Abstract
Genotoxic stress triggers the p53 tumor suppressor network to activate cellular responses that lead to cell cycle arrest, DNA repair, apoptosis or senescence. This network functions mainly through transactivation of different downstream targets, including cell cycle inhibitor p21, which is required for short-term cell cycle arrest or long-term cellular senescence, or proapoptotic genes such as p53 upregulated modulator of apoptosis (PUMA) and Noxa. However, the mechanism that switches from cell cycle arrest to apoptosis is still unknown. In this study, we found that mice harboring a hypomorphic mutant p53, R172P, a mutation that abrogates p53-mediated apoptosis while keeping cell cycle control mostly intact, are more susceptible to ultraviolet-B (UVB)-induced skin damage, inflammation, and immunosuppression than wild-type mice. p53R172P embryonic fibroblasts (MEFs) are hypersensitive to UVB and prematurely senesce after UVB exposure, in stark contrast to wild-type MEFs, which undergo apoptosis. However, these mutant cells are able to repair UV-induced DNA lesions, indicating that the UV hypersensitive phenotype results from the subsequent damage response. Mutant MEFs show an induction of p53 and p21 after UVR, while wild-type MEFs additionally induce PUMA and Noxa. Importantly, p53R172P MEFs failed to downregulate anti-apoptotic protein Bcl-2, which has been shown to play an important role in p53-dependent apoptosis. Taken together, these data demonstrate that in the absence of p53-mediated apoptosis, cells undergo cellular senescence to prevent genomic instability. Our results also indicate that p53-dependent apoptosis may play an active role in balancing cellular growth.
Introduction
In response to genomic DNA damage, cells activate a network of pathways that rapidly modulate many cellular activities. One of the central outcomes of the DNA damage response pathway is stabilization and activation of the tumor suppressor p53, which leads to cell cycle arrest and DNA repair, apoptosis, or cellular senescence. p53 is best characterized as a transcription factor. It transactivates many different genes to induce and regulate a diverse network of cellular functions that are crucial in protecting the cell against genome instability as well as maintaining the homeostasis of cell growth and differentiation.Citation1,Citation2 Initial genomic insults lead to the stabilization of p53 and nuclear localization where transient cell cycle arrest can be quickly activated, which allows the repair of damaged DNA prior to replication. Upon excessive and irreparable damage, p53 can further trigger a signaling cascade to induce programmed cell death through transcription of different pro-apoptotic genes, most notably p53-upregulated modulator of apoptosis (PUMA) and NoxaCitation3 as well as trans-repression of anti-apoptotic genes including Bcl-2.Citation4 Apoptosis is a direct mechanism that protects cells against the accumulation of genomic instability leading to tumorigenesis.
DNA damage may also induce an irreversible cell cycle arrest termed cellular senescence.Citation5–Citation7 One key p53 target playing an important role in cell cycle checkpoint regulation and induction of cellular senescence is the cyclin-dependent kinase inhibitor p21, that is commonly transactivated and induces G1 arrest after DNA damage by inhibiting the cyclinE/CDK2 complex.Citation7,Citation8 Cellular senescence can be induced by a variety of different stimuli including telomere shortening (replicative senescence), oncogenic signaling (oncogene-induced senescence), or stress/DNA damage independent of the previous two signals (premature senescence).Citation9 Regardless of the stimuli, cellular senescence plays a parallel function to apoptosis in preventing genomic instability thereby inhibiting tumor formation.Citation10 The cell fate decision after DNA damage to either activate apoptosis or cellular senescence occurs via the p53 pathway, but the trigger remains unclear—speculations include different stimuli and signal strength/persistence as well as cell type specific responses.Citation11
Fundamental to the initiation of most tumors is DNA damage, which, if inaccurately or inappropriately repaired, can lead to oncogene activation or deactivation of tumor suppressor genes that drive cell proliferation and/or survival in the absence of physiological stimuli.Citation12,Citation13 One carcinogen that directly damages genomic DNA is ultraviolet radiation (UVR).Citation14,Citation15 Excessive exposure to UVR is potentially hazardous to our health, since evidence links UVR exposure to several conditions such as skin inflammation, immunosuppression, gene mutations, multiple cancers and aging.Citation16–Citation18 Tumor suppressor p53 plays an essential role in mediating the cellular responses and DNA damage repair resulting from UVR.Citation19,Citation20 Previous work revealed that in the absence of one or both alleles of p53, mice exhibit extreme sensitivity to tumor induction by UVB and a drastic decrease of apoptosis in the dermis.Citation21,Citation22 More recent studies underline UVB as an inducer of p53-mediated premature cellular senescenceCitation23,Citation24 as well as p53-mediated apoptosis.Citation25 To directly decipher between p53 functions after UVR, we utilized a hypomorphic, separation-of-function p53 point mutation, R172P, retaining a partial cell cycle arrest capability but losing complete p53-mediated apoptosis.Citation26
In this study, we found that mutant p53 mice, hereafter referred to as p53P/P, are more susceptible to UV-induced skin damage, inflammation and immunosuppression than wildtype mice. While at the cellular level, p53P/P mouse embryonic fibroblasts (MEFs) show UV hypersensitivity and cannot transactivate pro-apoptotic genes PUMA and Noxa, but retain the ability to upregulate p21, which ultimately drives the premature cellular senescence and the sensitivity phenotype observed. Conversely, wild-type MEFs undergo apoptosis retaining the ability to induce these pro-apoptotic genes and die in a dose dependent manner. Collectively our studies indicate the importance of p53-mediated apoptosis in vivo and show that in its absence, cellular senescence is triggered as a fail-safe mechanism to protect cells against genomic instability but comes at a cost.
Results
Mutant p53R172P mice are hypersensitive to UVB radiation.
Tumor suppressor p53 plays a central role in the cellular response to UV radiation.Citation20 Previous studies have demonstrated that p53-deficient mice exposed to chronic UV radiation show an increase in skin tumor formation.Citation19 To investigate the cellular response to UVR, which is known to cause erythema and inflammation in the skin, we exposed hypomorphic p53 mutant mice (p53P/P) to UV radiation and analyzed the acute cutaneous inflammatory response. Because it is difficult to measure erythema in mice with pigmented skin, we determined the macroscopic inflammation in skin by measuring double-fold skin thickness after 4.5 kJ/m2 of UVB exposure. At 24 and 48 hours post UVB treatment, the skin fold thickness increased in the wild-type mice by 6% and 27% from baseline measurements, while p53P/P mice had an average of 27% and 74% increase, respectively. The mice were exposed to a second dose of UVB and measurements were taken 24 hours post the second exposure. Wild-type skin had increased to 33% from the baseline, while the p53P/P mice had a significant increase of up to 100% (). The skin fold swelling differences in the UV-irradiated p53P/+ mice were statistically indistinguishable from that observed in wild-type mice. To further investigate the inflammatory response, dorsal skin sections from mice that underwent acute UVR were stained with hematoxylin and eosin, which revealed that UVR not only increased the thickness of the epidermis, but also the dermal layer (). These data indicate that p53P/P mice are hypersensitive to UVB exposure.
