Abstract
In a series of discoveries over the preceding decade, a number of laboratories have unequivocally established that apoptotic proteins and pathways are well conserved cell fate determinants, which act independent of a cell death response. Within this context, the role for apoptotic proteins in the induction of cell differentiation has been widely documented. Despite these discoveries, little information has been forthcoming regarding a conserved mechanism by which apoptotic proteins achieve this non-death outcome. In the following discussion, we will explore the premise that the penultimate step in apoptosis, genome wide DNA damage/strand breaks act as a conserved genomic reprogramming event necessary for cell differentiation (Larsen et al., Proc Natl Acad Sci USA 2010; 107 (9):4230-5). Moreover, we hypothesis that directed DNA damage, as mediated by known apoptotic proteins, may participate in numerous forms of regulated gene expression.
Introduction
DNA damage is largely assumed to be a detrimental event and is frequently associated with impaired cell survival. In addition, DNA damage is a common molecular trigger for the development of oncogenic mutations. For example DNA damage/strand breaks precede a recombination of two distinct genetic loci to produce a hybrid gene with growth altering properties. Given the dire outcomes associated with DNA damage, the cell has evolved a number of DNA repair pathways which recognize and redact genome damage that occurs from a variety of external insults.
Despite the propensity to consider DNA damage as a solely negative phenomenon, a growing body of evidence suggests that focal DNA damage is in fact required for normal cell function. Specifically, controlled or non-random DNA damage appears to be a conserved mechanism which propagates alterations in gene expression. The best studied example in this regard is the regulated DNA breakage that propels adaptive immunity. Here, the Rag1/2 nuclease complex induces controlled DNA breaks at specific loci that are paired with variable recombination events to create the unique genes that underwrite the diversity of antibody production.Citation1 Another well documented example of tolerable or beneficial DNA breakage occurs during the exchange of genetic material between homologous chromosomes in meiotic crossover. In this instance, DNA breaks are induced by the topoisomerase II-like protein Spo11, which propels an exchange between large intergenic regions.Citation2
Additional studies suggest that directed DNA strand breaks may promote gene expression independent of follow-on recombination events. For example, work from the Rosenfeld Laboratory has shown that glucocorticoid induced gene expression is dependent on a topoisomerase IIbeta mediated DNA strand break. The strand breaks are directed to the promoter region of the glucocorticoid responsive gene, an alteration that prompts histone modifications which are favourable to initiating gene transcription.Citation3,Citation4 Undoubtedly, there is an inherent risk associated with such DNA damage yet these observations establish that limited DNA strand breaks can yield a notable beneficial outcome for a cell.Citation4
Recently, work from our laboratory has transformed the paradigm described above and shown that genome wide DNA strand breaks act as a key regulatory step to promote muscle cell differentiation.Citation5 The DNA strand breaks are formed by a transient activation of caspase activated DNase or CAD, an observation that suggests apoptotic proteins and pathways act as conserved genomic reprogramming factors.
Caspase 3/CAD Signaling: The Executioners
The caspase family of cysteine proteases signal through proteolytic cleavage, altering the activity of an extensive repertoire of substrates. Caspase 3 is an integration point for a variety of canonical cell death pathways, acting to target cytosolic and nuclear factors which in turn accelerate cellular demise.Citation6 One key feature of this potent signal is the ability to activate the primary nuclease involved in disseminating extensive DNA strand breaks, termed caspase activated DNase or CAD.Citation7 DNA damage in the form of genomic fragmentation is a well characterized component of cell death or apoptosis. Although early stage DNA fragmentation is not an absolute requirement for all early stage forms of apoptotic cell death, these nuclear events are assumed to improve the efficiency of the process.Citation8 Indeed, the release of nucleotides and presentation of nucleosomes at the cell surface promote the removal of degenerate cells through attraction of phagocytic cells.Citation9,Citation10
The principle regulation of CAD comes by formation of a restrictive complex with its inhibitor, ICAD, an interaction that physically restrains inactive CAD monomers from dimerizing into the active form. Two isoforms of ICAD are observed, ICAD-long (L) and ICAD-short (S), with the ICAD-L isoform having the most defined role in regulating CAD. Association by N-terminal CIDE domains begins when the CAD protein is synthesized, here ICAD-L acts as a specific chaperone along with Hsp70 and Hsp40 to properly fold CAD.Citation11,Citation12 Following translation the ICAD-L/CAD complex localizes to the nucleus, where it remains highly mobile; however, stable interactions with DNA have been noted suggesting that CAD can be activated not only in the free nuclear space but also in a DNA associated complex.Citation13–Citation15
