Abstract
The importance of the CDK4 protein in human cancer first became evident following the identification of a germ line mutation in the Cdk4 locus that predisposes humans to melanoma. This mutation results in substitution of arginine with cysteine at position 24 (R24C). In an earlier study, we introduced the R24C mutation into the Cdk4 locus of mice using Cre-loxP-mediated "knock-in" technology and observed a very low incidence of spontaneous melanomas in Cdk4R24C/R24C mice. This suggested that additional oncogenic mutations might be required for development of melanomas. Here we report an increased incidence of spontaneous cutaneous melanoma in mice expressing the oncogene HRAS(G12V) in melanocytes on a Cdk4R24C background. Treatment of Tyr-HRas:Cdk4R24C/R24C mice with the carcinogen, DMBA/TPA resulted in a further increase in the number of nevi and melanomas developed when compared with Tyr-HRas:Cdk4+/+ mice. In summary, in Tyr-HRas:Cdk4R24C/R24C mice, we observed that activated CDK4 cooperates with the oncogenic HRAS(G12V)protein to increase the susceptibility of melanoma development in vivo.
Introduction
Malignant transformation of melanocytes presents in the clinic as melanoma.Citation1 Melanoma is known for its aggressive clinical behavior and resistance to conventional therapies. Thus, there is a need to gain insight into the genes involved in the pathogenesis of this disease. Although mouse models have helped us to understand the molecular mechanisms that predispose individuals to melanoma, many questions remain unresolved in this area of research.
Some progress in our understanding of melanoma has come from the observation of deregulated cell cycle regulatory genes in melanoma tissue. Mutations of the Cdkn2a and Cdk4 genes are often observed in human familial melanoma.Citation2,Citation3 Approximately 20–40% of familial melanoma patients inherit Cdkn2a mutations.Citation4 The Cdkn2a gene encodes two distinct tumor-suppressor proteins, Ink4a and Arf (known as p14 in humans and p19 in mice). Homozygous deletion of this locus has been observed in cultured melanoma lines, such as SK-MEL.Citation5 CdkN2A (Ink4a−/−:Arf−/−) knockout mice do not develop melanoma, but do develop tumors such as fibrosarcoma and lymphoma.Citation6
The importance of the Rb pathway in melanoma was highlighted further with the identification of a germline Cdk4 mutation in familial melanoma patients.Citation2,Citation3 Two mutations have been observed in the 24th codon of Cdk4, R24C and R24H. Both of these mutations prevent binding of affected Cdk4 isoforms to Ink4a inhibitors, leading to loss of cell cycle regulation. Further, progeny from crosses of Ink4a−/−:Arf−/− mice with Tyr-HRas mice develop spontaneous cutaneous metastatic melanoma with a low incidence of metastatic optical melanoma.Citation7 In 1999 Chin et al. crossed a doxycycline-inducible Tyr-HRas mouse with an Ink4a−/−:Arf−/− mouse to demonstrate that persistent expression of HRas is required for both initiation and maintenance of melanoma.Citation8 In the case of Ink4a−/−:Arf−/−:Tyr-HRas mice, removal of doxycycline from the feed causes H-ras repression, resulting in regression of pre-existing melanomas.Citation8
Interestingly, activated Ras has also been observed in proliferative defects in human skin. In the melanocytes of Spitz nevi, for example, the H-ras coding sequence contains point mutations and the entire locus is amplified.Citation9 Activated N-Ras has been reported in 33% of primary human melanomas and in 26% of metastatic melanomas.Citation10 Recently, two point mutations in the Braf gene were observed in 66% of melanomas, resulting in increase in Braf kinase activity.Citation11 These observations suggest that members of the Ras pathway play an important role in melanoma development. Furthermore, Cdk4R24C/R24C mice treated with DMBA/TPA develop melanoma.Citation12 To determine whether cooperativity exists between the Cdk4-R24C and mutant HRas genes, we mated Tyr-HRas mice, with Cdk4R24C/R24C mice to study their respective roles in the development of melanoma.
Results
The Cdk4-R24C mutation contributes to the development of melanoma in Tyr-HRas mice.