Mice homozygous for p53R172P are more susceptible to UVR-induced immunosuppression.
It is well known that UV radiation causes systemic immune suppression, which is associated with an increased risk of skin cancer development.Citation27,Citation28 Previous studies have shown that UV-induced DNA damage initiates immune suppression in wild-type mice.Citation22,Citation29 To determine whether p53P/P mice exhibit differences in their immune status after UV exposure, we compared the extent of UVB-induced immune suppression among genotypes. Mice were exposed to UVB and then immunized with C. albicans 5 days later. The suppression of delayed-type hypersensitivity (DTH), as described previously,Citation30 was used to measure immune status. The results indicate that both wild-type and p53P/P non-irradiated mice were able to mount an effective DTH response (). Interestingly, p53P/P mice were significantly more sensitive to the immunosuppressive effects of UV radiation to relatively low UV doses, 0.5 and 2.5 kJ/m2 (p < 0.005; p53P/P vs. p53+/+). At 5 kJ/m2, which induced immune suppression in the wild-type mice, p53P/P mice exhibited significantly increased immune suppression (p < 0.005). No significant difference in suppression was observed at the highest dose (10 kJ/m2). These results indicate that the increased susceptibility of p53P/P mice to UV-induced immune suppression is in parallel with their hypersensitivity to UVR.
UVR induces p53-p21 driven cellular senescence in the skin of p53R172P mice.
UVR exposure induces p53-dependent apoptosis in wild-type mouse skin.Citation25 To determine whether UVR can also induce apoptosis in p53P/P mice, we performed a TUNEL assay. Apoptotic cells were detected in UV irradiated wild-type samples receiving 5 and 10 kJ/m2 of UVB (Fig. S1) while virtually no apoptosis was detected in p53P/P and non-irradiated wild-type samples. In contrast, exposure to 10 Gy of ionizing irradiation induced slight apoptosis in wild-type skin, which was absent in skin sections from p53P/P mice. This confirmed the apoptosis-deficient phenotype of p53P/P. To identify whether other cellular responses occurred after UVR, we stained skin sections for β-galactosidase (SA-β-gal), a marker indicative of cellular senescence, at 24 and 72 hours after UVR. As shown in and S2, senescent cells were observed specifically in the hair follicles of p53P/P mice 24 hours after 10 kJ/m2 of UVB treatment; the number of these cells increased significantly after 72 hours of UV exposure. Low levels of senescence, however, were observed in skin sections of p53P/+ mice and were absent in wildtype mice even after exposure to ionizing radiation (IR). These results indicate that in the absence of apoptosis, p53P/P skin cells undergo senescence in response to UVB damage. To identify if these responses in the skin were dependent on the expression of p53 and the downstream target, p21, we used immunofluorescent staining. Interestingly, skin sections from UV-irradiated p53P/P mice show an increased level of both p53 and p21 proteins, while the wild-type show a modest increase in p53 and p21 expression levels (). Our results indicated that UVB exposure induces p53 and p21 in both wild-type and mutant mice, but the mutant skin undergoes senescence instead of apoptosis, possibly due to the high levels of mutant p53 and nuclear p21, correlating with cell cycle inhibition activity.
p53R172P MEFs are specifically hypersensitive to UVR but not to IR.
To determine the sensitivity of p53P/P cells to UVB treatment, we exposed early passage p53P/P mouse embryonic fibroblasts (MEFs) to different UVB doses to compare the rate of cell survival in wild-type and p53−/− MEFs. As shown in , UVB doses of 50, 100 and 250 J/m2 were cytotoxic for wild-type cells (approximately 75, 55 and 20 percent survival respectively), but were not as cytotoxic to p53−/− MEFs. However, survival of p53P/P MEFs was significantly less when compared to wild-type and p53−/−, with the most significant difference observed at 100 J/m2, which accounted for just a 27% survival rate. These results indicate that hypomorphic p53P/P drives MEF hypersensitivity to UVB treatment. Non-irradiated p53P/P MEFs grow at a similar rate to that of the wild-type cells (data not shown), ruling out that the sensitivity observed in p53P/P MEFs was due to a growth disadvantage in untreated MEFs.Citation31 To determine if the hypomorphic mutation of p53 is also important for MEF survival after IR, we exposed MEFs to different doses of IR. As shown in , wild-type, p53P/P and p53−/− MEFs all show a relatively similar survival pattern in response to IR, indicating that p53 hypomorphic mutation does not effect the survival of MEFs in response to IR. A similar trend was observed after performing a colony formation assay with both UVR and IR, at similar doses (data not shown). Therefore, we conclude that p53P/P is specifically hypersensitive to UVB treatment and not to IR.
Mutant p53R172P cells are able to remove UV-induced damaged bases normally.
UV irradiation induces distortions to DNA strands by forming cyclobutane pyrimidine dimers (CPDs) or pyrimidine (6-4) pyrimidinone photoproduct [(6-4) photoproducts], which initiate the nucleotide excision repair (NER) pathway as well as cell cycle check-points.Citation32 To determine if the hypersensitivity of p53P/P MEFs result from a defect in the ability of the cells to repair UV-induced CPD and (6-4) photoproducts, DNA was isolated from UVB irradiated MEFs (200 J/m2) at 6, 24 and 48 hours post irradiation and were analyzed for CPD and (6-4) photoproducts by a radioimmunoassay.Citation33 The results shown in reveal that the rate of removal of both (6-4) photoproducts and CPD in p53P/P is as efficient as p53P/+ and wild-type cells at 48 hours after UVR; this result suggests that the removal and repair of damaged DNA bases is not, per se, affected by the p53P/P mutation.