To activate CAD, caspase 3 targets ICAD for proteolytic cleavage at 2 aspartic acids, D117 and D224. This caspase directed cleavage destabilizes the ICAD/CAD interaction and allows CAD dimerization. The CAD dimer aptly resembles an open scissor like structure, with the catalytic site of the dimer located in a crevice structure large enough to accommodate double stranded DNA.Citation16 The substrate DNA fully enters this structure and a double stranded DNA break is catalyzed. This protein conformation is believed to exclude nucleosome associated DNA, localizing the DNA break between nucleosomes. Under apoptotic conditions the extent of CAD activation is such that periodic DNA laddering is observed.Citation8
Caspase 3/CAD Mediated DNA Strand Breaks: A Vital Genome Reprogramming Event
In addition to a well characterized role in apoptosis, caspase 3 activation is also a highly conserved step in the induction of cell differentiation. Transient caspase 3 activity has been shown to be essential for differentiation of most somatic cell types studied and for the maturation of both ES and germs cells.Citation17 The extent of caspase 3 activation appears to control the balance between differentiation and apoptosis, with lower versus higher levels of activity controlling each cell fate respectively.Citation18 Defining the caspase 3 substrates that convey the differentiation signal has been of considerable interest, yet limited information has been forthcoming. Studies have suggested that caspase cleavage activation of select kinases propels the differentiation program whereas other studies have shown that cleavage inactivation of transcription factors establishes a permissive environment for the process.Citation19–Citation23 Although these specific caspase substrate interactions appear to be critical, these same catalytic events do not explain the ability of caspase 3 to induce the global genome reprogramming that typifies cell differentiation.
Recently, we have reported that caspase 3 activates CAD in healthy muscle cells and that this step is essential for completion of myogenic differentiation.Citation5 During early stages of differentiation, myoblast nuclei are subject to CAD dependent DNA strand breaks and inhibition of CAD activity (by limiting caspase 3 activation, repressing CAD expression or overexpressing a non cleavable version of the CAD inhibitor ICAD) leads to a near complete blockade in differentiation with a concurrent loss in the formation of strand breaks. The CAD mediated strand breaks are in part permissive, as the induction of the critical regulatory factor p21 is dependent on a caspase 3/CAD directed strand break within the p21 promoter.
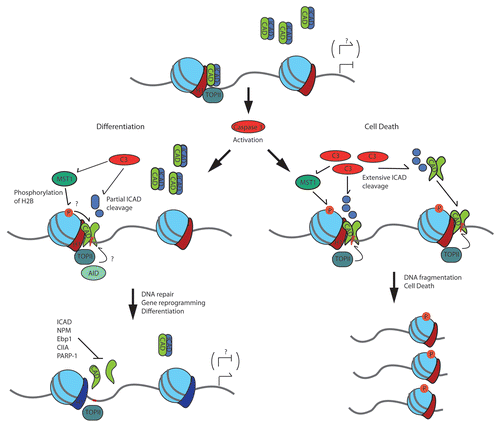
Nevertheless, like many interesting discoveries, our recent work opens the door to many more questions than was answered in the initial study. Paramount among the unresolved mechanisms, once CAD is activated how is the DNase restrained sufficiently to induce strand breaks yet not destroy the genome? We hypothesize that two independent mechanisms may contribute to the restrained activation of CAD. First, we anticipate that it is the DNA associated pool of CAD that is activated during differentiation. CAD has been shown to be complexed with ICAD in a nuclear position, a localization that would permit ready activation once caspase 3 was activated.Citation13,Citation14 In contrast, the excessive activation of caspase 3 during apoptosis would allow for targeting of both the DNA bound and unbound CAD/ICAD complexes, the later being free to inflict indiscriminate breaks throughout the genome. Consistent with this hypothesis we have noted that the p21 promoter is subject to consistently localized strand breaks during differentiation, while apoptotic muscle cells display widespread formation of strand breaks that are variable from experiment to experiment (see Fig. 4 in ref. Citation5). A confirmation of our hypothesis will require a definitive visualization of distinct ICAD/CAD pools and/or a mapping of the CAD targeted genome that is specific to each cell fate outcome. Second, we hypothesize that caspase targeting of ICAD during differentiation is directed at the D117 cleavage site, whereas the greatly elevated caspase activity associated with apoptosis would result in cleavage at both D117 and D224. In this model of CAD activation, caspase cleavage at the D117 site would lead to a partial release of CAD while caspase targeting of both sites would remove any ICAD mediated inhibition with a higher resulting level of CAD activity. We have noted an ICAD cleavage event during differentiation that is consistent with a D117 only deletion, and a mutation in this site that renders ICAD caspase resistant leads to a reduction in myoblast differentiation (see Fig. 2 in ref. Citation5).
