Abstract
Cellular senescence is an irreversible state of terminal growth arrest that requires functional p53. Acting to block tumor formation, induction of senescence has also been demonstrated to contribute to tumor clearance via the immune system following p53 reactivation.1, 2 The Hdm2-antagonist, Nutlin-3a, has been shown to reactivate p53 and induce a quiescent state in various cancer cell lines,3, 4 similar to the G1 arrest observed upon RNAi targeting of Hdm2 in MCF7 breast cancer.5 In the present study we show that HdmX, a negative regulator of p53, impacts the senescence pathway. Specifically, overexpression of HdmX blocks Ras mediated senescence in primary human fibroblasts. The interaction of HdmX with p53 and the re-localization of HdmX to the nucleus through Hdm2 association appear to be required for this activity. Furthermore, inhibiting HdmX in prostate adenocarcinoma cells expressing wild-type p53, mutant Ras and high levels of HdmX induced cellular senescence as measured by an increase in irreversible b-galactosidase staining. Together these results suggest that HdmX overexpression may contribute to tumor formation by blocking senescence and that targeting HdmX may represent an attractive anti-cancer therapeutic approach.
Introduction
Cellular senescence is a physiological program of terminal growth arrest. Both replicative (telomere shortening) and premature (stress induced) senescence are crucial anti-tumor processes that require a functional p53 pathway.Citation6 Not only is senescence critical to normal cellular aging and prevention of tumor formation, but also in the treatment of existing tumors. Induction of premature tumor cell senescence following treatment with genotoxic chemotherapeutics has been described.Citation7 Treatment-induced senescence has been associated with reactivation of the p53 pathway and is thought to reduce tumor volume via clearance of senescent cells by the immune system;Citation1,Citation2 therefore, senescence induction has become an attractive goal for anti-cancer therapy design.
The onset of oncogene-induced senescence is regulated by the tumor suppressor protein p53. This has been shown using mouse embryonic fibroblasts (MEFs) where oncogenic Ras and p53 cooperate to induce cellular senescence.Citation8 It has also been demonstrated that overexpression of MdmX, a homolog of Mdm2 that also represses p53 activity, can block oncogene induced senescence in mouse embryonic fibroblasts,Citation9 suggesting a role for MdmX in the senescence pathway.
Studies of senescence have been somewhat confounded by differences in the senescence pathway existing between human and mouse fibroblasts cell culture systems. While it appears that p53 and Rb are critical mediators of senescence induction and maintenance in both cell culture systems, other regulators of the senescence pathway are not as clearly defined. For example, the inactivation of p21 by homologous recombination is sufficient to prevent senescence in human cellsCitation10 while p21-null MEFs undergo senescence normally.Citation11 This difference implies that there is a missing link between the p53 and Rb pathways or other proteins can compensate for the loss of p21 in the mouse.
In the present study, we examined how deregulation of human MdmX (HdmX) impacted oncogene-induced senescence in primary non-transformed, human diploid fibroblasts and prostate cancer cells. In the approximately 50% of human cancers that retain wild-type p53, a significant percentage of these tumors overexpress HdmX.Citation12 We find that overexpression of HdmX in primary fibroblasts inhibits H-Ras induced cellular senescence. Using site-directed mutants, the binding of HdmX to p53 and Hdm2 both appear to be required for HdmX's inhibition of Ras-mediated senescence suggesting that both protein interactions may be critical for HdmX's contribution to tumor formation.
Given that HdmX blocks Ras-mediated senescence, we investigated whether reducing levels of HdmX in tumor cell lines harboring high HdmX, mutant Ras and wild-type p53 would lead to tumor cell senescence. Reduction of HdmX protein levels in LNCaP cells (high HdmX, mutant Ras, wild-type p53) led to an increase in irreversibly β-galactosidase positive cells and an increase in PAI-1, a senescence associated p53 target gene.Citation13 These findings demonstrate that release of p53 by inhibition of HdmX can trigger senescence in senescent capable human tumor cell lines and provide further support for targeting HdmX as an anticancer therapeutic.
Results
HdmX blocks Ras-mediated senescence in HDFs.