To determine whether Cdk4-R24C and HRAS(G12V) alleles cooperate in the development of melanoma, we crossed Tyr-HRas mice with Cdk4R24C/R24C and Cdk4+/+ mice to generate three different mouse strains: Tyr-HRas:Cdk4R24C/R24C, Tyr-HRas:Cdk4+/R24C and Tyr-HRas:Cdk4+/+. Because the Tyr-HRas(G12V) transgene is integrated on the Y chromosome, only males carry the transgene. Therefore, all the mice used in this study were males. We compared the development of melanomas in Tyr-HRas:Cdk4R24C/R24C, Tyr-HRas:Cdk4+/R24C and Tyr-HRas/Cdk4+/+ over a period of eighteen months. Tyr-HRas:Cdk4R24C/R24C and Tyr-HRas:Cdk4+/R24C began to develop cutaneous melanomas between the ages of seven and fifteen months (), while Tyr-HRas:Cdk4+/+ mice did not develop any tumors. We observed a tumor incidence of 30% in Tyr-HRas:Cdk4R24C/R24C and Tyr-HRas:Cdk4+/R24C mice. Interestingly, there was no significant difference in the tumor incidences in these two genotypes. Females (which do not carry activated HRAS transgene) were used as controls. As expected, we did not observe any spontaneous melanomas in the females.
Spontaneous cutaneous melanoma in Tyr-HRas:Cdk4.
The cutaneous melanomas that developed in Tyr-HRas:Cdk4R24C/R24C and Tyr-HRas:Cdk4+/R24C mice lacked pigmentation and therefore, were classified as amelanotic melanomas. These tumors were very similar to those observed in the original mouse model of Tyr-HRas mice.Citation7 The most common anatomical site for cutaneous melanomas was the ear pinna (96%) followed by the tail (3%) (). The tumors were locally invasive, and there was no evidence of macro-metastasis upon autopsy of the tumor-bearing mice. Upon sectioning and staining with hematoxylin and eosin, we observed hyper-proliferation of the dermis in all of the cutaneous melanomas (), with epidermal hyperplasia observed in some tumors (data not shown). The tumors were composed predominantly of spindle shaped cells () with a few epithelioid cells that expanded the dermis to form a tumor nodule. The tumor cells in the dermis were present in a storiform (cart wheel) pattern and showed mitotic figures as evidenced by the appearance of condensed chromosomes (see insert in ).
Expression of melanocyte-specific markers in Tyr-HRas: Cdk4 mice.
Expression of the HRAS(G12V) gene is expected to be restricted to melanocytes. We determined the cellular composition of tumor tissue with two melanocyte-specific markers, Tyrosinase Related Protein 2 (TRP-2) and S-100 protein in the immunostaining studies presented in . In the normal ear pinna, we observed S-100 and TRP-2-specific staining of melanocytes at the junction of the epidermis and dermis ( and g). We observed staining with anti-S-100 and anti-TRP-2 antibodies in melanocytes, throughout the dermis and (, f, h and i) in the melanoma sections of spontaneous tumors derived from Tyr-HRas:Cdk4R24C/R24C and Tyr-HRas: Cdk4+/R24C mice. TRP-2 staining was significantly lower than S-100 staining ( vs. f and h vs. e) for mice with a Tyr-HRas:Cdk4+/R24C or Tyr-HRas:Cdk4R24C/R24C genotype. Since expression of the HRAS(G12V) allele is expected to be restricted to melanocytes, and that the skin on the ear pinna and the tail contain a high numbers of melanocytes, it was not surprising that 96% of the tumors observed localized to these regions.
Nevi, melanoma and papilloma development in DMBA/TPA-treated Tyr-HRas:Cdk4 mice.
To determine whether co-expression of the CDK4-R24C and HRAS(G12V) alleles leads to increased incidence of melanoma following carcinogen exposure, we treated Tyr-HRas:Cdk4R24C/R24C and Tyr-HRas:Cdk4+/+ mice with DMBA/TPA according to the two-stage carcinogenesis protocol described by Rane et al.Citation12 Both mouse strains developed papillomas (benign skin tumors characterized by proliferation of cells in the epidermis) after 10–12 weeks of TPA treatment (). We observed no significant difference in the time of onset and the number of papillomas in the two genotypes. We also observed that mice of both genotypes developed nevi (clusters of melanocytes that result in a benign pigmented spot on the skin ). Tyr-HRas:Cdk4R24C/R24C mice developed significantly greater numbers of nevi (approximately 5–6 nevi per mouse) when compared to Tyr-HRas:Cdk4+/+ mice (approximately 1–2 nevi per mouse) (). The incidence of nevi in Tyr-HRas:Cdk4R24C/R24C was also significantly higher than that observed in Tyr-HRas:Cdk4+/+ mice (). The nevi were composed of spindle shaped cells with copious amounts of melanin pigment ( and E), but mitotic figures were not observed. Most nevi observed in the two genotypes did not grow in size. In approximately 33% of Tyr-HRas:Cdk4R24C/R24C mice, the nevi grew in size and progressed to develop melanoma ( and ). In contrast to the spontaneously developed melanomas, histological analysis revealed that the tumors arising in the DMBA/TPA-treated mice were composed of epithelioid and spindle shaped cells with copious melanin pigment within the dermis. ( vs. and F). We also examined the lymph nodes from the DMBA/TPA-treated mice. The lymph nodes of DMBA/TPA-treated Tyr-HRas:Cdk4R24C/R24C mice contained pigmented cells (). These cells did not form tumors but were admixed with histiocytes in a fashion reminiscent of dermatopathic lymphadenitis observed in humans.Citation13
Additional genetic alterations in melanomas derived from Tyr-HR as:Cdk4R24C mice.