Our observations demonstrate that caspase 3 activation of CAD (through ICAD cleavage and release of active CAD) is the primary step in promoting DNA damage/strand breaks during myoblast differentiation. Nevertheless, caspase 3 is known to target a wide variety of substrates during apoptosis and cell differentiation.Citation18 Therefore, we hypothesize that caspase 3 targets multiple proteins to ensure efficient activity of the CAD nuclease. Apoptotic signalling pathways modify chromatin ultra-structure prior to DNA damage and nuclear dissolution. In this regard, post-translational modifications of histones assume prominent regulatory roles. As such it is reasonable to assume that similar events pre-empt or assist CAD to promote DNA damage during cell differentiation. Specifically, we assert that the ste20-like kinase MST1 participates in the formation of caspase mediated nicks/strand breaks in differentiating myoblasts by phosphorylating and modifying the activity of chromatin regulatory proteins. This supposition is based on the following observations. First, MST1 was initially characterized as a pro-apoptotic, caspase 3 sensitive kinase and more recently has been demonstrated to directly phosphorylate histone H2B (at serine 14) leading to chromatin compaction and apoptosis.Citation24–Citation26 Secondly, our laboratory was the first to describe a non-apoptotic function for MST1, i.e., MST1 as a pro-differentiation kinase.Citation22 Subsequent to our observations, numerous groups reported a role for MST1 in limiting cell cycle progression in both mammalian and Drosophila systems (reviewed in Zeng and Hong 2008).Citation27 Our experiment was noteworthy in that an activated MST1 kinase (caspase 3 cleavage activated kinase domain) provided a partial rescue of differentiation in caspase 3 null myoblasts, yet prolonged expression of MST1 led to formation of picnotic nuclei and apoptosis.Citation22 Together, these observations suggest that MST1 may stimulate differentiation by modulating or by activating chromatin remodelling protein(s) to enhance CAD activity.
Caspase 3 can also target DNA binding proteins to modulate gene expression and several of these targets have the potential to modify CAD access to sensitive genomic elements. The matrix attachment protein, Special AT-rich binding protein 1 (SATB1), is a global organizer of chromatin that facilitates the interaction of DNA elements termed matrix attachment regions (MARs) to the nuclear matrix. SATB1 is a demonstrated cleavage target of caspase 3/6 under both apoptotic and non-apoptotic conditions.Citation28–Citation30 Cleavage of SATB1 releases MARs from the nuclear matrix, leading to chromatin disruption.Citation31 A specific role for caspase cleavage of SATB1 during differentiation has not been demonstrated, yet it is reasonable to suggest that removal of SATB1 may in some instances expose genomic elements that are targeted by CAD to coordinate gene expression.
CAD activity during cell differentiation may also be moderated by additional chromatin modifications independent of caspase function. CAD/ICAD association to DNA can be potentiated by association with the C-terminal of histone H1, and this interaction can further stimulate the nuclease activity of CAD.Citation32 Specifically, CAD has been reported to dynamically associate with the histone H1 variants, namely H1.5 and H1.0 in both healthy and apoptotic cells.Citation33 Moreover, H1 variants can affect specific gene expression by cooperating with additional regulatory factors.Citation34 This preferential association to histone variants could partly influence where in the genome CAD inflicts DNA strand breaks. Of interest, coordination between the H1.5 variant and the transcription factor Msx1 has been demonstrated to occur at the promoters of repressed genes in proliferating myoblasts.Citation35 For example, expression of the cell cycle inhibitor p21 (which we have identified as a bona fide CAD target) is impeded by Msx1 prior to myoblast differentiation.Citation36 Based on our observations a reasonable conjecture is that CAD may be directed to relieve this repressed state and engage p21 expression by interacting with histone H1.5 and thereby displacing Msx1. How operative this mechanism is and how extensive across the genome it may be will require further investigation.