Next we analyzed if the in vivo hypersensitivity in p53P/P mice is due to problems in repair of UV-induced CDP and (6-4) photoproducts by extracting DNA samples from mouse skin at various time points (0, 6 and 24 hours) post UVR. At 5.0 kJ/m2 of UVB, a dose which showed significant hypersensitivity and immunosuppression, the (6-4) photoproducts were removed rapidly in both genotypes, with only 53–67% remaining at 6 h and 19–43% still remaining after 24 hours, with no significant change in the rate of repair between genotypes. The removal of CPD was slightly slower with 51–71% remaining at 6 hours and 24–48% still remaining at 24 hours, but here again, the rate of repair was similar regardless of the genotype (). Nonetheless, these results indicate that the increased sensitivity of p53P/P mice to UV carcinogenesis is not due to defects in the repair of UV-induced DNA damage.
Wild-type MEFs undergo apoptosis after exposure to UV radiation.
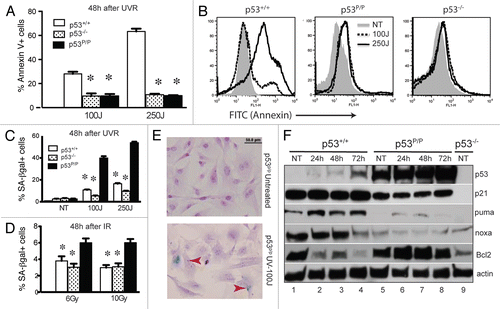
The prominent cellular response for wild-type MEFs after exposure to UV radiation is to undergo apoptosis and p53 plays an essential role in mediating this death pathway.Citation25,Citation34 The p53P/P mutation renders the cells resistant to p53-dependent apoptosis,Citation26,Citation35 so to determine the extent of apoptosis in wild-type and mutant MEFs after UVB radiation in our system, we stained irradiated MEFs with labeled AnnexinV and PI, then analyzed the samples by flow cytometry.Citation36 MEFs were treated with UVB (100 and 250 J/m2) and the cells were harvested 24 and 48 hours later. In response to UVB, 15 to 25% of wild-type MEFs undergo apoptosis at the dose of 100 J/m2, correlating with previous reports.Citation37 This percentage increased from 28 to 63% when these cells were treated with a higher dose ( and S3a). A typical profile is shown in . This apoptosis is dependent on p53, since a very low level of apoptosis (under 10%) was detected in the p53-deficient MEFs. As expected, p53P/P MEFs exhibited almost identical apoptotic levels as p53−/− MEF, further indicating that p53P/P MEFs are deficient in p53-mediated apoptosis.
In the absence of apoptosis, p53R172P MEFs undergo senescence after UV exposure.
In nonhomologous end-joining deficient p53P/P mice, accumulated DNA damage at the antigen receptor genes drive developing lymphocytes to undergo senescence with elevated amounts of p53 and p21,Citation10 and spontaneous DNA damage induces cellular senescence in pancreatic beta cells of these mutant mice.Citation38 We next asked if the initial DNA damage caused by UVR could drive p53P/P MEFs to undergo senescence. Wild-type, p53−/− and p53P/P MEFs were treated with UVB radiation at the doses of 100 and 250 J/m2, harvested 24 and 48 hours later and stained for senescence associated β-galactosidase. As shown in and S3B, a very low percentage of senescent cells was detected in both wild-type and p53−/− MEFs, while a high percentage of senescent cells was detected in p53P/P MEFs. The level exceeded 50% in p53P/P MEFs 48 hours after 250 J/m2 treatment compared to less than 20% found in wild-type p53−/− MEFs. A representative field of senescent p53P/P MEFs stained with SA-β-gal and counter-stained with H&E, is shown in . On the other hand, although the percentage of senescent cells was also higher in p53P/P MEFs after IR treatment compared to wild-type and p53−/− MEFs, the overall number is much lower after IR treatment (only 3–6%), indicating that MEFs do not rapidly undergo senescence after acute IR treatment ( and S3C). These results indicated that the p53R172P mutation responds to UV-induced DNA damage by progressively driving cells into senescence in a dose and time dependent manner, which explains the lower expansion rate after UV treatment in p53P/P MEFs. Collectively, these results revealed that p53R172P was activated in response to UV induced damage, but not gamma-irradiation, to drive cells into senescence, which is responsible for the slow growth of p53P/P MEFs.
p53R172P is unable to transactivate proapoptotic genes.
In response to DNA damage, p53 is stabilized and transactivates downstream targets to induce cellular responsive programs. To determine the difference between wild-type and mutant p53 in response to UVB irradiation, we analyzed the protein levels of known downstream apoptotic and cellular senescence markers 24, 48 and 72 hours post UVB irradiation. As shown in , S4 and S5, low levels of p53 were detected in non-treated MEFs as well as in MEFs that received 100 J/m2 of UVB. However, at 250 J/m2 the level of p53 in wild-type cells was increased initially (24h), but then decreased 48 and 72h post irradiation. In contrast, mutant p53P/P levels constantly remained high. Elevated expression of p53 corresponded to an increase in p21 levels in both wild-type and p53P/P MEFs after UVR at 250 J/m2 (Fig. S4). A slightly reduced level of p21 is also noticed in the mutant MEFs when compared to wild-type, which is consistent with previous reports of p53P/P mutant cells.Citation26,Citation31 Significantly, UVR upregulated the levels of proapoptotic proteins PUMA in wild-type MEFs, while PUMA levels, as well as another p53-dependent proapoptotic protein NOXA, remain very low in p53P/P cells (), which is consistent with these mutant cells being unable to trigger p53-mediated apoptosis. Therefore, deficient apoptosis in p53P/P is at least partially due to the mutant p53 being unable to transactivate proapoptotic factors that are important for inducing apoptosis. Additionally, anti-apoptotic protein Bcl-2 levels decreased in wild-type after UVB treatment, while in the mutant cells, Bcl-2 levels remained high and only slightly decreased at 72h in p53P/P. Additional apoptoti markers were analyzed between genotypes, which confirmed our results and ruled out the involvement of Bax and Bak proteins (Fig. S4) since these levels are not changed before or after the UVB treatment. These results demonstrated that the mutant p53 is unable to downregulate prosurvival protein Bcl-2 in response to UVB exposure, which is essential for the induction of apoptosis.
Discussion
Tumor suppressor p53 plays a central role in mediating cellular responses after UVR.Citation20,Citation39 Our study demonstrates that the p53R172P mutation leads to UVB radiation hypersensitivity both in vivo and in vitro. In the absence of p53-dependent apoptosis, mutant mice showed much less tolerance to UVB radiation. At very low doses, mutant mice suffered more severe skin swelling and immunosuppression compared to the wild-type mice. However, there is no evidence of changes in repairing UVR-induced DNA damage in p53P/P mice, indicating that the increased susceptibility results from UVR-induced cellular responses.
