Abstract
MYB is a leucine zipper transcription factor that is essential for hematopoesis and for renewal of colonic crypts. There is also ample evidence showing that MYB is leukemogenic in several animal species. However, it was not until recently that clear evidence was presented showing that MYB actually is an oncogene rearranged in human cancer. In a recent study, a novel mechanism of activation of MYB involving gene fusion was identified in carcinomas of the breast and head and neck. A t(6;9) translocation was shown to generate fusions between MYB and the transcription factor gene NFIB. The fusions consistently result in loss of the 3´-end of MYB, including several highly conserved target sites for microRNAs that negatively regulate MYB expression. Deletion of these target sites may disrupt the repression of MYB, leading to overexpression of MYB-NFIB transcripts and protein and to transcriptional activation of critical MYB target genes associated with apoptosis, cell cycle control, cell growth/angiogenesis, and cell adhesion. This study, together with previous and recent data showing rearrangements and copy number alterations of the MYB locus in T-cell leukemia and certain solid tumors, will be the main focus of this review.
Introduction: Fusion Oncogenes in Solid Tumors
Tumor-type specific chromosome rearrangements often result in gene fusions encoding potent oncoproteins. To date, close to 400 gene fusions have been identified in human cancer and an increasing number of these fusions are being recognized as important diagnostic and prognostic markers and as targets for new cancer therapies.Citation1,Citation2 Recent estimates have indicated that gene fusions account for 20% of human cancer morbidity. Most gene fusions have thus far been identified in leukemias and sarcomas and only a few in carcinomas.Citation1 This paucity is likely due to an inability to discover these rearrangements rather than a true lack of gene fusions in carcinomas. Recent data suggest that the mechanisms by which they are generated may be partly different from that in leukemias and sarcomas. Thus, previous cytogenetic studies have clearly demonstrated that recurrent balanced chromosome translocations are rare in carcinomas whereas they are common in leukemias and sarcomas.Citation2 Therefore, it is not surprising that several new gene fusions in carcinomas have been shown to result from intrachromosomal rearrangements or cryptic translocations/insertions that are not visible at the cytogenetic level.Citation3–Citation7 In line with this reasoning, it was recently shown that more than 50% of prostate cancers harbor androgen regulated gene fusions in which an ETS (E26 transformation specific) transcription factor gene (ERG or ETV1) is fused to the TMPRSS2 promoter region,Citation4 and subgroups of non-small-cell lung cancers have EML4-ALK, SLC34A2-ROS or CD74-ROS gene fusions.Citation5,Citation8 In addition, fusion oncogenes have been found in subsets of thyroid carcinomas, renal cell carcinomas, breast cancer and mucoepidermoid carcinomas of the salivary and bronchial glands.Citation1,Citation9,Citation10 Continued studies of cancer genomes using massively parallel sequencing and paired-end sequencing strategies will most likely lead to the identification of new gene fusions also in other common epithelial cancers.
Fusion genes are potent oncogenes as demonstrated by their ability to induce tumors in various transgenic mouse models. For example, the RET-PTC1 fusion induces tumors that are morphologically similar to human papillary thyroid carcinomas when specifically overexpressed in the thyroid.Citation11,Citation12 Similarly, the FUS-DDIT3 fusion causes myxoid liposarcomas when expressed under control of a ubiquitously expressed promoter,Citation13 the SYT-SSX2 fusion induces synovial sarcoma-like tumors when expressed in immature myoblasts,Citation14 whereas the PAX3-FKHR fusion induces alveolar rhabdomyosarcomas when targeted to Myf6-expressing skeletal muscle cells.Citation15 In the latter mice, the incidence of tumors is significantly increased by concurrent loss of p53 or Ink4a/Arf function.
Recent studies have also shown that the ETV6-NTRK3 fusion, typical of human secretory breast carcinoma, can induce breast tumors in mice through transformation of committed alveolar bipotent or CD61(+) luminal progenitor cells,Citation16 and that the EML4-ALK fusion induces lung adenocarcinomas in mice.Citation17 Similarly, studies of the oncogenic role of the TMPRSS2-ERG fusion have shown that ERG has an important role in mouse prostate cancer progression and cooperates with Pten haploinsufficiency to promote progression of high-grade prostatic intraepithelial neoplasia (HGPIN) to invasive prostatic adenocarcinoma and that transgenic TMPRSS2-ERG mice develop PIN, but only in the context of PI3-kinase pathway activation.Citation18,Citation19 Taken together, these studies clearly demonstrate that fusion oncogenes are pathogenetically critical genes in solid tumors and as such also key targets for therapy.
The majority of fusion oncogenes in solid tumors encode aberrant transcription factors while a minority express chimeric proteins that deregulate growth factor signaling.Citation20 Many fusion oncoproteins exert their effects through deregulation of genes encoding cell cycle proteins or by affecting the stability of these proteins. For example, the Ewing sarcoma associated EWSR1-FLI1 fusion protein has been shown to induce expression of MYC, CCND1 and CCNE1 both at the mRNA and protein levelsCitation21–Citation23 and also to downregulate CDKN1A (p21), CDKN1B (p27) and CDKN1C (p57), leading to a reduction of the corresponding CDKI (cyclin dependent kinase inhibitor) proteins.Citation21,Citation22,Citation24 Recently, Sanchez et al. also demonstrated that EWS-FLI1 upregulates a more oncogenic isoform of cyclin D1, termed D1b, by promoting the expression of an alternative splice form of the CCND1 transcript.Citation25 Cyclin D1 is also induced by several other fusion oncoproteins such as ETV6-NTRK3, SYT-SSX2 and RET-PTC1.Citation26–Citation28 The PAX3-FKHR fusion protein has been shown to increase proteasome-dependent degradation of p27,Citation29 and the FUS-DDIT3 fusion protein was recently shown to directly bind CDK2 through its DDIT3 part.Citation30 However, the biological consequence of this interaction is not yet fully understood.
Gene Fusions Involving Transcription Factors and Transcriptional Coactivators in Salivary Gland Tumors
Tumors of the salivary glands constitute up to 10% of the tumors of the head and neck region.Citation31 There are more than 30 histopathological subtypes, of which pleomorphic adenomas and Warthin tumors are the most common benign tumors and mucoepidermoid carcinomas and adenoid cystic carcinomas (ACC) are the most common malignant tumors.Citation32 Recent studies have shown that these tumor types are characterized by a highly specific pattern of chromosome translocations resulting in fusion oncogenes.Citation3,Citation6,Citation7,Citation9,Citation33,Citation34 The major targets of the translocations are transcription factors involved in growth factor signaling and cell cycle regulation, transcriptional coactivators and tyrosine kinase receptors.