Curbing the Caspase3/CAD Signal
The transient activity of caspase and CAD in differentiating cells suggests the deployment of a mechanism(s) to moderate or terminate this signal prior to inducing extensive DNA damage. Several studies have examined the signal cascades that activate and modulate caspase 3 during non-death responses, yet a definitive control mechanism has not been elucidated.Citation37–Citation39 Such inactivation could indirectly end CAD activity by discontinuing proteolysis of ICAD. Intact ICAD (both long and short isoforms) can disassemble the CAD dimer, moreover our observation that ICAD-L is only partial cleaved (with a large pool of intact ICAD-L remaining) suggests that this mechanism maybe operative to prevent extensive DNA fragmentation.Citation16
Intriguingly, alternate methods that block the nuclease activity of CAD have been noted. Admittedly, these cellular responses have been implicated primarily in response to caspase activity in apoptotic settings or to restrain CAD activity when this nuclease is activated as a by product of another molecular mechanism. Nevertheless, it is reasonable to suggest that these same factors moderate CAD activity during cell differentiation. Polyribosylation of CAD by PARP-1 can further restrict CAD nuclease activity, and a transient increase in PARP-1 activity is reported in skeletal muscle differentiation concurrent with the observed DNA strand break formation reported in Larsen et al.Citation5,Citation40,Citation41 We have not examined a role for PARP-1 in CAD mediated DNA strand breaks and repair, yet PARP-1 has been demonstrated to play a role in regulating gene expression that results from controlled DNA strand breaks in response to glucocorticoid stimulation. Nerve growth factor (NGF) may also provide a signal conduit to moderate CAD activity in both apoptotic and cell differentiation scenarios. For example, NGF protects neuronal cells from apoptosis through a number of mechanisms, two of these mechanisms involve inhibition of CAD. NGF stimulates nucleophosmin and Ebp1 to directly interact with CAD, diminishing its nuclease activity while not preventing caspase cleavage of ICAD itself.Citation42,Citation43 Given that caspase 3 has been shown to mediate neuronal differentiation, a valid interpretation of the above experiment may be that NGF restrains CAD activity to act as a moderating influence on gene expression during neuron differentiation (as in skeletal muscle cells) while limiting a global escalation of CAD activity.Citation21
CAD Induced DNA Strand Breaks as a General Component of Gene Expression
An intriguing question that arises from the study of Larsen et al. is whether CAD induced DNA strand breaks are simply a differentiation specific regulatory event or is CAD directed DNA damage a common mechanism for altering gene expression?
As noted above, the role of DNA strand breaks regulating changes in gene expression and genomic reprogramming has been characterized in a number of processes. However, in many instances the induction of the DNA breaks have been linked to proteins with weak or questionable nuclease activity. As such it is reasonable to assume that localized CAD activation may coordinate with the other DNA damage related proteins to promote break induced alterations in gene regulation. Glucocorticoid stimulated gene expression involves topoisomerase II dependent DNA strand breaks. These breaks are directed by the estrogen receptor α (ERα) and stimulate target gene expression through PARP-1 mediated exchange of histone H1 for HMGB1/2.Citation3,Citation44 A cooperation between CAD and topoisomerase II in stimulating the formation of glucocorticoid stimulated breaks was not examined in this study, yet such a coordination could effectively direct and stimulate the induction of DNA strand breaks. Indeed, an interplay between CAD and topoisomerase II has been reported in the initial induction of DNA strand breaks during apoptosis, and the proteins co-localize in both healthy and apoptotic nuclei.Citation45 These observations combined with weak inherent nuclease activity of topoisomerase II suggest that CAD may provide the operative strand break capacity in such forms of gene regulation.