Beside being a direct DNA damaging agent that causes genomic instability and mutagenesis, UVR also induces immunosuppression, which has been identified as a major risk factor for cancer induction.Citation40 It is well known that UV radiation causes specific immune suppression resulting in susceptibility to UV-induced skin tumors.Citation41,Citation42 Interestingly, p53P/P mice were much more susceptible to the immunosuppressive effects of UV radiation, in that significant immune suppression was observed at very low doses, doses of UVR that did not suppress DTH in wild-type mice. These results differ from those found in p53−/− mice, which did not display increased susceptibility to UVB-induced immune suppression compared to wild-type mice.Citation22 Since it is known that different strains of mice exhibit different levels of susceptibility to UV-induced immune suppression,Citation43,Citation44 it is possible that the strain of mice utilized in our study (C57BL/6 129/Sv mix) could explain the discrepancy between the results observed in p53−/− and p53R172P mice. However, this explanation is unlikely because the littermate controls in our study did not exhibit the same sensitivity as p53R172P mice. Because we did not observe defects in the repair of UV-induced DNA damage and/or other genetic alterations in Ras, p16INK4a and p19ARF genes in the p53P/P mice (data not shown), we conclude that the inability to repair DNA damage did not contribute to the increased susceptibility of p53P/P mice to UV carcinogenesis. Instead, the mutation on p53 leading to the lack of apoptosis and the strong induction of cell cycle arrest and senescence may be the main contributor to hypersensitivity to UVB. Consistent with this, a recent study linked chronic UV exposure to premature senescence in human skin fibroblasts,Citation23 which may have direct impact on UV induced photoaging. Further research is required to investigate the role of p53-dependent apoptosis in UV-induced inflammation and immunosuppression. Mutations in the p53 gene that may be linked to immune suppression are also directly connected to tumorigenesis, an area not yet well explored.
UV-induced DNA damage can be repaired by the NER pathway,Citation32 or alternatively, cells with severely damaged DNA undergo apoptosis. Tumor suppressor p53 plays a central role in both processes.Citation45 It can directly participate in DNA damage repair by NERCitation46,Citation47 and has a known function to mediate cell cycle arrest to provide time for cells to repair damaged DNA before re-entering the cell cycle. It is also required for inducing programmed cell death after DNA damage. Unlike p53-deficient fibroblasts, which are defective in gross NER, our study showed that the mutant p53P/P cells have a normal ability to repair DNA after UVR, indicating that DNA damage per se is not the sole cause of sensitivity to UVR. It is not completely understood how p53 participates during the NER process. Several lines of evidence show that p53 may directly bind to NER helicase factors XPD or XPB,Citation48 or function in NER via regulating gene expression, such as XPC or XPE genesCitation49,Citation50 as well as transactivating genes that encode for the initiation of global genome repair.Citation51 Our study indicates that the hypomorphic mutant p53R172P retains at least partial function in terms of repairing UV-induced DNA damage.
Our study reveals that two p53-dependent events are important for the induction of apoptosis after UVB exposure: upregulation of pro-apoptotic genes such as PUMA and Noxa and downregulation of the anti-apoptotic gene Bcl-2, through the inactivation of the transcriptional factor E2F1.Citation52,Citation53 The hypomorphic mutant p53 is defective in both events. These results are consistent with a previous report that loss of PUMA or Noxa renders MEFs partially resistant to UV-induced cell death.Citation25 Overexpression of Bcl-2 also inhibits UV-induced apoptosis, which mimics the mutant p53P/P MEFs after UVR. Interestingly, downregulation of Bcl-2 by RNA interference restores the apoptosis response after UVB exposure in p53−/− fibroblasts,Citation52 indicating that Bcl-2 plays a critical role in deciding the cell fate.
Reports showed that lack of apoptosis in PUMA−/− or NOXA−/− renders cells resistant to UV exposure as indicated by their higher survival rate compared to the wild-type.Citation25 This is not consistent with our results, which showed that p53P/P MEFs grew slower than the wild-type after UVB exposure. We consider that lack of apoptosis drives cells to senescence after DNA damage, which significantly slows down cell growth. A high expression of p21, a strong cell cycle kinase inhibitor and a typical downstream target of p53, was found in these mutant cells. Evidence indicated that p21 is a key component that drives cells into senescence.Citation54 However, it is intriguing to observe that p53R172P failed to transactivate PUMA/Noxa but retains the ability to transactivate p21 to almost normal levels. Although we don't know the mechanism, the clue maybe in the nature of promoters that p53 regulates. Morachis and colleagues recently showed that the kinetics of genes involved in cellular responses are expressed differently, cell cycle regulator promoters are “preloaded” and can be activated much faster than the apoptotic molecules which need to gather and activate the pre-initiation complex.Citation55 This raises interesting possibilities in regard to the inability of the mutant p53 to transactivate certain promoters due to either steric hindrance presented by the point mutation in the DNA binding domain or perhaps a lack of consensus sequences needed to bind properly. Regardless, further studies are needed to understand exactly why p53R172P cannot transactivate apoptotic molecules but can partially activate cell cycle regulators, which our data suggests is the main reason p53P/P cells undergo cellular senescence.
It is also interesting to observe that UVR exposure induces MEFs into apoptosis unlike IR exposure. UVR and IR exposure inflict different types of DNA damage, thus triggering a different response upstream of p53. UV mainly causes modification of chemical bonds on the DNA bases, especially on pyrimidine bases to form dimers or (6-4) photoproducts between C-T bases; such damage will cause distortions in the chromatin structure that might accentuate certain cellular responses that are very different from the DNA double strand breaks that IR causes. ATR and ATM are activated by different kinds of DNA damage, although they share some overlapping downstream targets in cell cycle checkpoint activation. ATM senses double-stranded DNA ends and ATR senses single-stranded DNA bound with RPA.Citation56 ATR is activated slowly in comparison to ATM, which is mainly due to the DNA breaks in those cells being recognized during the S and G2 phase of the cell cycle. Damage to genomic DNA by UVB can be sensed by ATR,Citation57 which mediates a signaling pathway via Chk1, to trigger cell cycle arrest, DNA repair, senescence or apoptosis. Relevant to this study, UV and IR radiation leads to different post-translational modifications to p53, most noticeably UV leads to phosphorylation at Ser389, while IR does not. This results in activating different downstream effectors. Interestingly, p53S389A mutant mice are more susceptible to tumors after chronic UV exposure but not after IR exposure.Citation58 Therefore, different types of DNA damage can modify p53 at different sites to trigger specific cellular responses.