Since p53 mutations are found in 50–55% of human cancersCitation14 we sought to determine whether p53 mutation contributes to the melanoma phenotype observed in our studies. We searched for such genetic alterations in both spontaneous and DMBA/TPA-induced melanomas. We therefore sequenced the DNA-binding domain from p53 transcripts isolated from isolated tumor tissue. The p53 DNA binding domain spans exons 4 to 9 of the transcript and corresponds to the region in which most cancer-associated mutations are observed.Citation15 cDNA from ten spontaneous melanomas and from four DMBA/TPA-induced melanomas were amplified and sequenced. We did not observe any mutations in this region of the p53 locus (data not shown).
Activation of the Ras-Erk pathway in spontaneous melanoma.
Since Tyr-HRas transgenic mice, carry the activated H-ras allele, we sought to determine whether the RAS-ERK pathway is also activated in Tyr-HRas:Cdk4 mice. Toward that end, we stained spontaneous melanomas with anti-H-ras and anti-phospho-Erk in immunofluoresence studies shown in . In addition, we examined the control normal ear pinna. Expression of H-ras was observed both in normal and in tumor samples of the ear. However, in the normal ear, staining was restricted to the layers of the epidermis (). In the tumor tissue, H-ras expression was detected in the epidermis (data not shown) and in the dermis (). We observed an increase in Erk-phosphorylation in all spontaneous tumor samples derived from Tyr-HRas:Cdk4+/R24C and Tyr-HRas:Cdk4R24C/R24C mice (). The normal ear pinna of these mice did not stain with phospho-Erk antibody (). These results suggest that the Ras-MAPK pathway is highly active in tumor cells compared to their normal cell counterparts.
Discussion
The existence cancer predisposition syndromes has provided basic researchers and clinicians with the opportunity to study the malignant transformation process in situ with some degree of predictability and with an eye towards mechanism. In the case of heritable melanoma, Wolfel et al.Citation16 reported a mutation in the CDK4 gene in which the arginine encoded at the 24th codon is replaced with cysteine. The mutated Cdk4R24C allele produces a protein that is refractory to negative regulation by Ink4 proteins, most notably, p16Ink4A. Unchecked CDK4 activity presumably leads to uncontrolled cell cycle progression through constitutive phosphorylation (and thus inactivation) of the pocket proteins p105Rb, p107 and p130. Inactivated pocket proteins are unable to check the trans-activation of cell proliferation-associated promoters by E2F family transcription factors. The net result is constitutive proliferation.
Modeling this genetic lesion in vivo became possible through loxP-Cre, “knock-in” technology, in which the R24C mutation is introduced through homologous recombination into the wild type CDK4 locus of mice. Development of the CDK4R24C mouse has enabled some fundamental observations in melanoma development. Rane et al.Citation12 isolated mouse embryo fibroblasts from Cdk4R24C mice. The CDK4 protein isoform from these cells exhibited deregulated kinase activity based upon findings from immune-complex kinase assays. The Cdk4R24C MEFs also escaped replicative sensence based upon cell proliferation assays using the 3T3 protocol. The growth properties of Cdk4R24C MEFs mimicked those of cells lacking all three RB family members. In this study, Rane et al. also observed ready transformation of Cdk4R24C MEFs by Ras, E1a and Myc.