DNA demethylation has been postulated to involve the formation of DNA strand breaks as a means to excise the methylated base followed by a DNA repair step. In this regard several proteins have been implicated that can also contribute to the formation of regulated DNA strand breaks. The cytidine deaminase AID has been reported to act as a demethylation agent in primordial germ cells and in the nuclear reprogramming of somatic cells via induced pluripotency (iPS).Citation46,Citation47 AID has been well studied for its role in antibody diversification where it is thought to stimulate a directed DNA strand break through deamination leading to activation of the base excision repair pathway.Citation48 AID has been shown to associate with methylated promoters of pluripotency genes in fibroblasts but not unmethylated promoters in undifferentiatied ES cells, and knockdown of AID gene expression impeded the demethylation of these promoters in heterokaryon fusions.Citation47 Interestingly global DNA demethylation is observed in skeletal muscle differentiation concurrent to the period of CAD induced DNA strand breaks.Citation49 Although we observed unaltered DNA methylation at the p21 promoter, these breaks may signal at other loci for the removal of the repressive mark, a step that may be dependent on a CAD like excision event. Alternatively, given the role of caspase/CAD in driving cell fate outcomes that are directly antagonistic to iPS (i.e., cell differentiation) achieving effective reprogramming may require a direct inhibition or limitation of caspase/CAD activity. As caspase mediated signal events are broadly conserved cell fate determinants, exploring the impact of a caspase/CAD signal nexus during nuclear reprogramming should be a priority.
Determining the genomic targets of caspase 3 activated CAD will assist in developing appreciation for the role these DNA strand breaks play in regulating gene expression (). Mapping these sites using rapidly developing next generation sequencing technology and determining protein interactions that direct CAD will begin to establish this understanding. Further examination of epigenetic changes that occur at these sites will provide insight into how the strand break is utilized to regulate gene expression not only in differentiation but potentially in other genomic reprogramming events.
Figures and Tables
Figure 1 Caspase 3 activation balances the dissemination of CAD induced DNA strand breaks for gene reprogramming or fragmentation. This model depicts several factors that may regulate CAD induced DNA strand breaks in physiologic and apoptotic conditions.
Acknowledgements
We thank the members of the Megeney Lab for their ongoing contributions to this work. B.D.L. is supported by a Computational Regulomics Training Program Graduate Student Fellowship from the Ontario Ministry of Research and Innovation Ontario Research Fund. L.A.M. is the Mach Gaensslen Chair in Cardiac Research. This work was supported by grants from the Canadian Institutes of Health Research and the Muscular Dystrophy Association (L.A.M.).
References
- Jung D, Alt FW. Unraveling V(D)J recombination; insights into gene regulation. Cell 2004; 116:299 - 311
- Li W, Ma H. Double-stranded DNA breaks and gene functions in recombination and meiosis. Cell Res 2006; 16:402 - 412
- Ju BG, Lunyak VV, Perissi V, Garcia-Bassets I, Rose DW, Glass CK, et al. A topoisomerase IIbeta-mediated dsDNA break required for regulated transcription. Science 2006; 312:1798 - 1802
- Lin C, Yang L, Tanasa B, Hutt K, Ju BG, Ohgi K, et al. Nuclear receptor-induced chromosomal proximity and DNA breaks underlie specific translocations in cancer. Cell 2009; 139:1069 - 1083
- Larsen BD, Rampalli S, Burns LE, Brunette S, Dilworth FJ, Megeney LA. Caspase 3/caspase-activated DNase promote cell differentiation by inducing DNA strand breaks. Proc Natl Acad Sci USA 2010; 107:4230 - 4235