To investigate whether HdmX could block oncogenic Ras-induced premature senescence in human cells in a fashion similar to that observed by MdmX in MEFs, IMR90s (human diploid fibroblasts) were transduced with oncogenic Ras (H-RasG12V) with or without HdmX. Following infection, cells were grown for 7 days in complete media then subjected to acidic β-galactosidase staining, a hallmark of senescent cells. Co-infection of Ras and HdmX significantly reduced senescence levels compared to Ras and GFP indicating that HdmX is capable of blocking oncogene induced premature senescence in human diploid fibroblasts ( and B). We also monitored senescence induction following co-infection of H-Ras and Hdm2. This is because Mdm2 overexpression has been observed to overcome growth arrest when co-introduced with other senescence inducers, such as H-Ras.Citation14 As expected, a decrease in the percentage of β-galactosidase positive cells was observed when IMR90s were co-infected with Hdm2 and Ras ().
HdmX binding to p53 and Hdm2 are required to block Ras-mediated senescence.
Binding of MdmX to p53 is required for MdmX's ability to block p53 transactivation and MdmX has been shown to localize to the nucleus more effectively upon binding with Mdm2;Citation15 therefore, we next determined if the p53 and/or Hdm2 (Ring finger) binding domains of HdmX are necessary for the ability of HdmX to block Ras-mediated senescence. MdmXG57A has been reported by Danovi et al. to render MdmX unable to bind p53 while MdmXC437G was described by Sharp et al. as being unable to bind Mdm2.Citation9,Citation16 As these amino acids are conserved in HdmX, site-directed mutagenesis was completed to create HdmXG57A and HdmXC437G. Co-immunoprecipitation assays confirmed the ability of HdmXG57A to bind Hdm2 but not p53 while HdmXC437G retained the ability to bind p53 but not Hdm2, as predicted (Suppl. Fig. 1). When co-infected with Ras, neither HdmXG57A nor HdmXC437G were able to block oncogene-induced premature senescence ().
HdmX must bind directly to p53 in order to inhibit p53 transactivation, which likely explains why HdmXG57A, deficient in this ability, did not block Ras-mediated senescence. Since HdmXC437G retains the ability to bind p53 yet, when overexpressed, failed to block Ras-mediated senescence; we hypothesized that HdmXC437G was mis-localized due to its inability to bind Hdm2. To address this hypothesis, nuclear and cytoplasmic fractionation was completed using IMR90 cells that were infected with wild-type HdmX, HdmXG57A or HdmXC437G along with either GFP or Hdm2 (). Wild-type HdmX, HdmXG57A and HdmXC437G were all primarily cytoplasmic in the presence of GFP. The addition of Hdm2 allowed for nuclear translocation of HdmX and HdmXG57A, however, HdmXC437G remained primarily cytoplasmic. These results suggest that HdmXC437G is unable to block Ras-mediated senescence, at least in part, due to its inability to bind Hdm2 and localize to the nucleus.
Reduction of HdmX in LNCaP cells induces senescence.
Since HdmX can block oncogenic Ras-induced senescence in non-transformed human diploid fibroblasts, we examined whether knocking down HdmX in tumor cells with high HdmX, mutant Ras and wild-type p53 would lead to tumor cell senescence. Using LNCaP cells, a prostate adenocarcinoma cell line containing wild-type p53, high levels of HdmX and mutant Ras, we silenced HdmX using shRNA interference and examined senescence levels using β-galactosidase staining. Western blot analysis of HdmX protein levels showed that infection with any shRNA led to a slight reduction of HdmX protein although shHdmX infection reduced HdmX protein to an undetectable level (). This off-target HdmX reduction seen following transduction of any shRNA is likely due to a p53 mediated stress response upon shRNA infection given the increase in p53 protein seen under all shRNA infection conditions (). Interestingly, the HdmX reduction following sh-LacZ infection translated into only 20% β-galactosidase positive cells compared to 8% in the GFP infected cells (). However, targeted HdmX knockdown to nearly undetectable protein levels () caused a dramatic induction of senescence (60%) (). This induction of senescence was specifically due to lowering HdmX protein levels because rescue of HdmX protein levels lowered the percentage of senescent cells to below GFP virus infected levels (<8%).
Recently, Huang and colleagues induced positive β-galactosidase staining in epithelial tumor cells by treatment with Nutlin-3a, a small molecule inhibitor of Hdm2.Citation4 However, upon drug removal, some cells were able to resume proliferation as determined by colony formation assays. Senescence, by definition, is an irreversible process; therefore, these authors questioned the use of β-galactosidase staining as a sole measurement of senescence.