Despite displaying clear properties of growth advantage in culture, homozygous CDK4R24C/R24C mice develop spontaneous melanoma at only a 2% frequency.Citation12 In their study, Sotillo et al.Citation17 made this same observation. High dose treatment of CDK4R24C mice with DMBA/TPA led to an increase in melanoma incidence to 95% of treated mice. It became clear from these observations that the CDK4R24C mutation might contribute to melanoma development, but it is not sufficient to induce malignant transformation. Subsequent genetic alterations seem to be required to observe the transformed phenotype.
Identification of the secondary tumor-promoting events led Sotillo et al.Citation17 to examine the role of the INK4 locus. DMBA/TPA treatment of mice nullizygous for p18Ink4c led to development of increased numbers of melanocytic lesions than did wild type controls. This observation suggests that cells acquiring loss-of-function mutations in p18Ink4c may also predispose individuals to developing melanoma.
Previous studies by Hacker et al.Citation18 suggested a role for Ras in UV-mediated melanomagenesis in their mouse model, Cdk4R24C/R24C/TPras. This strain expresses both the R24C mutation and the activated Ha-Ras (G12V) allele from the tyrosinase (a melanocyte-specific) promoter. In their report, UV treatment of Cdk4R24C/R24C/TPras mice results in increased incidence of melanoma, compared to non-UV-treated controls. From this study, it appears that both inactivation of the Ink4 genes (via the R24C mutation) coupled with activated Ras activity is sufficient to promote melanoma. This incidence is increased with the added stimulus of UV irradiation.
In the present study we describe our findings of melanoma development in a similar transgenic mouse model to that reported by Hacker, et al. We bred Cdk4R24C/R24C mice with Tyr-HRas mice. We observed a 30% incidence of melanomas in Tyr-HRas:Cdk4+/R24C and Tyr-HRas:Cdk4R24C/R24C mice, suggesting that a second mutation was indeed required for development of melanomas. We observed no significant difference in the incidence of spontaneous melanomas between Tyr-HRas:Cdk4+/R24C and Tyr-HRas:Cdk4R24C/R24C mice, suggesting that a mutation in one allele of Cdk4, along with a H-ras mutation is sufficient to increase the incidence of melanomas. These data stand in contrast to those reported by Hacker et al.Citation18 in which TPRas:Cdk4+/R24C mice did not develop any spontaneous melanomas (Tyr-HRas and TPRas are Tyrosinase-HRas mice). This may be attributed to the differences in the tyrosinase promoter constructs used to make the transgenic mice. The Tyr-HRas mice in our study had an additional enhancer region in their construct compared to the TPRas mice used in the Hacker, et al. study. In addition, the Tyr-HRas mice were on an FVB/N background whereas the TPRas mice were on C57BL/6XSJL/C3HeN background.Citation7,Citation19
Although spontaneous melanomas did arise in our Tyr-HRas:Cdk4R24C mice, the addition of DMBA/TPA or UV irradiation suggests the role for yet more genetic changes to occur for maximal penetrance and high level of tumor incidence. Since p53 mutations are found in 50–55% of human cancers we examined the p53 locus for mutation in the spontaneous and DMBA/TPA-induced melanoma. We did not observe loss of p53 expression or mutations in the p53 locus. Our data support the findings of Chin et al. and Sotillo et al. who also observed no mutations in the “hotspot” region of p53 locus in melanomas derived from Tyr-HRas:Ink4a−/− and Cdk4R24C/R24C mice.Citation7,Citation17 These studies correlate well with the low incidence of p53 mutations found in primary or metastatic human melanoma.Citation20
Activation of Ras/Raf/Mek/Erk pathway in melanoma can occur by mutations in BRAF and NRAS. Activation of MAP kinase pathway leads to (1) downregulation of E-cadherin expression freeing melanoma cells from keratinocyte-mediated control; (2) upregulation of integrin expression which promotes survival and invasive growth of melanoma cells in the Extracellular Matrix (ECM); and (3) increased expression and activity of proteolytic enzymes such as MMP-9 which results in the degradation of ECM.Citation21 Based on these observations, we investigated whether the MAP kinase pathway was active in spontaneous melanomas. Positive immunoreactivity with a phospho-Erk antibody in these tumors suggested that MAP kinase pathway confers a growth advantage to tumor cells derived from Tyr-HRas:Cdk4+/R24C and Tyr-HRas:Cdk4R24C/R24C mice.