- Fischer U, Janicke RU, Schulze-Osthoff K. Many cuts to ruin: a comprehensive update of caspase substrates. Cell Death Differ 2003; 10:76 - 100
- Widlak P, Garrard WT. Roles of the major apoptotic nuclease-DNA fragmentation factor-in biology and disease. Cell Mol Life Sci 2009; 66:263 - 274
- Samejima K, Earnshaw WC. Trashing the genome: the role of nucleases during apoptosis. Nat Rev Mol Cell Biol 2005; 6:677 - 688
- Elliott MR, Chekeni FB, Trampont PC, Lazarowski ER, Kadl A, Walk SF, et al. Nucleotides released by apoptotic cells act as a find-me signal to promote phagocytic clearance. Nature 2009; 461:282 - 286
- Radic M, Marion T, Monestier M. Nucleosomes are exposed at the cell surface in apoptosis. J Immunol 2004; 172:6692 - 6700
- McCarty JS, Toh SY, Li P. Study of DFF45 in its role of chaperone and inhibitor: Two independent inhibitory domains of DFF40 nuclease activity. Biochem Biophys Res Commun 1999; 264:176 - 180
- Sakahira H, Nagata S. Co-translational folding of caspase-activated DNase with Hsp70, Hsp40 and inhibitor of caspase-activated DNase. J Biol Chem 2002; 277:3364 - 3370
- Lechardeur D, Xu M, Lukacs GL. Contrasting nuclear dynamics of the caspase-activated DNase (CAD) in dividing and apoptotic cells. J Cell Biol 2004; 167:851 - 862
- Korn C, Scholz SR, Gimadutdinow O, Lurz R, Pingoud A, Meiss G. Interaction of DNA fragmentation factor (DFF) with DNA reveals an unprecedented mechanism for nuclease inhibition and suggests that DFF can be activated in a DNA-bound state. J Biol Chem 2005; 280:6005 - 6015
- Reh S, Korn C, Gimadutdinow O, Meiss G. Structural basis for stable DNA complex formation by the caspase-activated DNase. J Biol Chem 2005; 280:41707 - 41715
- Woo EJ, Kim YG, Kim MS, Han WD, Shin S, Robinson H, et al. Structural mechanism for inactivation and activation of CAD/DFF40 in the apoptotic pathway. Mol Cell 2004; 14:531 - 539
- Abdul-Ghani M, Megeney LA. Rehabilitation of a contract killer: caspase-3 directs stem cell differentiation. Cell Stem Cell 2008; 2:515 - 516
- Fernando P, Megeney LA. Is caspase-dependent apoptosis only cell differentiation taken to the extreme?. FASEB J 2007; 21:8 - 17
- Zeuner A, Eramo A, Testa U, Felli N, Pelosi E, Mariani G, et al. Control of erythroid cell production via caspase-mediated cleavage of transcription factor SCL/Tal-1. Cell Death Differ 2003; 10:905 - 913
- Fujita J, Crane AM, Souza MK, Dejosez M, Kyba M, Flavell RA, et al. Caspase activity mediates the differentiation of embryonic stem cells. Cell Stem Cell 2008; 2:595 - 601
- Fernando P, Brunette S, Megeney LA. Neural stem cell differentiation is dependent upon endogenous caspase 3 activity. FASEB J 2005; 19:1671 - 1673
- Fernando P, Kelly JF, Balazsi K, Slack RS, Megeney LA. Caspase 3 activity is required for skeletal muscle differentiation. Proc Natl Acad Sci USA 2002; 99:11025 - 11030
- Kanuka H, Kuranaga E, Takemoto K, Hiratou T, Okano H, Miura M. Drosophila caspase transduces Shaggy/GSK-3beta kinase activity in neural precursor development. EMBO J 2005; 24:3793 - 3806
- Cheung WL, Ajiro K, Samejima K, Kloc M, Cheung P, Mizzen CA, et al. Apoptotic phosphorylation of histone H2B is mediated by mammalian sterile twenty kinase. Cell 2003; 113:507 - 517
- Ahn SH, Cheung WL, Hsu JY, Diaz RL, Smith MM, Allis CD. Sterile 20 kinase phosphorylates histone H2B at serine 10 during hydrogen peroxide-induced apoptosis in S. cerevisiae. Cell 2005; 120:25 - 36
- Graves JD, Gotoh Y, Draves KE, Ambrose D, Han DK, Wright M, et al. Caspase-mediated activation and induction of apoptosis by the mammalian Ste20-like kinase Mst1. EMBO J 1998; 17:2224 - 2234
- Zeng Q, Hong W. The emerging role of the hippo pathway in cell contact inhibition, organ size control and cancer development in mammals. Cancer Cell 2008; 13:188 - 192
- Sun Y, Wang T, Su Y, Yin Y, Xu S, Ma C, et al. The behavior of SATB1, a MAR-binding protein, in response to apoptosis stimulation. Cell Biol Int 2006; 30:244 - 247