In order to determine if the β-galactosidase staining detected in LNCaP cells upon reduction of HdmX protein was reversible, we infected LNCaP cells with sh-HdmX as described in . Following 7 days of selection, instead of performing β-galactosidase staining, we elevated HdmX protein levels by reinfecting with an HdmX-rescue construct harboring a silent mutation that made the hdmX mRNA resistant to the hdmX short hairpin RNA. When β-galactosidase staining was completed on day 14, the percentage β-galactosidase positive cells was the same as sh-HdmX followed by GFP infection in the control condition (approximately 65%) (). We observed by reinfecting with lentiviral GFP that sh-HdmX selected cells, which have undergone senescence by day 7, can in fact be reinfected (). Also, HdmX was detected in sh-HdmX + HdmX-rescue co-infected cells at levels sufficient to block senescence ( and C). Finally, PAI-1 expression, a reported senescence-associated gene,Citation13 is elevated in LNCaP cells infected with shHdmX compared to shLacZ infected cells further suggesting that these cells have in fact been programmed for cellular senescence (). This data suggests that the increase in β-galactosidase positive LNCaP cells detected after lowering HdmX protein levels represents a permanent senescent state.
Discussion
Uncovering the specific genes and regulatory mechanisms responsible for the induction and maintenance of tumor cell senescence is an active area of research intended to guide the development of new anti-cancer therapeutics. At the nexus of senescence induction is the p53 tumor suppressor protein. This study describes how HdmX inactivation or mislocalization in cells can lead to the induction of cellular senescence.
Like MdmX in mouse embryonic fibroblasts, HdmX overexpression was demonstrated to block oncogenic Ras mediated senescence in human diploid fibroblasts (). Since the direct binding of HdmX to p53 and/or Hdm2 are known to play critical roles in the activity of HdmX, point mutations were made in HdmX aimed at inhibiting its ability to bind to either p53 or Hdm2 (HdmXG57A and HdmXC437G, respectively). When substituted for wild-type HdmX in combination with oncogenic Ras, neither mutant was able to block senescence suggesting that HdmX binding to p53 and Hdm2 are required to block oncogene induced senescence (). Nuclear cytoplasmic fractionation provided further evidence that HdmX binding to Hdm2 facilitates its nuclear localization, suggesting that the mis-localization of HdmX is, at least in part, responsible for HdmXC437G's inability to block senescence. While a recent publication details the first small molecule screen of inhibitors intended to block the interaction of HdmX with p53,Citation17 these results suggest that the development of small-molecules that trigger HdmX cytoplasmic localization or inhibit HdmX nuclear localization may also be effective at senescence induction in certain human tumor cell lines. Such inhibitors will likely need to target the HdmX:Hdm2 heterodimer RING finger domains given that disruption of this interaction has been reported to trigger p53 activation.Citation18,Citation19
Reports suggest that HdmX is overexpressed in several human tumor types, most of which maintain wild-type p53.Citation12 Approximately 40% of all tumor cell lines examined, including several prostate cancer cell lines, were found to overexpress HdmX. Taken with our data demonstrating that HdmX overexpression blocks Ras mediated senescence, we examined whether reduction of HdmX in a prostate adenocarcinoma cell line containing wild-type p53, mutant Ras and high levels of HdmX would induce senescence. The use of RNAi targeting HdmX induced senescence as indicated by an increase in irreversible β-galactosidase staining in these LNCaP cells.
When HdmX protein levels were elevated by co-infection of lentiviral HdmX-rescue with sh-HdmX expressing lentivirus, senescence was returned to baseline (), suggesting that the specific reduction of HdmX induces senescence in LNCaP cells. After we allowed HdmX reduction for 7 days, the re-expression of HdmX in LNCap cells was unable to inhibit cellular senescence (). These results suggest that even short term inhibition of HdmX may be sufficient clinically to trigger cellular senescence in the right genetic background. It should be noted that when similar experiments were performed in MCF7 breast cancer cell lines (wild-type p53 and Ras, high HdmX), we observed only cell cycle arrest,Citation5 without detectable cellular senescence (Heminger, personal communication). While we found this result initially perplexing, a recent publication suggests that simply inducing p53 may in fact suppress p21 and p16 induced cellular senescence.Citation20 Our studies suggest that tumors overexpressing HdmX that harbor mutant Ras and wild-type p53 maybe receptive to senescence induction.
HdmX as a target for anti-cancer drug design has been supported by a number of studies.Citation9,Citation18,Citation19,Citation21,Citation22 For example, treatment of tumors in mice that are overexpressing MdmX with siMdmX has been shown by Gilkes and colleagues to reduce tumor volume through an unknown mechanism.Citation21 This study provides data suggesting that HdmX overexpression may contribute to tumor formation by blocking senescence and that targeting HdmX in human tumors that retain wild-type p53 and harbor mutated Ras genes may lead to tumor cell senescence.