The studies presented here suggest that development of malignant melanoma appears to require at least two genetic changes. The observed heritable R24C mutation of Cdk4 seems sufficient to provide cells of the skin epithelium with a growth advantage. This growth advantage manifests itself in culture through loss of contact inhibition and escape from replicative senescence. In situ Cdk4R24C epithelia are hyperplastic, but invasiveness through basement membranes and entry into the vertical growth phase require an additional genetic change. That change is unlikely to be mutation of the p53 gene, as we and others have reported. Mutation of the Ras pathway and upregulation of the Ras/Raf/Mek/Erk cascade appears to be one mechanism by which cells can translate the growth advantage of the R24C mutation into the invasive behavior characteristic of melanoma.
Materials and Methods
Mouse strains.
Tyr-HRas mice that express mutant HRAS(G12V) in melanocytes under the control of the melanocyte-specific tyrosinase gene have been described.Citation7 These mice, on an FVB/N background, were obtained from the NCI (National Cancer Institute, Bethesda http://mouse.ncifcrf.gov). Genomic DNA from mouse-tails was isolated using the Puregene DNA isolation kit (Gentra systems) and the presence of the transgene was confirmed using PCR with specific primers as described on the NCI database http://mouse.ncifcrf.gov.
Generation of Cdk4R24C/R24C mice has been described.Citation12 PCR genotyping of Cdk4R24C/R24C and Cdk4+/+ mice was performed using the following primers: mCdk4-14 (5′-GCG TCG CTG GCT GAT TAT GGA AGG T-3′) and mCdk4-15 (5′-CCT TAC GGG TCC TAT TGT CCT CTC C-3′).
H&E staining and indirect immunoflourescence.
Melanomas and nevi were fixed in 10% buffered formalin and embedded in paraffin. Sections were either stained with Hematoxylin and Eosin (H&E) or processed for immunofluorescence. Sections were deparaffinized and antigen retrieval was carried out using citrate buffer (H-3300, Vector labs). Sections were blocked with 5% goat serum in phosphate buffered saline with 0.5% Tween-20 (PBST) for one hour and incubated overnight with various primary polyclonal antibodies in PBS with 1% Bovine Serum Albumin (BSA): anti-S-100 at a 1:400 dilution (Dako, United Kingdom), anti-Tyrosinase-related protein 2 at a 1:100 dilution (TRP-2 generously provided by Dr. Vincent Hearing, Laboratory of Cell Biology, National Cancer Institute, Bethesda, MD), anti-Hras at a 1:50 dilution (sc-520, Santa Cruz Biotechnology), anti-phosphoERK1/2 at a 1:100 dilution (9101, Cell Signaling Technology). This was followed by incubation in Cy-2 goat anti-rabbit secondary antibody at a 1:1,000 dilution in PBS-1%BSA for one hour (Jackson Immunoreserach). Sections were counter-stained with DAPI (Invitrogen) and visualized using a Nikon E800 microscope. Negative controls were stained with secondary antibody alone.
Two-stage chemical skin carcinogenesis.
Tyr-HRas:Cdk4+/+ (n = 17) and Tyr-HRas:Cdk4R24C/R24C (n = 14) mice were shaved at 4 weeks of age. 25 µg of 9,10-di-methyl-1,2-benz[a]anthracene (DMBA, Sigma) in 200 µl of acetone was applied to the skin in the shaved area for tumor initiation. This was followed by biweekly application of 2 µg of 12-O-Tetradecanoylphorbol-13-acetate (TPA, Sigma) in 200 µl of acetone for tumor promotion. The TPA treatment was carried out for 20 weeks and the mice were monitored for the development of papillomas, nevi and melanomas. At the end of 30 weeks, the mice were grouped into tumor-free and tumor-bearing categories and Kaplan-Meier survival curve analysis was performed.
p53 mutation analysis.
RNA was extracted from tumors using the Trizol reagent (Invitrogen). cDNA was synthesized using Superscript II reverse transcriptase (Invitrogen) and oligo (dT). PCR was performed using the primers, 5′-GCC CCT GTC ATC TTT TGT-3′ and 5′-GCA GGT GGG CAG CGC TC-3′, spanning the DNA binding domain of p53. The PCR reaction was carried out at 94°C for 30 sec, 60°C for 30 sec and 72°C for 4 minutes for 40 cycles. The amplified PCR products were sequenced using the above primers.
Statistical analysis.
The Kaplan-Meier survival curve was generated using Prizm 4 software (GraphPad). The Kaplan-Meier curve also included censored mice that died unexpectedly or were euthanized due to paralysis or skin infections. The difference between survival curves of the different genotypes was assessed using the log-rank test.