Pleomorphic adenomas are characterized by translocations resulting in gene fusions involving the transcription factor genes PLAG1 and HMGA2. PLAG1 is a developmentally regulated DNA-binding zinc finger protein that belongs to a family of cell cycle progression-related proteins (reviewed in ref. Citation9 and Citation35). Ectopic overexpression of PLAG1 due to promoter swapping with at least five other genes (), cause deregulation of PLAG1 target genes and activation of the IGF-II mitogenic signaling pathway in salivary gland pleomorphic adenomas.Citation36 PLAG1 is also involved in gene fusions with two other genes (HAS2 and COL1A2) in lipoblastomas (reviewed in ref. Citation9) and has been shown to induce acute myeloid leukemia in cooperation with the CBFB-MYH11 fusion protein in mice.Citation37
HMGA2 belongs to a family of non-histone nuclear proteins that orchestrate the assembly of nucleoprotein complexes (reviewed in ref. Citation38). They play important roles in gene transcription, recombination and chromatin structure. Genomic rearrangements of the 3′-part of HMGA2 due to gene fusions () or truncations in pleomorphic adenomas and benign mesenchymal tumors result in activation of the expression of HMGA2 and its cellular targets, including the cell cycle regulators CCNA1 and CCNB2.Citation39,Citation40 In a subset of tumors with HMGA2 fusions the gene fusion is amplified together with other closely linked genes such as MDM2.Citation7 The molecular mechanism by which HMGA2 is activated by chromosomal rearrangements is still unclear. Previous studies have suggested that separation of the DNA-binding domains from the acidic domain and the mRNA-destabilizing AUUUA motifs in the 3′-UTR (untranslated region) is the critical event leading to stabilization and overexpression of HMGA2 transcripts (reviewed in ref. Citation9). However, recent studies have indicated that HMGA2 may also be deregulated by loss of negatively regulating Let-7 microRNA (miRNA) targetsites in the 3′-UTR as a consequence of gene fusion/truncation.Citation41–Citation43
Fusion of the transcriptional coactivators MAML2 and CRTC1 (a.k.a. MECT1, TORC1 or WAMTP1) is a characteristic feature of low-grade mucoepidermoid carcinomas of the salivary, bronchial and thyroid glands ().Citation44–Citation46 MAML2 belongs to a family of Mastermind-like, nuclear proteins that functions as coactivators for Notch receptorsCitation47,Citation48 whereas CRTC1 belongs to a family of highly conserved CREB (cAMP response element-binding protein) coactivators.Citation49,Citation50 The CRTC1-MAML2 fusion encodes a chimeric protein consisting of the CREB-binding domain of CRTC1 fused to the transactivation domain of MAML2.Citation44,Citation45 Functional studies have shown that the N-terminal CREB-binding domain of the fusion is crucial for its transforming activity,Citation51,Citation52 and that CRTC1-MAML2 can activate transcription of cAMP/CREB target genes (Enlund et al. unpublished data and ref. 51 and Citation52). An identical CRTC1-MAML2 fusion has also been found in Warthin's tumors () and in clear cell hidradenomas of the skin,Citation45,Citation53,Citation54 indicating that the fusion is etiologically linked to benign and low-grade malignant, histogenetically related tumor types originating from diverse exocrine glands.
Much to our surprise we recently identified a second gene fusion in cutaneous hidradenomas and MEC. The fusion consists of the amino-terminal domain of EWSR1 and the DNA-binding, C-terminal domain of the transcription factor POU5F1 ().Citation33 POU5F1 is expressed during early development to maintain the pluripotent status of embryonic stem and germ cells. Interestingly, the morphology of the EWSR1-POU5F1 positive tumors were more immature compared to the CRTC1-MAML2 positive tumors, raising the question of whether gene fusion could be a mechanism of POU5F1 reactivation leading to a more undifferentiated, stem cell-like phenotype in these tumors.
Recently, a hitherto unknown type of salivary gland tumor, mammary analogue secretory carcinoma of salivary glands, was described.Citation55 These tumors show strong histomorphologic and immunohistochemical resemblance to secretory carcinoma of the breast. In addition, both tumor types show an identical ETV6-NTRK3 gene fusion (), thus further emphasizing the histogenetic similarities between breast and salivary glands.
Finally, a recent study by Persson and co-workers identified a novel mechanism of activation of the MYB oncogene involving gene fusion in adenoid cystic carcinomas of the breast and head and neck.Citation34 In this fusion, a major part of MYB is linked to the last coding exon(s) of the transcription factor gene NFIB (). The fusion results in overexpression of MYB-NFIB transcripts and protein as well as to transcriptional activation of critical MYB target genes. This study, together with previous and recent data showing rearrangements or copy number alterations of MYB in leukemias and certain solid tumors, will be the main focus of this review.
Identification of a Novel Mechanism of Activation of MYB Involving Gene Fusion
The c-Myb oncogene was first identified almost 30 years ago as the cellular homologue of the transforming v-Myb gene of two avian retroviruses that induce leukemia, avian myeloblastosis virus (AMV) and E26 leukemia virus.Citation56–Citation59 The v-myb oncogenes of AMV and E26 encode N- and C-terminally truncated myb proteins. The murine Myb locus is also a common site of retroviral insertional mutagenesis.Citation60 MYB belongs to a family of transcription factors that include the closely related family members MYBL1 (a.k.a. AMYB) and MYBL2 (a.k.a. BMYB). MYB proteins function as transcriptional regulators and contain three functional key domains, an N-terminal DNA-binding domain comprised of three tandem 50 amino acid myb repeats that specifically bind to the sequence PyAACG/TG, a centrally located transcription activation domain and a C-terminal negative regulatory domain (NRD) involved in transcriptional repression (reviewed in refs. Citation61 and Citation62). The latter contains a leucine zipper-like motif and an EVES-motif that can modulate the activity of MYB. Disruption of the leucine zipper-like motif enhances the transforming activity of Myb and the EVES-motif has been shown to regulate the activity of Myb by inter- and intramolecular interactions.Citation63 Myb may also be sumoylated at two sites within the NRD leading to a reduction in Myb activity.Citation64
The wild-type MYB-protein has a half-life of about 30 minutes and is post-translationally modified by phosphorylation, acetylation, sumoylation and ubiquitylation. These modifications may affect protein levels, DNA-binding and/or the transactivation capacity of MYB. MYB plays an important role in the control of cell proliferation, apoptosis and differentiation of in particular hematopoietic progenitor cells (reviewed in refs. Citation61 and Citation62). Loss of Myb function in mice results in embryonic lethality due to failure of fetal hepatic hematopoiesis and conditional Myb knockout in adult hematopoietic stem cells was recently shown to result in loss of self-renewal due to impaired proliferation and accelerated differentiation.Citation65,Citation66 More recent work has also demonstrated that MYB is important during colon development (reviewed in ref. Citation62). MYB is highly expressed in immature, proliferating epithelial, endothelial and hematopoietic cells and is downregulated as cells become more differentiated. High expression levels of MYB have also been found in pancreatic, colon and breast tumors as well as in most leukemias and lymphomas (reviewed in ref. Citation62). Overexpression of MYB in colorectal cancer has been attributed to frequent mutations in attenuation sequences in the first intron that are known to regulate the transcription of MYB. In breast cancer, ERα (estrogen receptor-α) relieves the attenuation leading to increased expression of MYB, a characteristic feature of most ERα+ breast cancers.
However, it was not until recently that clear evidence was presented showing that MYB actually is an oncogene rearranged in human cancer.Citation34,Citation60,Citation67 The most compelling evidence in support of this observation derives from studies of a recurrent t(6;9)(q22–23;p23–24) translocation in ACC.Citation34 This translocation was originally described in 1986 and was subsequently shown to be a primary and tumor-type specific aberration in ACC.Citation68,Citation69 The t(6;9) translocation is found in approximately one third of karyotypically abnormal ACCs. Positional cloning of the 6q22–23 and 9p23–24 breakpoint regions revealed a fusion between the two transcription factor genes MYB and NFIB.Citation34 Due to alternative splicing and variable breakpoints in MYB and NFIB, at least 11 fusion transcript variants have been identified. Fusions consisting of MYB exon 14 linked to NFIB exons 8c and/or 9 predominate (), suggesting that most breakpoints occur in MYB intron 14 and in NFIB intron 8. The majority of transcripts contained one or more of the alternatively spliced NFIB exons 8a, 8a alternative, 8b, and/or 8c and MYB exon 9a. Preliminary studies of these transcript variants, although admittedly still limited, do not indicate that they have prognostic significance (unpublished data). However, since the fusion was found in a high frequency of ACCs, irrespective of whether they were derived from the breast, salivary glands, lacrimal glands or ceruminal glands of the ear, but not in any of 25 non-ACC tumor samples, we believe that the MYB-NFIB fusion is an important new diagnostic biomarker for this tumor entity. These findings unequivocally identify gene fusion as a novel mechanism for activation of MYB in human cancer and add to the evidence that MYB is an important human oncogene.
