoechst 33342.
Figure 5 Association of MCL-1 and γ-H2AX is altered in ATR defective cells. (A) F02-98 cells (Seckel) or primary human fibroblasts (Hs68) were either untreated (0) or treated with etoposide (Etop) for 3 h or exposed to UVB radiation for 20 s and allowed to recover for 3 h. Total nuclear extracts were immunoprecipitated with anti-MCL-1 antibody and probed for γ-H2AX and MCL-1. Ab represents anti-MCL-1 antibody used in the absence of nuclear extracts. The lower parts indicate total nuclear extracts following immunoprecipitation, probed for non-immunoprecipitated γ-H2AX (Post-IP) and the Input Control was taken from the total nuclear extracts prior to immunoprecipitation and probed for Oct1 expression. (B) HT-144 cells (ATM-deficient) were either untreated (0) or treated with Etoposide (Etop) for 3 h or exposed to UVB radiation for 20 sec and allowed to recover for 3 h. Total nuclear extracts were immunoprecipitated with anti-MCL-1 antibody and probed for γ-H2AX and MCL-1. Ab represents anti-MCL-1 antibody used in the absence of nuclear extracts.
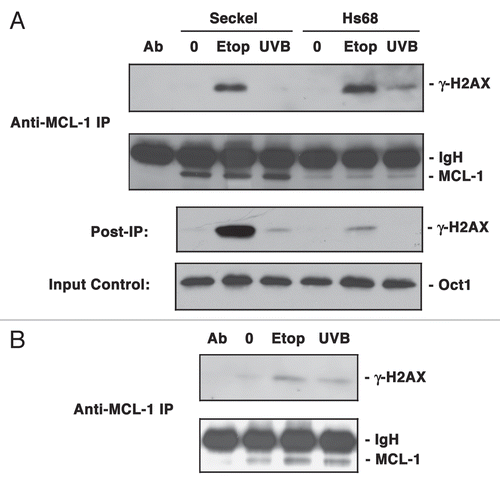
Figure 6 MCL-1 co-immunoprecipitates with NBS1 following DNA damage. (A) HeLa cells were either untreated (0) or treated with Etoposide (Etop) for 3 h or exposed to UVB radiation (UV) for 20 s and allowed to recover for 3 h. Total nuclear extracts were immunoprecipitated with anti-MCL-1 antibody or anti-NBS1 antibody and probed for MCL-1, NBS1 or γ-H2AX. (B) HeLa cells were untreated or treated with etoposide for 3 h. Cells were fixed and stained with anti-MCL-1 or anti-NBS1 antibodies and visualized by confocal microscopy using goat anti-rabbit Alexa Fluor 488 (green; for MCL-1) or goat anti-mouse Alexa Fluor 594 (red; for NBS1). Nuclei were stained with Hoechst 33342.
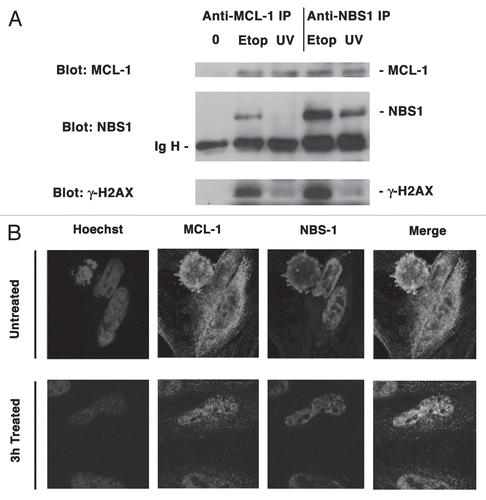
Figure 7 Chromatin immunoprecipitation demonstrates presence of MCL-1 at sites of DSB. (A) HeLa cells were retrovirally transduced to express HA-ER-I-Ppo1. Cells were not induced (0) or induced with 4-OHT for 2 h or 16 h. Cytosolic or total nuclear proteins were separated and blots probed with anti-MCL-1 or with anti-vinculin or anti-Oct1 as loading controls for cytosol or nucleus, respectively. Samples were resolved on the same gel, with vertical lines indicating the removal of irrelevant intervening lanes. (B) HeLa cells were retrovirally transduced to express HA-ER-I-Ppo1. Cells were not induced (0) or induced with 4-OHT for 2 h or 16 h. Following crosslinking and shearing of DNA, samples were immunoprecipitated using anti-NBS1 antibody. Ab indicates mock immunoprecipitation with no DNA added. Purified DNA was subjected to PCR as described in the Materials and Methods. The lower part shows PCR products of DNA samples taken from the respective extracts prior to immunoprecipitation. C indicates non-specific control for PCR. (C) HeLa cells were treated exactly as in (B), except for the use of anti-MCL-1 antibody for immunoprecipitation.