Figures and Tables
Figure 1 Contribution of the Cdk4R24C mutation in the development of melanoma in Tyr-HRas mice. (A) Crosses performed to produce the required transgenic mice represented by dotted rectangles. Male mice are represented by squares and female mice are represented by ovals. (B) Mice of three genotypes, Tyr-HRas:Cdk4+/R24C (squares), Tyr-HRas:Cdk4R24C/R24C (triangles) and Tyr-HRas:Cdk4+/+ (crosses) were examined for tumor incidence over a period of 18 months (n = 26 for each genotype).
Figure 2 Tyr-HRas:Cdk4R24C mice develop spontaneous cutaneous melanomas. (A) photograph of 11-month old Try-HRas:Cdk4+/R24C mice with cutaneous amelanotic melanoma on the ear pinna (indicated by the white arrow). (B) 40x magnification of Hematoxylin and eosin (H&E) stained section of normal ear pinna. (C) 40x magnification of an H&E, stained section of cutaneous ear melanoma. (D and E) are 400x magnified H&E sections of normal ear pinna and of the cutaneous ear melanoma, respectively. Note the atypical spindle shaped cells in the dermis of cutaneous ear melanoma in (E) (indicated by black arrow). Inset in (E) showing mitotic cell with condensed chromosomes (1,000x) seen in the cutaneous ear melanoma section.
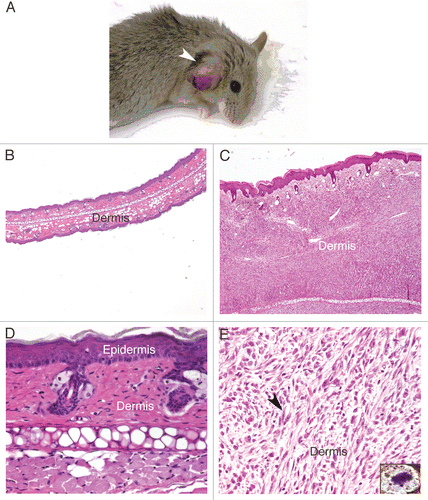
Figure 3 Expression of melanoma-specific protein markers in Tyr-HRas:Cdk4 mice. (a–c) are 40x magnification of H&E stained sections of normal ear pinna (a), a cutaneous mealanoma from Try-HRas:Cdk4+/R24C mouse (b) and a cutaneous melanoma from Try-HRas:Cdk4R24C/R24C mouse (c). (d–f) are immunofluorescence studies to determine expression of cytoplasmic melanoma tumor marker, S-100. Cells are stained with DAPI to determine the location of nuclei. S-100 expressing cells fluoresce orange color. (g–i) are immunofluorescence studies to determine expression of cytoplasmic melanoma marker, Tyrosinase Related Protein-2 (TRP-2). Cells are stained with DAPI to determine the location of nuclei. TRP-2 expressing cells fluoresce orange. (d, f, g and h) contain insets showing S-100 and TRP-2 expressing cells at higher magnifications. (j and k) are immunofluorescence studies using DAPI and anti-Rabbit IgG as negative control in normal (j) and tumor (k) ear samples. Magnification from d–k is 600X. E, epidermis; D, dermis.
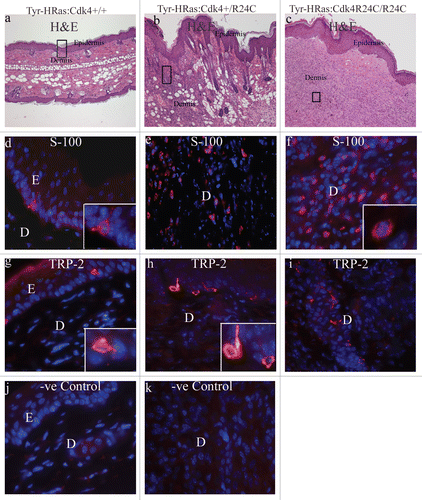
Figure 4 Nevi, melanoma and papilloma development in DMBA/TPA-treated Tyr-HRas:Cdk4 mice. Representative pictures were taken after 25 weeks of TPA treatment. Tyr-HRas:Cdk4R24C/R24C mice develop nevi (black arrow) (A). Tyr-HRas:Cdk4R24C/R24 mice develop melanoma (yellow arrow) (B). Tyr-HRas:Cdk4+/+ mice develop papillomas (white arrows) (C).