Molecular Consequences of the MYB-NFIB Gene Fusion
The expression of wild-type MYB is low in normal adult salivary gland and breast tissues.Citation34 In contrast, the MYB-NFIB fusion is highly expressed in translocation-positive tumors; the expression of the 5′-part of MYB is significantly higher than that of the 3′-part, consistent with disruption of the 3′-part of the gene as a consequence of fusion to NFIB. This observation indicates that the increased MYB expression is not due to expression of the non-rearranged wild-type MYB allele. The MYB-NFIB protein is also highly expressed in fusion-positive ACCs (). The immunoreactivity was, however, somewhat variable between tumors expressing similar levels of fusion transcript. The reason for this is unclear but could for example be due to heterogeneous expression of the fusion protein in different tumor cell populations or to fixation artifacts of the tissue specimens. Transient transfection of CHEF/18,Citation70 and NIH-3T3 cells with a GFP-tagged MYB-NFIB construct confirmed the nuclear localization of the fusion oncoprotein. A diffuse nuclear staining was observed in both cell types ().
The predicted MYB-NFIB fusion protein retains the DNA-binding and transactivation domains of wild-type MYB and is therefore expected to activate MYB target genes. Indeed, 14 of 16 confirmed MYB targets were overexpressed in fusion-positive tumors relative to normal salivary gland tissue ().Citation34 These included genes associated with cell cycle control (CCNB1, CDC2 and MAD1L1), apoptosis (API5, BCL2, BIRC3, HSPA8 and SET), cell growth and angiogenesis (MYC, KIT, VEGFA, FGF2, CD53) and cell adhesion (CD34). Several of these genes have previously also been reported to be overexpressed in ACC (reviewed in ref. Citation34), thus further emphasizing their potential role in the pathogenesis of ACC. Continued studies of these and other transcriptional targets of the MYB-NFIB fusion will provide important new insights into their potential role as new diagnostic biomarkers and therapeutic targets for ACC.
The molecular mechanism by which MYB is activated by the t(6;9) translocation is still obscure. Since the minimal common part of MYB lost due to gene fusion with NFIB is exon 15, including the 3′-UTR, it has been proposed that MYB may be deregulated as a result of loss of binding sites for negatively regulating miRNAs.Citation34
Is the MYB-fusion Another Example of Disruption of miRNA-Directed Repression of an Oncogene Activated by Chromosomal Translocation?
As previously indicated, MYB expression is tightly regulated by attenuation sequences located in the first intron of the gene and by a leucine-rich NRD in the C-terminus (see above). Mutation of the attenuation sequences or truncation/disruption of the NRD correlate with elevated expression levels of MYB in both human and experimentally induced neoplasms. However, recent studies have also indicated that MYB is negatively regulated by several miRNAs. The perhaps most compelling evidence of the latter mechanism derives from studies of miR-150, a miRNA selectively expressed in mature, resting B and T cells, but not in their progenitors. miR-150 directly regulates MYB expression in vivo through two conserved target sites in the 3′-UTR of MYB mRNA, and recent studies have shown that Myb is highly expressed during B-cell activation in Mir150-/- knockout mice and conversely that Myb expression is significantly reduced in mice overexpressing miR-150.Citation71 These findings have subsequently been confirmed by others showing that miR-150 indeed is an important regulator of MYB expression during differentiation.Citation72–Citation75 In addition to miR-150, there are at least three other miRNAs, miR-15a, miR-16 and miR-34a, with target sites in the 3′-UTR of MYB that have been shown to negatively regulate MYB expression in vitro.Citation76–Citation78 Thus, several lines of evidence point to an intricate miRNA-mediated regulation of MYB expression during development and differentiation.
Since the minimal common part of MYB lost due to gene fusion with NFIB is exon 15 (encoding the last 38 amino acids of the MYB protein), including the 3′-UTR, it has been proposed that MYB may be deregulated as a result of loss of binding sites for negatively regulating miRNAs in the 3′-UTR.Citation34 This hypothesis is supported by the observation that the most common fusion types do not disrupt the C-terminal NRD of MYB encoded by exons 10–13.Citation61,Citation79–Citation81 Forced overexpression of miR-150, miR-15a and miR-16 in short-term cultured fusion-positive ACC cells as well as in a T-cell acute lymphoblastic leukemia (T-ALL) cell line with MYB activation due to genomic duplicationCitation67 resulted in a 30% downregulation of MYB mRNA in T-ALL cells after 45 hrs, whereas transfection of primary ACC cells did not significantly alter the expression of MYB. The reduction in MYB mRNA in the control T-ALL cells is comparable to previously published data.Citation73 The miRNAs miR-150 and miR-15a/16 are also expressed in fusion-positive ACCs as well as in normal salivary gland and breast tissues.Citation34 Taken together, these findings support the hypothesis that deletion of miRNA target sites in the 3′-UTR of MYB through gene fusion may disrupt the repression of MYB by miRNAs. Whether this is the major mechanism for activation of MYB in ACC or if also sequences in the 3′-UTR of NFIB (exon 9 only encodes the last five amino acids of the NFIB protein) and/or infrequent disruption of the MYB NRD may contribute to the deregulation of MYB remains to be elucidated.
In this respect MYB is reminiscent of HMGA2, another oncogene frequently activated by chromosomal translocations. Recent studies have shown that deregulation of HMGA2 by loss of negatively regulating Let-7 miRNA target sites in the 3′-UTR due to gene fusion, is an important pathogenetic mechanism in several tumor types, including pleomorphic salivary gland adenomas.Citation9,Citation41–Citation43 Interestingly, in a subset of the latter cases the activation is due to fusions of the last five amino acids of the NFIB protein to the DNA-binding domains of HMGA2,Citation9,Citation82 that is the same part of NFIB that is fused to MYB in ACC. Taken together available data, although admittedly still limited, suggest that MYB-NFIB may be another example of disruption of miRNA-directed repression of an oncogene activated by chromosomal translocations. However, additional studies are necessary to clarify the molecular mechanism underlying the activation of MYB by the t(6;9) and the potential contribution of NFIB.