The cellular response to UVR is also cell type-dependent.Citation59 Recent evidence showed that engagement of ATR-ATRIP with the TOPBP1 molecule triggers a DNA damage response and induces cells to undergo senescence in a p53-dependent fashion.Citation60 Toledo and co-workers found that a short transient signaling resulted in cell cycle arrest, whereas longer persistent signaling led to senescence. In our study, UVR treatment leads to apoptosis in wild-type mice in a dose-dependent fashion, but induced senescence in an apoptosis-deficient mutant. These results indicated that UVB may activate more than the TOPBP1-dependent pathway and these alternate pathways may be required for triggering apoptosis. Another mutant p53 mouse was reported recently where deletion of the proline rich domain of p53, important for the induction of apoptosis, eliminates the apoptosis pathway but not cell cycle arrest.Citation61 This mutation changes the tumor spectrum, where the mutant mice exhibit abnormal B lymphocyte development and succumb to B cell lymphoma in comparison with p53 null mice, which succumb to mostly thymic lymphomas. Most importantly, our work suggests that p53-dependent apoptosis, along with DNA repair and cell cycle arrest in response to UVR, work together to control the proliferation of cells and maintain the genomic integrity. These responses are necessary to ensure cell homeostasis. A deficiency in one component, in our case, the apoptotic pathway, may lead to unbalanced cell homeostasis that might propagate into pathogenic consequences if not suppressed.
Our results demonstrated that a p53-dependent cellular response to UVR plays a major role in influencing the rate of cell proliferation. Apoptosis is an essential physiological process that is important for maintaining homeostasis of cells and for eliminating damaged cells to maintain genomic integrity. Our results also indicate that in the absence of p53-dependent apoptosis, cells may undergo senescence, a process that inhibits cell growth, which is also essential for preventing genomic instability. In parallel, senescence may also be connected with hypersensitivity to UVB and immunosuppression and possibly aging. Our study is a clear example of p53-dependent cellular responses, rather than DNA repair activity per se, which plays a dominant role in regulating cell growth and proliferation. Furthermore, abnormal functions of p53 due to mutations could introduce adverse effects to the cell or organism even in the presence of normal DNA damage repair.
Materials and Methods
Animals.
Mice maintenance has been previously described.Citation38 The protocols were approved by the Institutional Animal Care and Use Committee at The University of Texas M.D. Anderson Cancer Center.
Acute irradiation.
For acute studies, mice were shaved on the dorsal area and irradiated once at indicated doses from a 137Cs γ-ray source (JLS Shepard & Associates, Glendale, CA), or UV using four FS40 sunlamps (National Biological, Twinsburg, OH).Citation30,Citation62 The UVB output of the sunlamps was measured with an IL 1700 radiometer. At indicated time points post irradiation, the dorsal skin fold was measured with calipers and skin samples from areas of irradiation were frozen in liquid nitrogen and a portion fixed for further studies. DNA was extracted from the frozen samples. Radioimmunoassay was performed to determine the extent of cyclopyrimidine dimer and (6-4) photoproduct formation.Citation33
Immune suppression.
To measure the effect of UVB on the immune response, a delayed-type hypersensitivity reaction was used, as described.Citation30 Mice were shaved on the dorsal side and the following day they were given indicated amounts of UVB radiation. Five days later they were immunized with formalin-fixed Candida albicans. Delayed-type hypersensitivity, as determined by an increase in footpad swelling in response to a challenge with C. albicans-specific antigen (Alerchek, Portland, ME), was measured 10 days post immunization. The mean change in footpad thickness (left foot + right foot ÷ 2) was calculated for each animal in the group (N = 5). The change in thickness ± the SEM was then calculated for the group.
Skin histology and immunofluorescence.
Skin was fixed in Bouin solution (Ricca Chemical Company) overnight, dehydrated through ethanol and embedded in paraffin for histological analysis. Sections (6 µm) were incubated with antibodies against p53 (Cell Signaling) and p21 (Santa Cruz). Secondary antibodies were applied according to the manufacturer's recommendations and incubated with Vectashield Hard Set with DAPI (Vector). Slides were analyzed by fluorescent microscopy (Olympus BX41). Additionally, a transferase-mediated dUTP nick-end labeling (TUNEL) assay kit (Promega) was used to detect apoptosis with DNase-1 treated sections as positive controls.Citation38
Cellular senescence.
Senescence-associated β-galactosidase (SA-β-gal) activity was detected in the skin by a staining kit (Cell Signaling). SA-β-gal activity in MEFs was detected as described.Citation63 Slides were subjected to hematoxylin-eosin (H&E) staining and examined by light microscopy (Olympus BX41). MEFs positive for SA-β-gal activity were scored against total cells using ImageJ (National Institutes of Health, Bethesda, MD).
Cell culture and UV irradiation.
Murine embryonic fibroblasts ∼day 13.5 were grown in Dulbecco's modified Eagle's medium (Mediatech) supplemented with 10% fetal bovine serum (SAFC Biosciences) and 100 U/mL penicillin (Cambrex). Cells were plated in 10 cm dishes in an incubator at 37°C with 5% CO2. An FS40 sunlamp was used for irradiation; UV output was measured with an IL 1700 radiometer. Upon subconfluency, cells were exposed to different UVB doses in a thin layer of PBS. Control cells were kept in the same culture conditions without UVB.
Radiation sensitivity assay.
Briefly, MEFs were seeded onto 6 cm dishes and later irradiated with differing dosages of ionizing radiation by using a 137Cs γ-ray source. After irradiation, the medium was replaced and cultures were incubated for 7 days at 37°C with a change in media at day 3. At the end of the assay, MEFs were trypsinized and viable cells were counted. Additionally, MEFs were plated onto 10 cm dishes and irradiated with UVB as described above and viability was assessed 3 days after irradiation.
Apoptosis and cell cycle analysis.
MEFs were irradiated either with ionizing radiation or UVB as stated above and collected at indicated time points. Apoptosis was analyzed using a FITC-Annexin V Apoptosis detection kit (BD Pharmingen). Data were collected using a flow cytometer (FACScalibur, Becton Dickinson) and analyzed by FlowJo software (Tree Star, OR).
Western blot analysis.