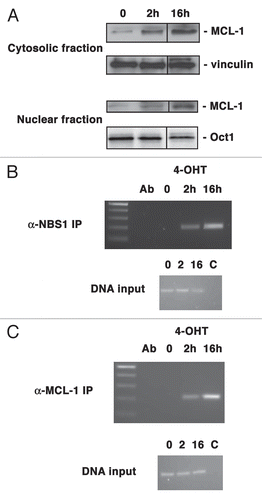
Table 1 Analysis of metaphase spreads from two independent experiments (A and B)
Acknoweledgements
We thank Dr. Peter Lansdorp and Ms. Liz Chavez for technical assistance in preparing and analyzing metaphase spreads for chromosomal aberrations. We thank Dr. Andrew Sandford for critical review of the manuscript. This work was supported by a grant to V.D. from the Canadian Institutes of Health Research (MOP-74481).
References
- Bartek J, Bartkova J, Lukas J. DNA damage signalling guards against activated oncogenes and tumour progression. Oncogene 2007; 26:7773 - 7779
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39 - 85
- Kobayashi J. Molecular mechanism of the recruitment of NBS1/hMRE11/hRAD50 complex to DNA double-strand breaks: NBS1 binds to gamma-H2AX through FHA/BRCT domain. J Radiat Res (Tokyo) 2004; 45:473 - 478
- Robison JG, Dixon K, Bissler JJ. Cell cycle-and proteasome-dependent formation of etoposide-induced replication protein A (RPA) or Mre11/Rad50/Nbs1 (MRN) complex repair foci. Cell Cycle 2007; 6:2399 - 2407
- Rogakou EP, Pilch DR, Orr AH, Ivanova VS, Bonner WM. DNA double-stranded breaks induce histone H2AX phosphorylation on serine 139. J Biol Chem 1998; 273:5858 - 5868
- Rogakou EP, Boon C, Redon C, Bonner WM. Megabase chromatin domains involved in DNA double-strand breaks in vivo. J Cell Biol 1999; 146:905 - 916
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366 - 374
- Friedberg EC. DNA damage and repair. Nature 2003; 421:436 - 440
- Lisby M, Rothstein R. Localization of checkpoint and repair proteins in eukaryotes. Biochimie 2005; 87:579 - 589
- Lisby M, Barlow JH, Burgess RC, Rothstein R. Choreography of the DNA damage response: spatiotemporal relationships among checkpoint and repair proteins. Cell 2004; 118:699 - 713
- Petrini JH, Stracker TH. The cellular response to DNA double-strand breaks: defining the sensors and mediators. Trends Cell Biol 2003; 13:458 - 462
- Costanzo V, Paull T, Gottesman M, Gautier J. Mre11 assembles linear DNA fragments into DNA damage signaling complexes. PLoS Biol 2004; 2:110
- Falck J, Coates J, Jackson SP. Conserved modes of recruitment of ATM, ATR and DNA-PKcs to sites of DNA damage. Nature 2005; 434:605 - 611
- Jazayeri A, Falck J, Lukas C, Bartek J, Smith GC, Lukas J, et al. ATM- and cell cycle-dependent regulation of ATR in response to DNA double-strand breaks. Nat Cell Biol 2006; 8:37 - 45
- Dzikiewicz-Krawczyk A. The importance of making ends meet: mutations in genes and altered expression of proteins of the MRN complex and cancer. Mutat Res 2008; 659:262 - 273
- Jamil S, Mojtabavi S, Hojabrpour P, Cheah S, Duronio V. An essential role for MCL-1 in ATR-mediated CHK1 phosphorylation. Mol Biol Cell 2008; 19:3212 - 3220
- Jamil S, Sobouti R, Hojabrpour P, Raj M, Kast J, Duronio V. A proteolytic fragment of Mcl-1 exhibits nuclear localization and regulates cell growth by interaction with Cdk1. Biochem J 2005; 387:659 - 667
- Kamer I, Sarig R, Zaltsman Y, Niv H, Oberkovitz G, Regev L, et al. Proapoptotic BID is an ATM effector in the DNA-damage response. Cell 2005; 122:593 - 603
- Zinkel SS, Hurov KE, Ong C, Abtahi FM, Gross A, Korsmeyer SJ. A role for proapoptotic BID in the DNA-damage response. Cell 2005; 122:579 - 591
- Wang Q, Gao F, May WS, Zhang Y, Flagg T, Deng X. Bcl2 negatively regulates DNA double-strand-break repair through a nonhomologous end-joining pathway. Mol Cell 2008; 29:488 - 498