Materials and Methods
RT-PCR.
Total RNA was isolated using the e.Z.N.A. Total RNA kit (Omega Bio-Tek) according to the manufacturer's instructions. RNA quality and quantity was assessed using NanoDrop (Thermo Scientific) with a target 260:280 of 2.0–2.2. One microgram of total RNA was used as a template for cDNA synthesis using q-Script™ cDNA SuperMix (Quanta Biosciences). Taqman based PCR was performed in triplicate using Assay on Demand probe sets (Applied Biosystems) and an Applied Biosystems 7900 Sequence Detection System. GAPDH was used as an endogenous control. The Assay on Demand probe, Serpine1/PAI-1 used in these experiments was a validated gene target.
Cell lines.
H1299 cells are derived from non-small-cell lung carcinoma devoid of p53 and IMR90 cells are primary human diploid fibroblasts, both purchased from American Type Culture Collection. LNCaP cells are a prostate adenocarcinoma cell line (wild-type p53, high HdmX, mutant Ras) were a generous gift from Madhavi Kadakia. All cell line passages in these experiments were grown for no longer than three months.
Immunoblotting.
Whole cell extracts and western blotting were performed as previously described with the following modifications.Citation23 Cell pellets were lysed in three freeze-thaws using single lysis buffer (50 mM Tris, pH 8.0, 150 mM NaCl, 1% NP40) containing a protease inhibitor cocktail (Sigma). 12% SDS-PAGE gels were used to separate 50–100 micrograms of protein extract. Antibodies obtained from Calbiochem (p53, Ab-4 and Ab-6), Bethyl Laboratories, Inc., (HdmX), Invitrogen (GFP), Sigma (actin), Santa Cruz Biotechnology (Hdm2, SMP14; α-Tubulin, DM1A) and BD Biosciences (Lamin A/C) were used as indicated.
Lentiviral production and titering.
Lentivirus was produced by cotransfection of 293FT cells with a pLenti vector and lentiviral packaging mix (Invitrogen) according to the manufacturer's instructions. Lentivirus-containing supernatant was harvested at 48 hours post transfection, purified by centrifugation and stored at −80°C. For titering, H1299 cells (100,000 cells/well) were transduced with Lentivirus in a serial dilution pattern followed by incubation in complete medium for 24 hours. Infections were carried out overnight in the presence of 4 µg/mL Polybrene (Sigma). Following transductions, cells were selected with appropriate antibiotic selection for 15 days and then stained with 1% crystal violet in 70% methanol for 5 minutes. Cells were washed with PBS for 5 minutes, colonies counted and then photographed at 1X magnification. Infectious viral particles per mL (IVP/mL) were then calculated.
Senescence associated beta-galactosidase staining.
Cells were processed using a Senescence beta-galactosdiase Staining Kit (Cell Signaling Technology) according to the manufacturer's instructions and visualized on an Olympus 1X70 fluorescence microscope. At least 100 cells were counted and scored per treatment condition, which were completed in biological triplicate.
Nuclear cytoplasmic fractionation.
IMR90 cells were transduced and selected as indicated. After approximately 8 days the cells were rinsed and pelleted. Using reagents supplied in the Nuclear/Cytoplasmic Fractionation Kit (Pierce), nuclear and cytoplasmic fractions were generated. Protein concentration was determined by Bradford method and SDS-PAGE gel and immunoblotting completed as previously described.
Figures and Tables
Figure 1 p53- and -Hdm2 binding domains of HdmX are essential for blocking oncogenic Ras induced premature senescence. IMR90 cells were transduced at an MOI of 5 with the indicated lentivirus: (1) mock infection (Mock), (2) H-Ras (Ras), (3) GFP, (4) Ras + GFP, (5) Ras + HdmX, (6) Ras + Hdm2, (7) Ras + HdmXG57A (XG57A) or (8) Ras + HdmXC437G (XC437G), followed by incubation in complete media for 7 days at which time β-galactosidase staining was completed. (A) Histogram represents average ± SeM of % beta-galactosidase positive cells normalized to transduction efficiency (based on GFP). over 100 cells were counted per treatment and each infection was repeated in biological triplicate. (B) Representative images for infections as described in (A) are shown. Bright-field or fluorescence images were taken at same exposure time and 100X magnification. (C) Western blot demonstrating expression of the indicated proteins.