- Gotzmann J, Meissner M, Gerner C. The fate of the nuclear matrix-associated-region-binding protein SATB1 during apoptosis. Cell Death Differ 2000; 7:425 - 438
- Tan JA, Sun Y, Song J, Chen Y, Krontiris TG, Durrin LK. SUMO conjugation to the matrix attachment region-binding protein, special AT-rich sequence-binding protein-1 (SATB1), targets SATB1 to promyelocytic nuclear bodies where it undergoes caspase cleavage. J Biol Chem 2008; 283:18124 - 18134
- Liu QY, Ribecco-Lutkiewicz M, Carson C, Testolin L, Bergeron D, Kohwi-Shigematsu T, et al. Mapping the initial DNA breaks in apoptotic Jurkat cells using ligation-mediated PCR. Cell Death Differ 2003; 10:278 - 289
- Widlak P, Kalinowska M, Parseghian MH, Lu X, Hansen JC, Garrard WT. The histone H1 C-terminal domain binds to the apoptotic nuclease, DNA fragmentation factor (DFF40/CAD) and stimulates DNA cleavage. Biochemistry 2005; 44:7871 - 7878
- Ninios YP, Sekeri-Pataryas KE, Sourlingas TG. Histone H1 subtype preferences of DFF40 and possible nuclear localization of DFF40/45 in normal and trichostatin A-treated NB4 leukemic cells. Apoptosis 2010; 15:128 - 138
- Izzo A, Kamieniarz K, Schneider R. The histone H1 family: specific members, specific functions?. Biol Chem 2008; 389:333 - 343
- Lee H, Habas R, Abate-Shen C. MSX1 cooperates with histone H1b for inhibition of transcription and myogenesis. Science 2004; 304:1675 - 1678
- Odelberg SJ, Kollhoff A, Keating MT. Dedifferentiation of mammalian myotubes induced by msx1. Cell 2000; 103:1099 - 1109
- Hunter AL, Zhang J, Chen SC, Si X, Wong B, Ekhterae D, et al. Apoptosis repressor with caspase recruitment domain (ARC) inhibits myogenic differentiation. FEBS Lett 2007; 581:879 - 884
- Murray TV, McMahon JM, Howley BA, Stanley A, Ritter T, Mohr A, et al. A non-apoptotic role for caspase-9 in muscle differentiation. J Cell Sci 2008; 121:3786 - 3793
- Koto A, Kuranaga E, Miura M. Temporal regulation of Drosophila IAP1 determines caspase functions in sensory organ development. J Cell Biol 2009; 187:219 - 231
- West JD, Ji C, Marnett LJ. Modulation of DNA fragmentation factor 40 nuclease activity by poly(ADP-ribose) polymerase-1. J Biol Chem 2005; 280:15141 - 15147
- Farzaneh F, Shall S, Johnstone AP. The dynamic nature of DNA-strand breaks present in differentiating muscle cells and quiescent lymphocytes. FEBS Lett 1985; 189:62 - 66
- Ahn JY, Liu X, Cheng D, Peng J, Chan PK, Wade PA, et al. Nucleophosmin/B23, a nuclear PI(3,4,5)P(3) receptor, mediates the antiapoptotic actions of NGF by inhibiting CAD. Mol Cell 2005; 18:435 - 445
- Ahn JY, Liu X, Liu Z, Pereira L, Cheng D, Peng J, et al. Nuclear Akt associates with PKC-phosphorylated Ebp1, preventing DNA fragmentation by inhibition of caspase-activated DNase. EMBO J 2006; 25:2083 - 2095
- Ju BG, Rosenfeld MG. A breaking strategy for topoisomerase IIbeta/PARP-1-dependent regulated transcription. Cell Cycle 2006; 5:2557 - 2560
- Durrieu F, Samejima K, Fortune JM, Kandels-Lewis S, Osheroff N, Earnshaw WC. DNA topoisomerase IIalpha interacts with CAD nuclease and is involved in chromatin condensation during apoptotic execution. Curr Biol 2000; 10:923 - 926
- Popp C, Dean W, Feng S, Cokus SJ, Andrews S, Pellegrini M, et al. Genome-wide erasure of DNA methylation in mouse primordial germ cells is affected by AID deficiency. Nature 2010; 463:1101 - 1105
- Bhutani N, Brady JJ, Damian M, Sacco A, Corbel SY, Blau HM. Reprogramming towards pluripotency requires AID-dependent DNA demethylation. Nature 2010; 463:1042 - 1047
- Petersen-Mahrt SK, Harris RS, Neuberger MS. AID mutates E. coli suggesting a DNA deamination mechanism for antibody diversification. Nature 2002; 418:99 - 103
- Jost JP, Oakeley EJ, Zhu B, Benjamin D, Thiry S, Siegmann M, et al. 5-Methylcytosine DNA glycosylase participates in the genome-wide loss of DNA methylation occurring during mouse myoblast differentiation. Nucleic Acids Res 2001; 29:4452 - 4461