Cells were irradiated as stated above and collected in lysis buffer. 40 µg of protein was loaded onto a 10% polyacrylamide gel. After transfer, the PVDF membrane (Biorad) was incubated with antibodies for p53, PUMA (Cell Signaling), p21 (BD Pharmingen), PARP and Noxa (Abcam), Bcl-2 and Bax (Santa Cruz), Bak (Calbiochem) and β-Actin (Thermo Scientific). Horseradish peroxidase conjugated secondary antibodies were applied per the manufacturers' recommendations. Signals were detected with an enhanced chemiluminescence kit (Perkin Elmer).
Statistical analysis.
Results are presented as the mean ± SEM. Differences were determined using a two-tailed, unpaired Student t test with CI of 95%. A p value less than 0.05 was denoted as statistically significant (GraphPad Prism Software, GraphPad, San Diego, CA).
Figures and Tables
Figure 1 Mutant p53P/P mice are hypersensitive to UVB radiation. (A) Mice were shaved and irradiated with 4.5 kJ/m2 of UVB at 0 and 48 h. Skin swelling was determined by measuring double skin-fold thickness of the dorsal skin at indicated time points. N = 5 per group. Data are the mean ± SEM, *indicates p < 0.05 vs. wild-type. (B) Representative H&E counterstaining of irradiated dorsal skin sections after indicated time points. Magnification, 20X. (C) UVB induced suppression of DTH. Mice were treated with UVB and later were immunized with Candida albicans. Hind footpads were measured 10 days later then challenged with an injection of Candida antigen. DTH was measured by footpad swelling 24 hours post-challenge. Negative controls were measured in mice not immunized but challenged. Positive controls were non-irradiated, but immunized and challenged mice. N > 5 per group. Data are the mean ± SEM, *indicates p < 0.005 vs. wild-type.
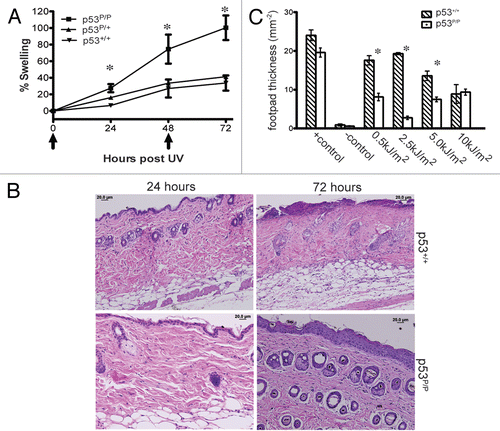
Figure 2 Induction of p53/p21 induced cellular senescence in p53R172P skin after UVB. (A) Representative skin sections from p53P/P, p53P/+ and p53+/+ mice 24 and 72 hours after 5 kJ/m2 of UVB, (top and middle panels), or 72 hours post 10 Gy of IR (bottom panels). All samples were stained with SA-β-gal followed by H&E counterstaining. Magnification, 20X. (B) Representative immunofluorescent staining of skin sections from p53P/P and p53+/+ mice; p53 (green) and p21 (red). Magnification, 40X.
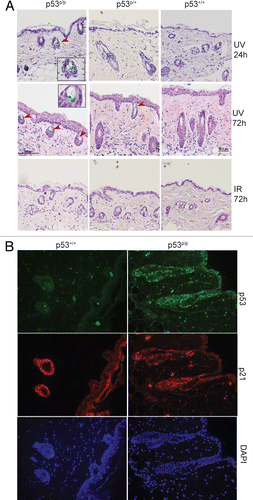
Figure 3 p53R172P MEFs are specifically hypersensitive to UVR but not IR irradiation. (A) Growth curves of p53+/+, p53P/P and p53−/− MEFs after different doses of UVB. (B) Growth curves after different doses of IR. Survival is determined by the percentage of viable cells at each treated dose over the total viable cells in untreated conditions. Representative results are presented; each experiment was repeated independently more than 5 times. Data are the mean ± SEM, *indicates p < 0.001 vs. wild-type. (C) DNA was extracted from MEFs 0, 6, 24 and 48 h post UVB exposure (200 J/m2). Photoproducts were measured by RIA using antibodies specific for CPDs or (6-4) photoproducts (6-4 PP). Representative results are presented; each experiment was repeated independently more than 3 times. (D) DNA from dorsal skin of mice 0, 6 and 24 h post UVB exposure (5 kJ/m2); photoproducts were measured as described above. N = 5 mice per time point.
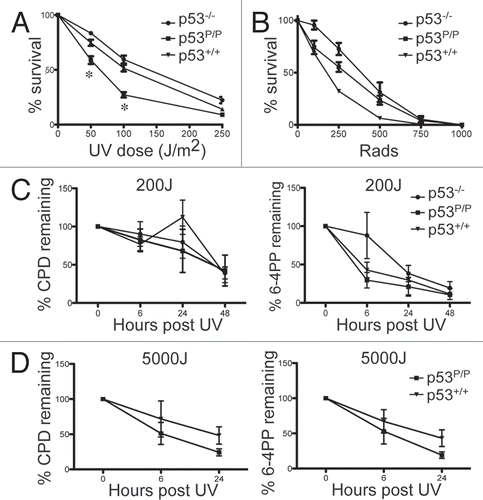
Figure 4 Wild-type MEFs undergo apoptosis while p53R172P MEFs trigger cellular senescence after UVB. (A) Cells were harvested 48 hours post UVR (100 and 250 J/m2) and apoptosis was measured using Annexin V and PI staining followed by FACS analysis. Error bars represent mean ± SEM. Differences determined using a two-tailed, unpaired student t-test. *indicates p < 0.001 vs. wild-type. (B) FACS profiles after Annexin V staining from different genotypes 48 h after UVR (100 and 250 J/m2). Representative results are presented; each experiment was repeated independently 3 times. (C) Quantification of SA-β-gal positive cells 48 h after UVB (100 and 250 J/m2). (D) Quantification of SA-β-gal 48 h after IR (6 and 10 Gy). Error bars represent mean ± SEM. *indicates p < 0.005 vs. wild-type. (e) p53P/P MEFs, untreated (upper part) and those exposed to 100 J/m2 of UVB radiation (lower part) were stained with SA-β-gal and counterstained with H&E. Magnification 40x. (F) Western blot analyses of p53, p21, PUMA, Noxa and Bcl-2 in p53+/+ and p53P/P MEFs that were exposed to 100 J/m2 of UVB and harvested 24, 48 and 72 h post irradiation, using untreated p53−/− MeFs as a control, β-actin is shown as a loading control.