Copy Number Alterations and Genomic Rearrangements of MYB in T-ALL and Epithelial Cancers
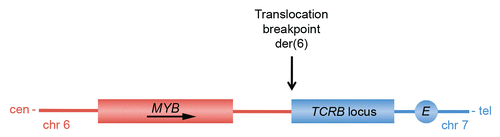
As predicted from the early studies demonstrating that Myb is leukemogenic in several animal species, it has now become clear that MYB is also rearranged in human leukemias. High-resolution comparative genomic hybridization array studies have revealed that a subset of T-ALL show selective amplification and overexpression of MYB (MYBdup).Citation60,Citation67 Interestingly, knockdown of MYB expression in T-ALL cell lines induces T-cell differentiation but has only limited effects on viability and cell proliferation.Citation67 However, γ-secretase inhibitors were shown to synergize with MYB knockdown to inhibit the growth of MYBdup T-ALL cells in cases with concomitant NOTCH1 mutations, suggesting that MYB indeed may be an interesting target for therapy in T-ALL. Studies on the mechanism by which the MYB duplications occur in T-ALL indicate that they are mediated somatically by homologous recombination between Alu-elements flanking the MYB locus.Citation83 In all cases analyzed, MYB was tandemly duplicated on one chromosome 6. The Myb locus is also a frequent insertion site in retrovirally-induced leukemias in mice; there are multiple known integration sites located both up- and downstream of Myb (reviewed in ref. Citation60). Recent studies have also identified a novel subgroup of T-ALL with t(6;7)(q23;q34) translocations that target MYB.Citation60 In these cases MYB and TCRB (T-cell receptor beta) are juxta-posed on the derivative chromosome 6, leading to activation of MYB expression (). The translocation breakpoints in 6q23 maps to two clusters located up to more than 50 kilobases telomeric of MYB. The TCRB-MYB translocation, which does not result in a true fusion gene, defines a new subgroup of T-ALL associated with very young age and a proliferation/mitosis signature.
There are also examples of solid tumors in which MYB has been identified as a target for gene amplification. Thus, about one third of BRCA1 mutated hereditary breast cancers show amplification of MYB and a corresponding overexpression of MYB mRNA and protein.Citation84 In contrast, MYB amplification is only found in 2% of sporadic breast cancers. Furthermore, selective MYB amplification has been found in approximately 10% of pancreatic cancersCitation85 as well as in two colorectal cancer cell linesCitation86 and two glioblastoma cell lines.Citation87 The above-mentioned studies, together with our recent findings of MYB-NFIB gene fusions in ACC, provide ample evidence that MYB indeed is an important human oncogene that is rearranged in a variety of human neoplasias originating from multiple cell lineages. Large-scale sequencing is currently changing our perceptions of the cancer genome, and by using high-resolution copy number arrays in combination with paired-end transcriptome sequencing strategies,Citation88,Citation89 we expect that additional MYB gene rearrangements, including gene fusions, will be found also in other tumor types.
Perspectives: Towards Development of New Diagnostic and Therapeutic Strategies Based on the MYB-NFIB Fusion Oncogene
Previous studies of ACC has been seriously hampered by a number of factors, including a limited knowledge of the cellular and molecular biology of ACC as well as of the pathogenetic mechanisms leading to tumorigenesis, a lack of relevant in vitro and animal models to study disease pathogenesis and treatment, and a paucity of critical targets for development of new therapy. In particular, the fact that six commonly used ACC cell lines recently were shown to be cross-contaminated or misidentified has prompted a re-evaluation of published data regarding signaling pathways and new targets for therapy in ACC.Citation90 The recent discovery of the MYB-NFIB gene fusion provides important clues to the molecular pathogenesis of this highly malignant tumor and opens up new avenues for improved diagnosis and identification of novel targets for pharmaceutical intervention.
The finding of the MYB-NFIB fusion in a high frequency of ACCs suggests that it may be used as a new diagnostic biomarker. We have recently developed a robust RT-PCR screening method for the fusion in formalin-fixed paraffin embedded tumor material (unpublished data) which should be particularly useful in the differential diagnosis of ACC. FISH using MYB-NFIB specific probes and immunohistochemistry using antibodies to the MYB or MYB-NFIB proteins will also be powerful adjunctive diagnostic tools. Efforts are also underway to develop noninvasive methods for detection of MYB-NFIB fusion transcripts in saliva from patients with primary salivary gland ACC. Similar studies of ERG gene fusions in urine samples from patients with prostate cancer has recently proved to be useful for noninvasive detection of prostate cancer.Citation91,Citation92
ACC is a slow-growing tumor with an often protracted clinical course. The poor long-term prognosis for these patients is mainly due to local recurrences and late onset of distant metastases.Citation93 To date, there is no effective systemic therapy available that inhibits the progression of the disease and improves the survival of ACC patients. Therefore, new therapeutic approaches are needed. As previously indicated, the MYB-NFIB fusion is a promising target for development of new therapeutic strategies for patients with this disease.Citation34 However, targeting transcription factors has proven to be notoriously difficult and accordingly there are few options available to directly target MYB. One such possibility was recently described by Williams and co-workersCitation94 who used a DNA vaccine encoding a fusion protein in which MYB was flanked by two tetanus toxin peptides. This vaccine was shown to significantly suppress tumor growth of an aggressive colon cancer syngenic transplant. It would be interesting to see whether such a vaccine also has similar effects against fusion-positive ACC xenografts in mice. In addition, clinical trials using antisense MYB oligodeoxinucleotides (ODN) have been conducted in patients with chronic myelogenous leukemia and other refractory leukemias.Citation95
An alternative therapeutic approach for fusion-positive ACCs would be to target the genes that are regulated by the MYB-NFIB fusion. Studies are now in progress to identify new transcriptional targets and to verify those that were recently suggested (). Among the latter are several that have recently been subjected to pharmaceutical intervention. For example, VEGFA may be inhibited by bevacizumab, a humanized VEGF-antibodyCitation96 as well as by several VEGFR inhibitors,Citation97 and BCL2 is inhibited by the small molecules ABT-737 and the related orally active ABT-263.Citation98 Moreover, the tyrosine kinase inhibitor (TKI) PD173074 inhibits FGF2 mediated proliferation through interaction with FGFR-1 and FGFR-2,Citation99,Citation100 and the pan histone deacetylase inhibitor LBH589 (panobinostat) has been shown to downregulate both MYC and MAD1L1Citation101 whereas BIRC3 (CIAP2) and BCL2 may be downregulated by for example fucoxantin and fucoxantinol.Citation102 KIT, which is a tyrosine kinase receptor for stem cell factor, is effectively inhibited by the TKI imatinib mesylate. However, despite the fact that KIT is highly overexpressed both at the RNA and protein levels in most cases of ACC, clinical studies have shown that imatinib has no major effects on advanced ACC of the head and neck.Citation103,Citation104 With the exception of imatinib, the abovementioned drugs and drugs with similar target specificities are interesting examples of potential MYB target gene inhibitors that deserve to be tested as single agents or in combination in in vitro and/or xenograft models to evaluate their efficacy in the treatment of ACC. Such efforts will hopefully lead to the emergence of potent therapies that will improve the survival of patients affected by this disease.
Abbreviations
| ETS | = | E26 transformation specific |
| HGPIN | = | high-grade prostatic intraepithelial neoplasia |
| CDKI | = | cyclin dependent kinase inhibitor |
| ACC | = | adenoid cystic carcinoma |
| UTR | = | untranslated region |
| miRNA | = | microRNA |
| CREB | = | cAMP response element-binding protein |
| AMV | = | avian myeloblastosis virus |
| NRD | = | negative regulatory domain |
| DBD | = | DNA binding domain |
| TAD | = | transactivation domain |
| ERα | = | estrogen receptor-α |
| T-ALL | = | T-cell acute lymphoblastic leukemia |
| TCRB | = | T-cell receptor beta |
| ODN | = | oligodeoxinucleotides |
| TKI | = | tyrosine kinase inhibitor |
| HDAC | = | histone deacetylase |
| DAPI | = | 4′,6′-diamidino-2′-phenylindole dihydrochloride |
| cen | = | centromere |
| tel | = | telomere |
Figures and Tables
Figure 1 Gene fusions in salivary gland tumors. (A) PLAG1- and HMGA2-fusions in pleomorphic adenomas. (B) MAML2- and POU5F1-fusions in mucoepidermoid carcinomas and Warthin's tumors (metaplastic variant). (C) ETV6-NTRK3 fusions in mammary analogue secretory carcinoma of salivary glands and (D) the recently identified novel MYB-NFIB fusion in adenoid cystic carcinoma.