- Rinkenberger JL, Horning S, Klocke B, Roth K, Korsmeyer SJ. Mcl-1 deficiency results in peri-implantation embryonic lethality. Genes Development 2000; 14:23 - 27
- Opferman JT, Iwasaki H, Ong CC, Suh H, Mizuno S, Akashi K, et al. Obligate role of anti-apoptotic MCL-1 in the survival of hematopoietic stem cells. Science 2005; 307:1101 - 1104
- Opferman JT, Letai A, Beard C, Sorcinelli MD, Ong CC, Korsmeyer SJ. Development and maintenance of B and T lymphocytes requires antiapoptotic MCL-1. Nature 2003; 426:671 - 676
- Kitada S, Reed JC. MCL-1 promoter insertions dial-up aggressiveness of chronic leukemia. J Natl Cancer Inst 2004; 96:642 - 643
- Le Gouill S, Podar K, Harousseau JL, Anderson KC. Mcl-1 regulation and its role in multiple myeloma. Cell Cycle 2004; 3:1259 - 1262
- van Maanen JM, Retel J, de Vries J, Pinedo HM. Mechanism of action of antitumor drug etoposide: a review. J Natl Cancer Inst 1988; 80:1526 - 1533
- Johansson F, Lagerqvist A, Erixon K, Jenssen D. A method to monitor replication fork progression in mammalian cells: nucleotide excision repair enhances and homologous recombination delays elongation along damaged DNA. Nucleic Acids Res 2004; 32:157
- Nijhawan D, Fang M, Traer E, Zhong Q, Gao W, Du F, et al. Elimination of Mcl-1 is required for the initiation of apoptosis following ultraviolet irradiation. Genes Dev 2003; 17:1475 - 1486
- O'Driscoll M, Ruiz-Perez VL, Woods CG, Jeggo PA, Goodship JA. A splicing mutation affecting expression of ataxia-telangiectasia and Rad3-related protein (ATR) results in Seckel syndrome. Nat Genet 2003; 33:497 - 501
- Berkovich E, Monnat RJ Jr, Kastan MB. Roles of ATM and NBS1 in chromatin structure modulation and DNA double-strand break repair. Nat Cell Biol 2007; 9:683 - 690
- Wu L, Luo K, Lou Z, Chen J. MDC1 regulates intra-S-phase checkpoint by targeting NBS1 to DNA double-strand breaks. Proc Natl Acad Sci USA 2008; 105:11200 - 11205
- Berkovich E, Monnat RJ Jr, Kastan MB. Assessment of protein dynamics and DNA repair following generation of DNA double-strand breaks at defined genomic sites. Nat Protoc 2008; 3:915 - 922
- Muscarella DE, Ellison EL, Ruoff BM, Vogt VM. Characterization of I-Ppo, an intron-encoded endonuclease that mediates homing of a group I intron in the ribosomal DNA of Physarum polycephalum. Mol Cell Biol 1990; 10:3386 - 3396
- Flick KE, Jurica MS, Monnat RJ Jr, Stoddard BL. DNA binding and cleavage by the nuclear intron-encoded homing endonuclease I-PpoI. Nature 1998; 394:96 - 101
- Schonn I, Hennesen J, Dartsch DC. Cellular responses to etoposide: cell death despite cell cycle arrest and repair of DNA damage. Apoptosis 2010; 15:162 - 172
- Paull TT, Rogakou EP, Yamazaki V, Kirchgessner CU, Gellert M, Bonner WM. A critical role for histone H2AX in recruitment of repair factors to nuclear foci after DNA damage. Curr Biol 2000; 10:886 - 895
- Fujise K, Zhang D, Liu J, Yeh ET. Regulation of apoptosis and cell cycle progression by MCL1. Differential role of proliferating cell nuclear antigen. J Biol Chem 2000; 275:39458 - 39465
- Liu H, Peng HW, Cheng YS, Yuan HS, Yang-Yen HF. Stabilization and enhancement of the antiapoptotic activity of mcl-1 by TCTP. Mol Cell Biol 2005; 25:3117 - 3126
- Sun Z, Hsiao J, Fay DS, Stern DF. Rad53 FHA domain associated with phosphorylated Rad9 in the DNA damage checkpoint. Science 1998; 281:272 - 274