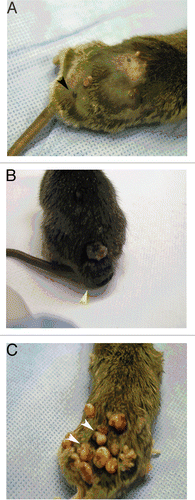
Figure 5 Incidence of nevi and melanoma in DMBA/TPA treated Tyr-HRas:Cdk4 mice. Incidence of nevi formation after DMBA/TPA treatment in Tyr-HRas:Cdk4+/+ and Tyr-HRas:Cdk4R24C/R24C mice is represented over number of weeks after TPA treatment (A). Average number of nevi per mouse in DMBA/TPA treated Tyr-HRas:Cdk4+/+ and Tyr-HRas:Cdk4R24C/R24C mice (B). Percentage of DMBA/TPA treated Tyr-HRas:Cdk4+/+ and Tyr-HRas:Cdk4R24C/R24C mice that developed melanoma (C). Tyr-HRas:Cdk4+/+ (n = 20) and Tyr-HRas:Cdk4R24C/R24C (n = 15).
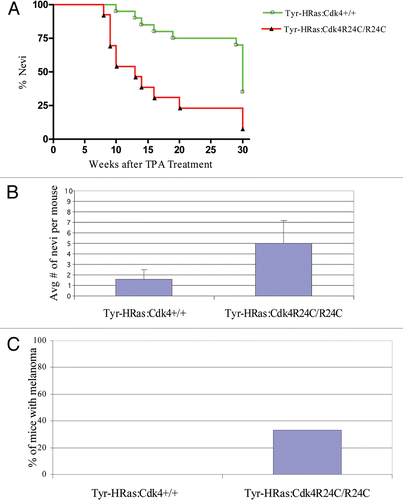
Figure 6 Organization of epithelia in Tyr-HRas:Cdk4 mice. (A–C) are 40x magnified H&E sections of normal skin, nevi and melanoma observed in Tyr-HRas:Cdk4+/+, Tyr-HRas:Cdk4+/+, Tyr-HRas:Cdk4R24C/R24C mice respectively. (D–F) are higher magnification (200x) views of the same sections shown in (A–C). (G) is a 200x magnification of papilloma observed in Tyr-HRas:Cdk4+/+ mice. (H and I) are 40x maginified H & E sections of lymph nodes obtained from Tyr-HRas:Cdk4+/+ and Tyr-HRas:Cdk4R24C/R24C mice respectively. (I) has a inset of higher magnification (200x) of the lymph nodes obtained from Tyr-HRas:Cdk4R24C/R24C mice.
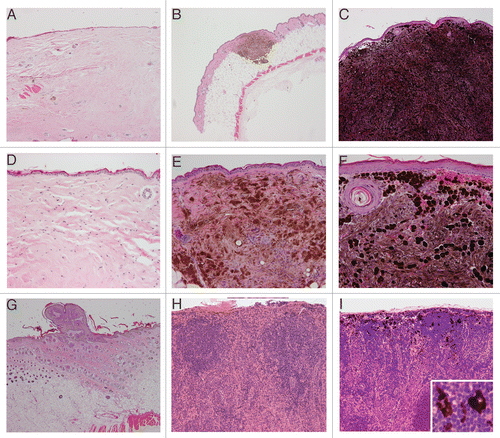
Figure 7 Expression of H-ras and phospho-Erk in sponataneous melanomas. (A and B) are representative immunofluorescent staining of H-ras in either normal ear pinna and spontaneous cutaneous melanomas derived from Tyr-HRas:Cdk4R24C/R24C mice respectively. Representative immunofluorescent staining with phospho-Erk in normal ear pinna (C). (D) shows immunofluorescent staining with phospho-Erk of spontaneous cutaneous melanoma derived from Tyr-HRas:Cdk4+/R24C mice. Magnification of (A, C and D) is 600x and (B) is 400x.
Acknowledgements
This work was supported by NIH grants PO1 CA95569 and RO1 AG22022 to E.P.R. The authors would like to thank Dr. Vincent Hearing, National Cancer Institute (NCI, Bethesda, MD) for providing us the TRP-2 antibody.