Additional material
Download Zip (23.4 MB)Acknowledgements
The authors would like to thank Dr. G. Lozano for the p53P/P mouse, Dr. David L. Mitchell, for his help in analyzing the repair of UV-induced DNA lesions and Peter Wolf, M.D. for his assistance determining the skin fold thickness. Supported by the American Cancer Society-Bonnie Kies Foundation (to C.Z.) and Grants CA116933 (to C.Z.), CA 112660 and CA131207 (to S.E.U.) from the National Cancer Institute and Grant ES07784 from the National Institute of Environmental Health Sciences. C.L.B. is supported in part by Grant P50 CA093459 from the National Institutes of Health.
References
- Vousden KH, Prives C. Blinded by the light: The growing complexity of p53. Cell 2009; 137:413 - 431
- Meek DW. Tumour suppression by p53: a role for the DNA damage response?. Nat Rev Cancer 2009;
- Roos WP, Kaina B. DNA damage-induced cell death by apoptosis. Trends Mol Med 2006; 12:440 - 450
- Zuckerman V, Wolyniec K, Sionov RV, Haupt S, Haupt Y. Tumour suppression by p53: the importance of apoptosis and cellular senescence. J Pathol 2009; 219:3 - 15
- Morachis JM, Murawsky CM, Emerson BM. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev 2009; 24:135 - 147
- Cheng Q, Chen J. Mechanism of p53 stabilization by ATM after DNA damage. Cell Cycle 2010; 9
- Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer 2009; 9:400 - 414
- Harper JW, Adami GR, Wei N, Keyomarsi K, Elledge SJ. The p21 Cdk-interacting protein Cip1 is a potent inhibitor of G1 cyclin-dependent kinases. Cell 1993; 75:805 - 816
- Campisi J. Senescent cells, tumor suppression and organismal aging: good citizens, bad neighbors. Cell 2005; 120:513 - 522
- Van Nguyen T, Puebla-Osorio N, Pang H, Dujka ME, Zhu C. DNA damage-induced cellular senescence is sufficient to suppress tumorigenesis: a mouse model. J Exp Med 2007; 204:1453 - 1461
- Junttila MR, Evan GI. p53—a Jack of all trades but master of none. Nat Rev Cancer 2009; 9:821 - 829
- Felsher DW. Oncogene addiction versus oncogene amnesia: perhaps more than just a bad habit?. Cancer Res 2008; 68:3081 - 3086
- Negrini S, Gorgoulis VG, Halazonetis TD. Genomic instability—an evolving hallmark of cancer. Nat Rev Mol Cell Biol 2010; 11:220 - 228
- Sinha RP, Hader DP. UV-induced DNA damage and repair: a review. Photochem Photobiol Sci 2002; 1:225 - 236
- Sancar A, Lindsey-Boltz LA, Unsal-Kacmaz K, Linn S. Molecular mechanisms of mammalian DNA repair and the DNA damage checkpoints. Annu Rev Biochem 2004; 73:39 - 85
- Halliday GM. Inflammation, gene mutation and photoimmunosuppression in response to UVR-induced oxidative damage contributes to photocarcinogenesis. Mutat Res 2005; 571:107 - 120
- Yarosh DB. DNA repair, immunosuppression and skin cancer. Cutis 2004; 74:10 - 13
- Hussein MR. Ultraviolet radiation and skin cancer: molecular mechanisms. J Cutan Pathol 2005; 32:191 - 205
- Benjamin CL, Ananthaswamy HN. p53 and the pathogenesis of skin cancer. Toxicol Appl Pharmacol 2007; 224:241 - 248
- Latonen L, Laiho M. Cellular UV damage responses—functions of tumor suppressor p53. Biochim Biophys Acta 2005; 1755:71 - 89
- Ziegler A, Jonason AS, Leffell DJ, Simon JA, Sharma HW, Kimmelman J, et al. Sunburn and p53 in the onset of skin cancer. Nature 1994; 372:773 - 776
- Jiang W, Ananthaswamy HN, Muller HK, Ouhtit A, Bolshakov S, Ullrich SE, et al. UV irradiation augments lymphoid malignancies in mice with one functional copy of wild-type p53. Proc Natl Acad Sci USA 2001; 98:9790 - 9795
- Chen W, Kang J, Xia J, Li Y, Yang B, Chen B, et al. p53-related apoptosis resistance and tumor suppression activity in UVB-induced premature senescent human skin fibroblasts. Int J Mol Med 2008; 21:645 - 653
- Borlon C, Vankoningsloo S, Godard P, Debacq-Chainiaux F, Toussaint O. Identification of p53-dependent genes potentially involved in UVB-mediated premature senescence of human skin fibroblasts using siRNA technology. Mech Ageing Dev 2008; 129:109 - 119
- Naik E, Michalak EM, Villunger A, Adams JM, Strasser A. Ultraviolet radiation triggers apoptosis of fibroblasts and skin keratinocytes mainly via the BH3-only protein Noxa. J Cell Biol 2007; 176:415 - 424
- Liu G, Parant JM, Lang G, Chau P, Chavez-Reyes A, El-Naggar AK, et al. Chromosome stability, in the absence of apoptosis, is critical for suppression of tumorigenesis in Trp53 mutant mice. Nat Genet 2004; 36:63 - 68
- Kripke ML, Lofgreen JS, Beard J, Jessup JM, Fisher MS. In vivo immune responses of mice during carcinogenesis by ultraviolet irradiation. J Natl Cancer Inst 1977; 59:1227 - 1230
- Kripke ML. Immunology of UV-induced skin cancer. Photochem Photobiol 1980; 32:837 - 839
- Kripke ML, Cox PA, Alas LG, Yarosh DB. Pyrimidine dimers in DNA initiate systemic immunosuppression in UV-irradiated mice. Proc Natl Acad Sci USA 1992; 89:7516 - 7520
- Nghiem DX, Walterscheid JP, Kazimi N, Ullrich SE. Ultraviolet radiation-induced immunosuppression of delayed-type hypersensitivity in mice. Methods 2002; 28:25 - 33
- Barboza JA, Liu G, Ju Z, El-Naggar AK, Lozano G. p21 delays tumor onset by preservation of chromosomal stability. Proc Natl Acad Sci USA 2006; 103:19842 - 19847
- Nouspikel T. DNA repair in mammalian cells: Nucleotide excision repair: variations on versatility. Cell Mol Life Sci 2009; 66:994 - 1009