- Yaffe MB, Elia AE. Phosphoserine/threonine-binding domains. Curr Opin Cell Biol 2001; 13:131 - 138
- Yu X, Chini CC, He M, Mer G, Chen J. The BRCT domain is a phospho-protein binding domain. Science 2003; 302:639 - 642
- Hekmat-Nejad M, You Z, Yee MC, Newport JW, Cimprich KA. Xenopus ATR is a replication-dependent chromatin-binding protein required for the DNA replication checkpoint. Curr Biol 2000; 10:1565 - 1573
- Costanzo V, Shechter D, Lupardus PJ, Cimprich KA, Gottesman M, Gautier J. An ATR- and Cdc7-dependent DNA damage checkpoint that inhibits initiation of DNA replication. Mol Cell 2003; 11:203 - 213
- Tauchi H, Kobayashi J, Morishima K, van Gent DC, Shiraishi T, Verkaik NS, et al. Nbs1 is essential for DNA repair by homologous recombination in higher vertebrate cells. Nature 2002; 420:93 - 98
- Tauchi H, Matsuura S, Kobayashi J, Sakamoto S, Komatsu K. Nijmegen breakage syndrome gene, NBS1 and molecular links to factors for genome stability. Oncogene 2002; 21:8967 - 8980
- Montecucco A, Biamonti G. Cellular response to etoposide treatment. Cancer Lett 2007; 252:9 - 18
- Mirzoeva OK, Petrini JH. DNA damage-dependent nuclear dynamics of the Mre11 complex. Mol Cell Biol 2001; 21:281 - 288
- Desai-Mehta A, Cerosaletti KM, Concannon P. Distinct functional domains of nibrin mediate Mre11 binding, focus formation and nuclear localization. Mol Cell Biol 2001; 21:2184 - 2191
- D'Amours D, Jackson SP. The Mre11 complex: at the crossroads of dna repair and checkpoint signalling. Nat Rev Mol Cell Biol 2002; 3:317 - 327
- Stracker TH, Theunissen JW, Morales M, Petrini JH. The Mre11 complex and the metabolism of chromosome breaks: the importance of communicating and holding things together. DNA Repair (Amst) 2004; 3:845 - 854
- Calonge TM, O'Connell MJ. Turning off the G2 DNA damage checkpoint. DNA Repair (Amst) 2008; 7:136 - 140
- Zhou BB, Elledge SJ. The DNA damage response: putting checkpoints in perspective. Nature 2000; 408:433 - 439
- Millar JB, Blevitt J, Gerace L, Sadhu K, Featherstone C, Russell P. p55CDC25 is a nuclear protein required for the initiation of mitosis in human cells. Proc Natl Acad Sci USA 1991; 88:10500 - 10504
- Latif C, den Elzen NR, O'Connell MJ. DNA damage checkpoint maintenance through sustained Chk1 activity. J Cell Sci 2004; 117:3489 - 3498
- Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 2000; 6:839 - 849
- Chini CC, Chen J. Human claspin is required for replication checkpoint control. J Biol Chem 2003; 278:30057 - 30062
- Kurose A, Tanaka T, Huang X, Traganos F, Darzynkiewicz Z. Synchronization in the cell cycle by inhibitors of DNA replication induces histone H2AX phosphorylation: an indication of DNA damage. Cell Prolif 2006; 39:231 - 240
- Lapenna S, Giordano A. Cell cycle kinases as therapeutic targets for cancer. Nat Rev Drug Discov 2009; 8:547 - 566
- Hong Y, Stambrook PJ. Restoration of an absent G1 arrest and protection from apoptosis in embryonic stem cells after ionizing radiation. Proc Natl Acad Sci USA 2004; 101:14443 - 14448
- Opferman JT. Life and death during hematopoietic differentiation. Curr Opin Immunol 2007; 19:497 - 502
- Wheaton K, Riabowol K. Protein kinase C delta blocks immediate-early gene expression in senescent cells by inactivating serum response factor. Mol Cell Biol 2004; 24:7298 - 7311
- Shiio Y, Eisenman RN, Yi EC, Donohoe S, Goodlett DR, Aebersold R. Quantitative proteomic analysis of chromatin-associated factors. J Am Soc Mass Spectrom 2003; 14:696 - 703