References
- Tucker MA, Goldstein AM. Melanoma etiology: where are we?. Oncogene 2003; 22:3042 - 3052
- Soufir N, Avril MF, Chompret A, Demenais F, Bombled J, Spatz A, et al. Prevalence of p16 and Cdk4 germline mutations in 48 melanoma-prone families in France. The French Familial Melanoma Study Group. Hum Mol Genet 1998; 7:209 - 216
- Zuo L, Weger J, Yang Q, Goldstein AM, Tucker MA, Walker GJ, et al. Germline mutations in the p16INK4a binding domain of Cdk4 in familial melanoma. Nat Genet 1996; 12:97 - 99
- Hayward N. New developments in melanoma genetics. Curr Oncol Rep 2000; 2:300 - 306
- Walker GJ, Flores JF, Glendening JM, Lin AH, Markl ID, Fountain JW. Virtually 100% of melanoma cell lines harbor alterations at the DNA level within CDKN2A, CDKN2B or one of their downstream targets. Genes Chromosomes Cancer 1998; 22:157 - 163
- Serrano M, Lee H, Chin L, Cordon-Cardo C, Beach D, DePinho RA. Role of the INK4a locus in tumor suppression and cell mortality. Cell 1996; 85:27 - 37
- Chin L, Pomerantz J, Polsky D, Jacobson M, Cohen C, Cordon-Cardo C, et al. Cooperative effects of INK4a and ras in melanoma susceptibility in vivo. Genes Dev 1997; 11:2822 - 2834
- Chin L, Tam A, Pomerantz J, Wong M, Holash J, Bardeesy N, et al. Essential role for oncogenic Ras in tumour maintenance. Nature 1999; 400:468 - 472
- Bastian BC, LeBoit PE, Pinkel D. Mutations and copy number increase of HRAS in Spitz nevi with distinctive histopathological features. Am J Pathol 2000; 157:967 - 972
- Demunter A, Stas M, Degreef H, De Wolf-Peeters C, van den Oord JJ. Analysis of N- and K-ras mutations in the distinctive tumor progression phases of melanoma. J Invest Dermatol 2001; 117:1483 - 1489
- Davies H, Bignell GR, Cox C, Stephens P, Edkins S, Clegg S, et al. Mutations of the BRAF gene in human cancer. Nature 2002; 417:949 - 954
- Rane SG, Cosenza SC, Mettus RV, Reddy EP. Germ line transmission of the Cdk4(R24C) mutation facilitates tumorigenesis and escape from cellular senescence. Mol Cell Biol 2002; 22:644 - 656
- Cangiarella J, Symmans WF, Shapiro RL, Roses DF, Cohen JM, Chhieng D, et al. Aspiration biopsy and the clinical management of patients with malignant melanoma and palpable regional lymph nodes. Cancer 2000; 90:162 - 166
- Hollstein M, Rice K, Greenblatt MS, Soussi T, Fuchs R, Sorlie T, et al. Database of p53 gene somatic mutations in human tumors and cell lines. Nucleic Acids Res 1994; 22:3551 - 3555
- Levine AJ. p53, the cellular gatekeeper for growth and division. Cell 1997; 88:323 - 331
- Wolfel T, Hauer M, Schneider J, Serrano M, Wolfel C, Klehmann-Hieb E, et al. A p16INK4a-insensitive CDK4 mutant targeted by cytolytic T lymphocytes in a human melanoma. Science 1995; 269:1281 - 1284
- Sotillo R, Garcia JF, Ortega S, Martin J, Dubus P, Barbacid M, et al. Invasive melanoma in Cdk4-targeted mice. Proc Natl Acad Sci USA 2001; 98:13312 - 13317
- Hacker E, Muller HK, Irwin N, Gabrielli B, Lincoln D, Pavey S, et al. Spontaneous and UV radiation-induced multiple metastatic melanomas in Cdk4R24C/R24C/TPras mice. Cancer Res 2006; 66:2946 - 2952
- Kramer TR, Powell MB, Wilson MM, Salvatore J, Grossniklaus HE. Pigmented uveal tumours in a transgenic mouse model. Br J Ophthalmol 1998; 82:953 - 960
- Meier F, Satyamoorthy K, Nesbit M, Hsu MY, Schittek B, Garbe C, et al. Molecular events in melanoma development and progression. Front Biosci 1998; 3:1005 - 1010
- Meier F, Schittek B, Busch S, Garbe C, Smalley K, Satyamoorthy K, et al. The RAS/RAF/MEK/ERK and PI3K/AKT signaling pathways present molecular targets for the effective treatment of advanced melanoma. Front Biosci 2005; 10:2986 - 3001