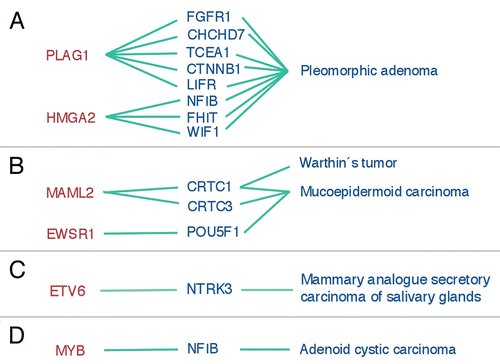
Figure 2 Schematic illustration of the MYB and NFIB genes and of two MYB-NFIB fusion variants (coding exons are shown i darker red and blue colors) and the resulting fusion proteins. Translocation breakpoints are shown by vertical arrows and miRNA binding sites for miR-15a/16 and miR-150 in the 3′-UTR of MYB are indicated by asteriks. The alternatively spliced MYB exon 9a and NFIB exons 8a, 8b and 8c are indicated. DBD, DNA binding domain; TAD, transactivation domain; NRD, negative regulatory domain.
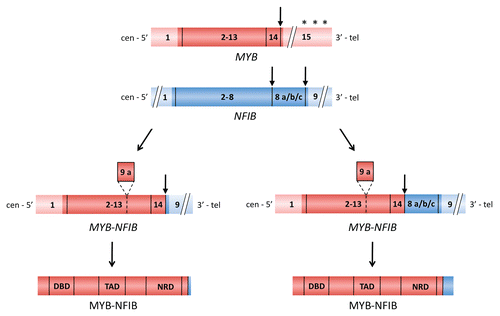
Figure 3 MYB-NFIB is a nuclear protein. (A) Immunostaining of the MYB-NFIB fusion protein in a translocation-positive ACC. Note the predominant nuclear staining of tumor cells whereas stromal cells are negative. (B) Chinese hamster embryo fibroblasts (CHEF/18 cells) and NIH-3T3 cells (inset) transfected with a MYB-NFIB-GFP expression construct. Nuclei are counterstained in blue with 4′,6′-diamidino-2′-phenylindole dihydrochloride (DAPI). Fluorescent cells were fixed on glass slides and imaged using a Zeiss LSM510 META confocal microscope system.
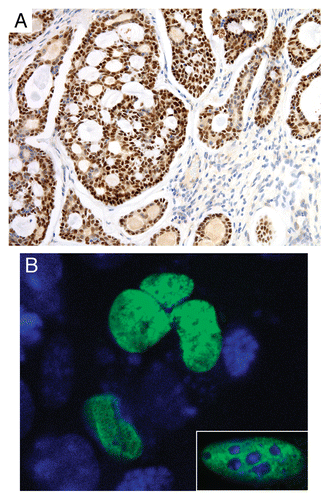
Figure 4 Confirmed MYB downstream target genes that are overexpressed in fusion-positive ACCs relative to normal salivary gland tissue.
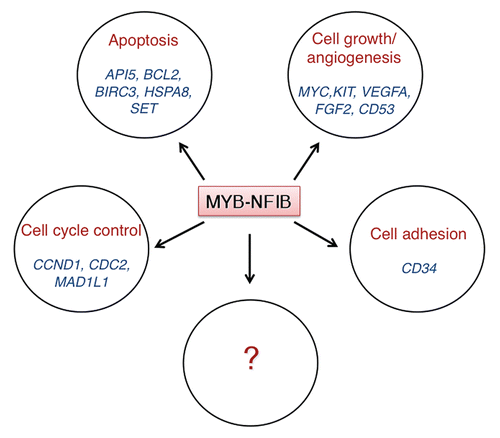
Figure 5 Schematic illustration of the translocation breakpoint region on the der(6) marker chromosome generated by the t(6;7)(q23;q34) translocation in T-ALL. Note that this translocation, unlike the t(6;9) in ACC, does not result in a MYB gene fusion. cen, centromere; tel, telomere. Adapted from reference Citation60.
Acknowledgements
This work was supported by the Swedish Cancer Society and the IngaBritt and Arne Lundberg Research Foundation. We thank Ulric Pedersen for help preparing the illustrations. The authors have declared that no competing interests exist.
References
- Mitelman F, Johansson B, Mertens F. The impact of translocations and gene fusions on cancer causation. Nat Rev Cancer 2007; 7:233 - 245
- Mitelman F, Johansson B, Mertens F. Mitelman Database of Chromosome Aberrations and Gene Fusions in Cancer: http://cgap.nci.nih.gov/Chromosomes/Mitelman 2010;
- Asp J, Persson F, Kost-Alimova M, Stenman G. CHCHD7-PLAG1 and TCEA1-PLAG1 gene fusions resulting from cryptic, intrachromosomal 8q rearrangements in pleomorphic salivary gland adenomas. Genes Chromosomes Cancer 2006; 45:820 - 828
- Tomlins SA, Laxman B, Dhanasekaran SM, Helgeson BE, Cao X, Morris DS, et al. Distinct classes of chromosomal rearrangements create oncogenic ETS gene fusions in prostate cancer. Nature 2007; 448:595 - 599
- Soda M, Choi YL, Enomoto M, Takada S, Yamashita Y, Ishikawa S, et al. Identification of the transforming EML4-ALK fusion gene in non-small-cell lung cancer. Nature 2007; 448:561 - 566
- Persson F, Winnes M, Andren Y, Wedell B, Dahlenfors R, Asp J, et al. High-resolution array CGH analysis of salivary gland tumors reveals fusion and amplification of the FGFR1 and PLAG1 genes in ring chromosomes. Oncogene 2008; 27:3072 - 3080
- Persson F, Andren Y, Winnes M, Wedell B, Nordkvist A, Gudnadottir G, et al. High-resolution genomic profiling of adenomas and carcinomas of the salivary glands reveals amplification, rearrangement and fusion of HMGA2. Genes Chromosomes Cancer 2009; 48:69 - 82
- Rikova K, Guo A, Zeng Q, Possemato A, Yu J, Haack H, et al. Global survey of phosphotyrosine signaling identifies oncogenic kinases in lung cancer. Cell 2007; 131:1190 - 1203
- Stenman G. Fusion oncogenes and tumor type specificity—insights from salivary gland tumors. Semin Cancer Biol 2005; 15:224 - 235
- Edwards PA. Fusion genes and chromosome translocations in the common epithelial cancers. J Pathol 2010; 220:244 - 254
- Jhiang SM, Sagartz JE, Tong Q, Parker-Thornburg J, Capen CC, Cho JY, et al. Targeted expression of the ret/PTC1 oncogene induces papillary thyroid carcinomas. Endocrinology 1996; 137:375 - 378
- Santoro M, Chiappetta G, Cerrato A, Salvatore D, Zhang L, Manzo G, et al. Development of thyroid papillary carcinomas secondary to tissue-specific expression of the RET/PTC1 oncogene in transgenic mice. Oncogene 1996; 12:1821 - 1826
- Perez-Losada J, Pintado B, Gutierrez-Adan A, Flores T, Banares-Gonzalez B, del Campo JC, et al. The chimeric FUS/TLS-CHOP fusion protein specifically induces liposarcomas in transgenic mice. Oncogene 2000; 19:2413 - 2422
- Haldar M, Hancock JD, Coffin CM, Lessnick SL, Capecchi MR. A conditional mouse model of synovial sarcoma: insights into a myogenic origin. Cancer Cell 2007; 11:375 - 388
- Keller C, Arenkiel BR, Coffin CM, El-Bardeesy N, DePinho RA, Capecchi MR. Alveolar rhabdomyosarcomas in conditional Pax3:Fkhr mice: Cooperativity of Ink4a/ARF and Trp53 loss of function. Genes Dev 2004; 18:2614 - 2626
- Li Z, Tognon CE, Godinho FJ, Yasaitis L, Hock H, Herschkowitz JI, et al. ETV6-NTRK3 fusion oncogene initiates breast cancer from committed mammary progenitors via activation of AP1 complex. Cancer Cell 2007; 12:542 - 558
- Soda M, Takada S, Takeuchi K, Choi YL, Enomoto M, Ueno T, et al. A mouse model for EML4-ALK-positive lung cancer. Proc Natl Acad Sci USA 2008; 105:19893 - 19897
- Carver BS, Tran J, Gopalan A, Chen Z, Shaikh S, Carracedo A, et al. Aberrant ERG expression cooperates with loss of PTEN to promote cancer progression in the prostate. Nat Genet 2009; 41:619 - 624
- King JC, Xu J, Wongvipat J, Hieronymus H, Carver BS, Leung DH, et al. Cooperativity of TMPRSS2-ERG with PI3-kinase pathway activation in prostate oncogenesis. Nat Genet 2009; 41:524 - 526