- Mitchell DL. Quantification of photoproducts in mammalian cell DNA using radioimmunoassay. Methods in molecular biology 2006; 314:Clifton, NJ 239 - 249
- Zhang G, Njauw CN, Park JM, Naruse C, Asano M, Tsao H. EphA2 is an essential mediator of UV radiation-induced apoptosis. Cancer Res 2008; 68:1691 - 1696
- Rowan S, Ludwig RL, Haupt Y, Bates S, Lu X, Oren M, et al. Specific loss of apoptotic but not cell cycle arrest function in a human tumor derived p53 mutant. EMBO J 1996; 15:827 - 838
- Vermes I, Haanen C, Steffens-Nakken H, Reutelingsperger C. A novel assay for apoptosis. Flow cytometric detection of phosphatidylserine expression on early apoptotic cells using fluorescein labelled Annexin V. J Immunol Methods 1995; 184:39 - 51
- Peters AC, Young LC, Maeda T, Tron VA, Andrew SE. Mammalian DNA mismatch repair protects cells from UVB-induced DNA damage by facilitating apoptosis and p53 activation. DNA Repair (Amst) 2003; 2:427 - 435
- Tavana O, Puebla-Osorio N, Sang M, Zhu C. Absence of p53-dependent apoptosis combined with nonhomologous end-joining deficiency leads to a severe diabetic phenotype in mice. Diabetes 2009;
- Lavin MF, Gueven N. The complexity of p53 stabilization and activation. Cell Death Differ 2006; 13:94150
- Beissert S, Loser K. Molecular and cellular mechanisms of photocarcinogenesis. Photochem Photobiol 2008; 84:29 - 34
- Kripke ML. Immunologic mechanisms in UV radiation carcinogenesis. Adv Cancer Res 1981; 34:69 - 106
- Kripke ML, Morison WL. Modulation of immune function by UV radiation. J Invest Dermatol 1985; 85:62 - 66
- Hart PH, Grimbaldeston MA, Swift GJ, Jaksic A, Noonan FP, Finlay-Jones JJ. Dermal mast cells determine susceptibility to ultraviolet B-induced systemic suppression of contact hypersensitivity responses in mice. J Exp Med 1998; 187:2045 - 2053
- Noonan FP, Hoffman HA. Susceptibility to immunosuppression by ultraviolet B radiation in the mouse. Immunogenetics 1994; 39:29 - 39
- Chang YC, Jan KY, Cheng CA, Liao CB, Liu YC. Direct involvement of the tumor suppressor p53 in nucleotide excision repair. DNA Repair (Amst) 2008; 7:751 - 761
- Ford JM, Hanawalt PC. Li-Fraumeni syndrome fibroblasts homozygous for p53 mutations are deficient in global DNA repair but exhibit normal transcription-coupled repair and enhanced UV resistance. Proc Natl Acad Sci USA 1995; 92:8876 - 8880
- Ford JM, Baron EL, Hanawalt PC. Human fibroblasts expressing the human papillomavirus E6 gene are deficient in global genomic nucleotide excision repair and sensitive to ultraviolet irradiation. Cancer Res 1998; 58:599 - 603
- Wang XW, Yeh H, Schaeffer L, Roy R, Moncollin V, Egly JM, et al. p53 modulation of TFIIH-associated nucleotide excision repair activity. Nat Genet 1995; 10:188 - 195
- Adimoolam S, Ford JM. p53 and DNA damage-inducible expression of the xeroderma pigmentosum group C gene. Proc Natl Acad Sci USA 2002; 99:12985 - 12990
- Hwang BJ, Ford JM, Hanawalt PC, Chu G. Expression of the p48 xeroderma pigmentosum gene is p53-dependent and is involved in global genomic repair. Proc Natl Acad Sci USA 1999; 96:424 - 428
- Helton ES, Chen X. p53 modulation of the DNA damage response. J Cell Biochem 2007; 100:883 - 896
- Knezevic D, Zhang W, Rochette PJ, Brash DE. Bcl-2 is the target of a UV-inducible apoptosis switch and a node for UV signaling. Proc Natl Acad Sci USA 2007; 104:11286 - 11291
- Wikonkal NM, Remenyik E, Knezevic D, Zhang W, Liu M, Zhao H, et al. Inactivating E2f1 reverts apoptosis resistance and cancer sensitivity in Trp53-deficient mice. Nat Cell Biol 2003; 5:655 - 660
- Jung YS, Qian Y, Chen X. Examination of the expanding pathways for the regulation of p21 expression and activity. Cell Signal 2010;
- Morachis JM, Murawsky CM, Emerson BM. Regulation of the p53 transcriptional response by structurally diverse core promoters. Genes Dev 24:135 - 147
- Cimprich KA, Cortez D. ATR: an essential regulator of genome integrity. Nat Rev Mol Cell Biol 2008; 9:616 - 627
- Unsal-Kacmaz K, Makhov AM, Griffith JD, Sancar A. Preferential binding of ATR protein to UV-damaged DNA. Proc Natl Acad Sci USA 2002; 99:6673 - 6678
- Bruins W, Zwart E, Attardi LD, Iwakuma T, Hoogervorst EM, Beems RB, et al. Increased sensitivity to UV radiation in mice with a p53 point mutation at Ser389. Mol Cell Biol 2004; 24:8884 - 8894
- D'Errico M, Lemma T, Calcagnile A, Proietti De Santis L, Dogliotti E. Cell type and DNA damage specific response of human skin cells to environmental agents. Mutat Res 2007; 614:37 - 47
- Toledo LI, Murga M, Gutierrez-Martinez P, Soria R, Fernandez-Capetillo O. ATR signaling can drive cells into senescence in the absence of DNA breaks. Genes Dev 2008; 22:297 - 302
- Slatter TL, Ganesan P, Holzhauer C, Mehta R, Rubio C, Williams G, et al. p53-mediated apoptosis prevents the accumulation of progenitor B cells and B-cell tumors. Cell Death Differ 2010; 17:540 - 550
- Sreevidya CS, Fukunaga A, Khaskhely NM, Masaki T, Ono R, Nishigori C, et al. Agents that reverse UV-induced immune suppression and photocarcinogenesis affect DNA repair. J Invest Dermatol 2010; 130:1428 - 1437
- Dimri GP, Lee X, Basile G, Acosta M, Scott G, Roskelley C, et al. A biomarker that identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 1995; 92:9363 - 9367