- Helman LJ, Meltzer P. Mechanisms of sarcoma development. Nat Rev Cancer 2003; 3:685 - 694
- Dauphinot L, De Oliveira C, Melot T, Sevenet N, Thomas V, Weissman BE, et al. Analysis of the expression of cell cycle regulators in Ewing cell lines: EWS-FLI-1 modulates p57KIP2 and c-Myc expression. Oncogene 2001; 20:3258 - 3265
- Matsumoto Y, Tanaka K, Nakatani F, Matsunobu T, Matsuda S, Iwamoto Y. Downregulation and forced expression of EWS-Fli1 fusion gene results in changes in the expression of G(1)regulatory genes. Br J Cancer 2001; 84:768 - 775
- Li X, Tanaka K, Nakatani F, Matsunobu T, Sakimura R, Hanada M, et al. Transactivation of cyclin E gene by EWS-Fli1 and antitumor effects of cyclin dependent kinase inhibitor on Ewing's family tumor cells. Int J Cancer 2005; 116:385 - 394
- Nakatani F, Tanaka K, Sakimura R, Matsumoto Y, Matsunobu T, Li X, et al. Identification of p21WAF1/CIP1 as a direct target of EWS-Fli1 oncogenic fusion protein. J Biol Chem 2003; 278:15105 - 15115
- Sanchez G, Bittencourt D, Laud K, Barbier J, Delattre O, Auboeuf D, et al. Alteration of cyclin D1 transcript elongation by a mutated transcription factor upregulates the oncogenic D1b splice isoform in cancer. Proc Natl Acad Sci USA 2008; 105:6004 - 6009
- Tognon C, Garnett M, Kenward E, Kay R, Morrison K, Sorensen PH. The chimeric protein tyrosine kinase ETV6-NTRK3 requires both Ras-Erk1/2 and PI3-kinase-Akt signaling for fibroblast transformation. Cancer Res 2001; 61:8909 - 8916
- Xie Y, Skytting B, Nilsson G, Gasbarri A, Haslam K, Bartolazzi A, et al. SYT-SSX is critical for cyclin D1 expression in synovial sarcoma cells: A gain of function of the t(X;18)(p11.2;q11.2) translocation. Cancer Res 2002; 62:3861 - 3867
- Cassinelli G, Favini E, Degl'Innocenti D, Salvi A, De Petro G, Pierotti MA, et al. RET/PTC1-driven neoplastic transformation and proinvasive phenotype of human thyrocytes involve Met induction and beta-catenin nuclear translocation. Neoplasia 2009; 11:10 - 21
- Zhang L, Wang C. PAX3-FKHR transformation increases 26 S proteasome-dependent degradation of p27Kip1, a potential role for elevated Skp2 expression. J Biol Chem 2003; 278:27 - 36
- Bento C, Andersson MK, Aman P. DDIT3/CHOP and the sarcoma fusion oncoprotein FUS-DDIT3/TLS-CHOP bind cyclin-dependent kinase 2. BMC Cell Biol 2009; 10:89
- Shah JP, Ihde JK. Salivary gland tumors. Curr Probl Surg 1990; 27:775 - 883
- Barnes L, Eveson JW, Reichart P, Sidransky D. World health organization classification of tumours. Pathology and genetics of head and neck tumours 2005; Lyon IARC Press 209 - 274
- Moller E, Stenman G, Mandahl N, Hamberg H, Molne L, van den Oord JJ, et al. POU5F1, encoding a key regulator of stem cell pluripotency, is fused to EWSR1 in hidradenoma of the skin and mucoepidermoid carcinoma of the salivary glands. J Pathol 2008; 215:78 - 86
- Persson M, Andren Y, Mark J, Horlings HM, Persson F, Stenman G. Recurrent fusion of MYB and NFIB transcription factor genes in carcinomas of the breast and head and neck. Proc Natl Acad Sci USA 2009; 106:18740 - 18744
- Kas K, Voz ML, Roijer E, Astrom AK, Meyen E, Stenman G, et al. Promoter swapping between the genes for a novel zinc finger protein and beta-catenin in pleiomorphic adenomas with t(3;8)(p21;q12) translocations. Nat Genet 1997; 15:170 - 174
- Voz ML, Mathys J, Hensen K, Pendeville H, Van Valckenborgh I, Van Huffel C, et al. Microarray screening for target genes of the proto-oncogene PLAG1. Oncogene 2004; 23:179 - 191
- Landrette SF, Kuo YH, Hensen K, Barjesteh van Waalwijk van Doorn-Khosrovani S, Perrat PN, Van de Ven WJ, et al. Plag1 and Plagl2 are oncogenes that induce acute myeloid leukemia in cooperation with Cbfb-MYH11. Blood 2005; 105:2900 - 2907
- Ashar HR, Chouinard RA Jr, Dokur M, Chada K. In vivo modulation of HMGA2 expression. Biochim Biophys Acta 2010; 1799:55 - 61
- Tessari MA, Gostissa M, Altamura S, Sgarra R, Rustighi A, Salvagno C, et al. Transcriptional activation of the cyclin A gene by the architectural transcription factor HMGA2. Mol Cell Biol 2003; 23:9104 - 9116
- De Martino I, Visone R, Wierinckx A, Palmieri D, Ferraro A, Cappabianca P, et al. HMGA proteins upregulate CCNB2 gene in mouse and human pituitary adenomas. Cancer Res 2009; 69:1844 - 1850
- Lee YS, Dutta A. The tumor suppressor micro-RNA let-7 represses the HMGA2 oncogene. Genes Dev 2007; 21:1025 - 1030
- Mayr C, Hemann MT, Bartel DP. Disrupting the pairing between let-7 and Hmga2 enhances oncogenic transformation. Science 2007; 315:1576 - 1579
- Klemke M, Meyer A, Hashemi Nezhad M, Belge G, Bartnitzke S, Bullerdiek J. Loss of let-7 binding sites resulting from truncations of the 3′ untranslated region of HMGA2 mRNA in uterine leiomyomas. Cancer Genet Cytogenet 2010; 196:119 - 123
- Tonon G, Modi S, Wu L, Kubo A, Coxon AB, Komiya T, et al. t(11;19)(q21;p13) translocation in mucoepidermoid carcinoma creates a novel fusion product that disrupts a Notch signaling pathway. Nat Genet 2003; 33:208 - 213
- Enlund F, Behboudi A, Andren Y, Oberg C, Lendahl U, Mark J, et al. Altered Notch signaling resulting from expression of a WAMTP1-MAML2 gene fusion in mucoepidermoid carcinomas and benign Warthin's tumors. Exp Cell Res 2004; 292:21 - 28
- Behboudi A, Enlund F, Winnes M, Andren Y, Nordkvist A, Leivo I, et al. Molecular classification of mucoepidermoid carcinomas-prognostic significance of the MECT1-MAML2 fusion oncogene. Genes Chromosomes Cancer 2006; 45:470 - 481
- Lin SE, Oyama T, Nagase T, Harigaya K, Kitagawa M. Identification of new human mastermind proteins defines a family that consists of positive regulators for notch signaling. J Biol Chem 2002; 277:50612 - 50620
- Wu L, Sun T, Kobayashi K, Gao P, Griffin JD. Identification of a family of mastermind-like transcriptional coactivators for mammalian notch receptors. Mol Cell Biol 2002; 22:7688 - 7700
- Conkright MD, Canettieri G, Screaton R, Guzman E, Miraglia L, Hogenesch JB, et al. TORCs: Transducers of regulated CREB activity. Mol Cell 2003; 12:413 - 423
- Iourgenko V, Zhang W, Mickanin C, Daly I, Jiang C, Hexham JM, et al. Identification of a family of cAMP response element-binding protein coactivators by genome-scale functional analysis in mammalian cells. Proc Natl Acad Sci USA 2003; 100:12147 - 12152
- Coxon A, Rozenblum E, Park YS, Joshi N, Tsurutani J, Dennis PA, et al. Mect1-Maml2 fusion oncogene linked to the aberrant activation of cyclic AMP/CREB regulated genes. Cancer Res 2005; 65:7137 - 7144
- Wu L, Liu J, Gao P, Nakamura M, Cao Y, Shen H, et al. Transforming activity of MECT1-MAML2 fusion oncoprotein is mediated by constitutive CREB activation. EMBO J 2005; 24:2391 - 2402
- Fehr A, Roser K, Belge G, Loning T, Bullerdiek J. A closer look at Warthin tumors and the t(11;19). Cancer Genet Cytogenet 2008; 180:135 - 139
- Winnes M, Molne L, Suurkula M, Andren Y, Persson F, Enlund F, et al. Frequent fusion of the CRTC1 and MAML2 genes in clear cell variants of cutaneous hidradenomas. Genes Chromosomes Cancer 2007; 46:559 - 563
- Skalova A, Vanecek T, Sima R, Laco J, Weinreb I, Perez-Ordonez B, et al. Mammary analogue secretory carcinoma of salivary glands, containing the ETV6-NTRK3 fusion gene: a hitherto undescribed salivary gland tumor entity. Am J Surg Pathol 2010; 34:599 - 608
- Duesberg PH, Bister K, Moscovici C. Genetic structure of avian myeloblastosis virus, released from transformed myeloblasts as a defective virus particle. Proc Natl Acad Sci USA 1980; 77:5120 - 5124
- Souza LM, Strommer JN, Hillyard RL, Komaromy MC, Baluda MA. Cellular sequences are present in the presumptive avian myeloblastosis virus genome. Proc Natl Acad Sci USA 1980; 77:5177 - 5181
- Baluda MA, Reddy EP. Anatomy of an integrated avian myeloblastosis provirus: structure and function. Oncogene 1994; 9:2761 - 2774
- Lipsick JS, Wang DM. Transformation by v-Myb. Oncogene 1999; 18:3047 - 3055
- Clappier E, Cuccuini W, Kalota A, Crinquette A, Cayuela JM, Dik WA, et al. The C-MYB locus is involved in chromosomal translocation and genomic duplications in human T-cell acute leukemia (T-ALL), the translocation defining a new T-ALL subtype in very young children. Blood 2007; 110:1251 - 1261
- Oh IH, Reddy EP. The myb gene family in cell growth, differentiation and apoptosis. Oncogene 1999; 18:3017 - 3033
- Ramsay RG, Gonda TJ. MYB function in normal and cancer cells. Nat Rev Cancer 2008; 8:523 - 534
- Dash AB, Orrico FC, Ness SA. The EVES motif mediates both intermolecular and intramolecular regulation of c-Myb. Genes Dev 1996; 10:1858 - 1869
- Molvaersmyr AK, Saether T, Gilfillan S, Lorenzo PI, Kvaloy H, Matre V, et al. A SUMO-regulated activation function controls synergy of c-Myb through a repressor-activator switch leading to differential p300 recruitment. Nucleic Acids Res 2010; 38:4970 - 4984
- Mucenski ML, McLain K, Kier AB, Swerdlow SH, Schreiner CM, Miller TA, et al. A functional c-myb gene is required for normal murine fetal hepatic hematopoiesis. Cell 1991; 65:677 - 689
- Lieu YK, Reddy EP. Conditional c-myb knockout in adult hematopoietic stem cells leads to loss of self-renewal due to impaired proliferation and accelerated differentiation. Proc Natl Acad Sci USA 2009; 106:21689 - 21694
- Lahortiga I, De Keersmaecker K, Van Vlierberghe P, Graux C, Cauwelier B, Lambert F, et al. Duplication of the MYB oncogene in T cell acute lymphoblastic leukemia. Nat Genet 2007; 39:593 - 595
- Stenman G, Sandros J, Dahlenfors R, Juberg-Ode M, Mark J. 6q- and loss of the Y chromosome—two common deviations in malignant human salivary gland tumors. Cancer Genet Cytogenet 1986; 22:283 - 293
- Nordkvist A, Mark J, Gustafsson H, Bang G, Stenman G. Non-random chromosome rearrangements in adenoid cystic carcinoma of the salivary glands. Genes Chromosomes Cancer 1994; 10:115 - 121
- Stenman G, Delorme EO, Lau CC, Sager R. Transfection with plasmid pSV2gptEJ induces chromosome rearrangements in CHEF cells. Proc Natl Acad Sci USA 1987; 84:184 - 188
- Xiao C, Calado DP, Galler G, Thai TH, Patterson HC, Wang J, et al. MiR-150 controls B cell differentiation by targeting the transcription factor c-Myb. Cell 2007; 131:146 - 159
- Barroga CF, Pham H, Kaushansky K. Thrombopoietin regulates c-Myb expression by modulating micro RNA 150 expression. Exp Hematol 2008; 36:1585 - 1592
- Lin YC, Kuo MW, Yu J, Kuo HH, Lin RJ, Lo WL, et al. c-Myb is an evolutionary conserved miR-150 target and miR-150/c-Myb interaction is important for embryonic development. Mol Biol Evol 2008; 25:2189 - 2198
- Tan LP, Wang M, Robertus JL, Schakel RN, Gibcus JH, Diepstra A, et al. miRNA profiling of B-cell subsets: specific miRNA profile for germinal center B cells with variation between centroblasts and centrocytes. Lab Invest 2009; 89:708 - 716
- Venugopal SK, Jiang J, Kim TH, Li Y, Wang SS, Torok NJ, et al. Liver fibrosis causes downregulation of miRNA-150 and miRNA-194 in hepatic stellate cells and their overexpression causes decreased stellate cell activation. Am J Physiol Gastrointest Liver Physiol 2009; 298:G101 - G106
- Chung EY, Dews M, Cozma D, Yu D, Wentzel EA, Chang TC, et al. c-Myb oncoprotein is an essential target of the dleu2 tumor suppressor microRNA cluster. Cancer Biol Ther 2008; 7:1758 - 1764
- Navarro F, Gutman D, Meire E, Caceres M, Rigoutsos I, Bentwich Z, et al. miR-34a contributes to megakaryocytic differentiation of K562 cells independently of p53. Blood 2009; 114:2181 - 2192
- Zhao H, Kalota A, Jin S, Gewirtz AM. The c-myb proto-oncogene and microRNA-15a comprise an active autoregulatory feedback loop in human hematopoietic cells. Blood 2009; 113:505 - 516
- Sakura H, Kanei-Ishii C, Nagase T, Nakagoshi H, Gonda TJ, Ishii S. Delineation of three functional domains of the transcriptional activator encoded by the c-myb protooncogene. Proc Natl Acad Sci USA 1989; 86:5758 - 5762
- Dubendorff JW, Whittaker LJ, Eltman JT, Lipsick JS. Carboxy-terminal elements of c-Myb negatively regulate transcriptional activation in cis and in trans. Genes Dev 1992; 6:2524 - 2535
- Press RD, Reddy EP, Ewert DL. Overexpression of C-terminally but not N-terminally truncated Myb induces fibrosarcomas: a novel nonhematopoietic target cell for the myb oncogene. Mol Cell Biol 1994; 14:2278 - 2290
- Geurts JM, Schoenmakers EF, Roijer E, Astrom AK, Stenman G, van de Ven WJ. Identification of NFIB as recurrent translocation partner gene of HMGIC in pleomorphic adenomas. Oncogene 1998; 16:865 - 872
- O'Neil J, Tchinda J, Gutierrez A, Moreau L, Maser RS, Wong KK, et al. Alu elements mediate MYB gene tandem duplication in human T-ALL. J Exp Med 2007; 204:3059 - 3066
- Kauraniemi P, Hedenfalk I, Persson K, Duggan DJ, Tanner M, Johannsson O, et al. MYB oncogene amplification in hereditary BRCA1 breast cancer. Cancer Res 2000; 60:5323 - 5328
- Wallrapp C, Muller-Pillasch F, Solinas-Toldo S, Lichter P, Friess H, Buchler M, et al. Characterization of a high copy number amplification at 6q24 in pancreatic cancer identifies c-myb as a candidate oncogene. Cancer Res 1997; 57:3135 - 3139
- Alitalo K, Winqvist R, Lin CC, de la Chapelle A, Schwab M, Bishop JM. Aberrant expression of an amplified c-myb oncogene in two cell lines from a colon carcinoma. Proc Natl Acad Sci USA 1984; 81:4534 - 4538
- Welter C, Henn W, Theisinger B, Fischer H, Zang KD, Blin N. The cellular myb oncogene is amplified, rearranged and activated in human glioblastoma cell lines. Cancer Lett 1990; 52:57 - 62
- Maher CA, Kumar-Sinha C, Cao X, Kalyana-Sundaram S, Han B, Jing X, et al. Transcriptome sequencing to detect gene fusions in cancer. Nature 2009; 458:97 - 101
- Maher CA, Palanisamy N, Brenner JC, Cao X, Kalyana-Sundaram S, Luo S, et al. Chimeric transcript discovery by paired-end transcriptome sequencing. Proc Natl Acad Sci USA 2009; 106:12353 - 12358
- Phuchareon J, Ohta Y, Woo JM, Eisele DW, Tetsu O. Genetic profiling reveals cross-contamination and misidentification of 6 adenoid cystic carcinoma cell lines: ACC2, ACC3, ACCM, ACCNS, ACCS and CAC2. PLoS One 2009; 4:6040
- Laxman B, Tomlins SA, Mehra R, Morris DS, Wang L, Helgeson BE, et al. Noninvasive detection of TMPRSS2:ERG fusion transcripts in the urine of men with prostate cancer. Neoplasia 2006; 8:885 - 888
- Rice KR, Chen Y, Ali A, Whitman EJ, Blase A, Ibrahim M, et al. Evaluation of the ETS-related gene mRNA in urine for the detection of prostate cancer. Clin Cancer Res 2010; 16:1572 - 1576
- Spiro RH. Salivary neoplasms: overview of a 35-year experience with 2,807 patients. Head Neck Surg 1986; 8:177 - 184
- Williams BB, Wall M, Miao RY, Williams B, Bertoncello I, Kershaw MH, et al. Induction of T cell-mediated immunity using a c-Myb DNA vaccine in a mouse model of colon cancer. Cancer Immunol Immunother 2008; 57:1635 - 1645
- Gewirtz AM. Myb targeted therapeutics for the treatment of human malignancies. Oncogene 1999; 18:3056 - 3062
- Presta LG, Chen H, O'Connor SJ, Chisholm V, Meng YG, Krummen L, et al. Humanization of an anti-vascular endothelial growth factor monoclonal antibody for the therapy of solid tumors and other disorders. Cancer Res 1997; 57:4593 - 4599
- Ivy SP, Wick JY, Kaufman BM. An overview of small-molecule inhibitors of VEGFR signaling. Nat Rev Clin Oncol 2009; 6:569 - 579
- Vogler M, Dinsdale D, Dyer MJ, Cohen GM. Bcl-2 inhibitors: small molecules with a big impact on cancer therapy. Cell Death Differ 2009; 16:360 - 367
- Bansal R, Magge S, Winkler S. Specific inhibitor of FGF receptor signaling: FGF-2-mediated effects on proliferation, differentiation and MAPK activation are inhibited by PD173074 in oligodendrocyte-lineage cells. J Neurosci Res 2003; 74:486 - 493
- Pardo OE, Latigo J, Jeffery RE, Nye E, Poulsom R, Spencer-Dene B, et al. The fibroblast growth factor receptor inhibitor PD173074 blocks small cell lung cancer growth in vitro and in vivo. Cancer Res 2009; 69:8645 - 8651
- Prystowsky MB, Adomako A, Smith RV, Kawachi N, McKimpson W, Atadja P, et al. The histone deacetylase inhibitor LBH589 inhibits expression of mitotic genes causing G2/M arrest and cell death in head and neck squamous cell carcinoma cell lines. J Pathol 2009; 218:467 - 477
- Ishikawa C, Tafuku S, Kadekaru T, Sawada S, Tomita M, Okudaira T, et al. Anti-adult T-cell leukemia effects of brown algae fucoxanthin and its deacetylated product, fucoxanthinol. Int J Cancer 2008; 123:2702 - 2712
- Hotte SJ, Winquist EW, Lamont E, MacKenzie M, Vokes E, Chen EX, et al. Imatinib mesylate in patients with adenoid cystic cancers of the salivary glands expressing c-kit: a Princess Margaret Hospital phase II consortium study. J Clin Oncol 2005; 23:585 - 590
- Pfeffer MR, Talmi Y, Catane R, Symon Z, Yosepovitch A, Levitt M. A phase II study of Imatinib for advanced adenoid cystic carcinoma of head and neck salivary glands. Oral Oncol 2007; 43:33 - 36