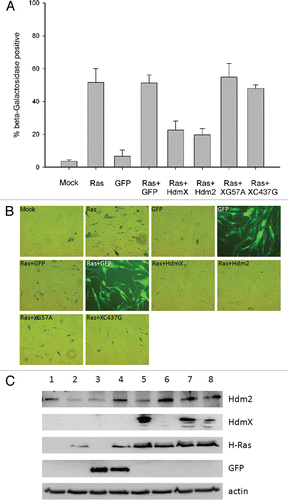
Figure 2 HdmX nuclear localization is dependent on association with Hdm2. IMR90 cells were transduced with lentivirus expressing wild-type HdmX, HdmXC437G or HdmXG57A in addition to GFP (left) or Hdm2 (right). Cells were selected and then nuclear and cytoplasmic protein extracts separated on an SDS-PAGE gel. Localization of Lamin A/C (a nuclear protein) and Tubulin (a cytoplasmic protein) confirm the extract fractionation was effective.
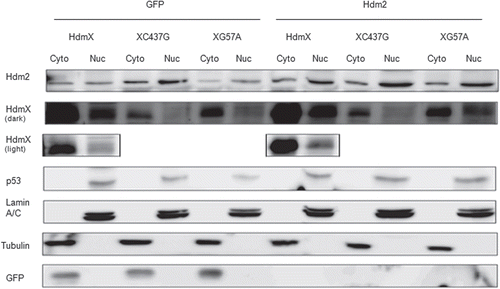
Figure 3 Loss of HdmX induces senescence in LNCaps. (A) LNCap cells were transduced with no virus (Mock), GFP, shLacZ (shL), HdmX, HdmX-Rescue (X/R), shHdmX (shX), shL + X/R, shX + X/R or shX + GFp, selected for 7 days and subjected to western blotting using the indicated antibodies. (B) Average % β-galactosidase positive cells normalized to transduction efficiency (based on GFP) ±SEM is reported where >100 cells were counted per treatment (same as in A) in three biological replicates, except Mock, which is in duplicate.
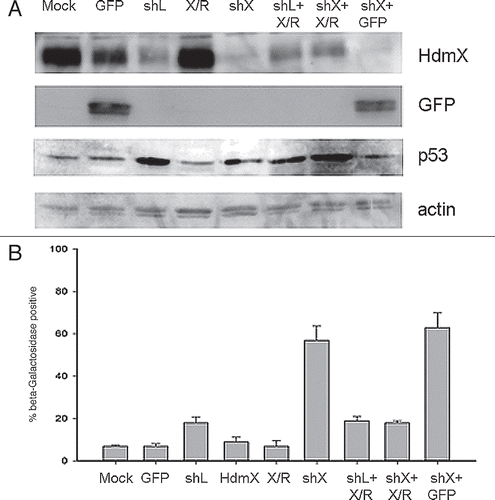
Figure 4 HdmX knockdown induces beta-galactosidase activity in LNCaps that cannot be reversed by HdmX overexpression post-senescence. LNCaps were transduced with no virus (Mock), GFP, shLacZ (shL), HdmX-Rescue (X/R), shL + X/R, shHdmX (shX) + X/R, shL, shX or shX and selection applied for 7 days. At day 7, shL infected cells were additionally infected with X/R (shL: X/R), shX infected cells with X/R (shX: X/R) and shX infected cells with GFP (shX: GFP). Cells were selected and incubated for an additional 7 days before β-galactosidase staining. (A) Average % beta-galactosidase positive cells normalized to transduction efficiency (based on GFP) ±SEM is reported where >100 cells were counted per treatment in biological triplicate. (B) Representative images of each treatment condition. All images taken at 100X magnification and the same exposure time for brightfield or fluorescence images. (C) Western blot confirming HdmX knock-down and rescue in additional treatment conditions not present in . (D) Rt-PCR demonstrating relative expression of PAI-1 normalized to GAPDH in shLacZ or shHdmX infected LNCaP cells at day 14. Bars represent 95% confidence intervals.
Additional material
Download Zip (81.3 KB)Acknowledgements
We would like to thank Mr. Maiorano for assistance with virus preparations, Dr. Madhavi Kadakia for the LNCaP cells and Dr. David Hitch for his input. These studies were supported by the National Cancer Institute (CA66430 to S.J.B.).
References
- Ventura A, Kirsch DG, McLaughlin ME, Tuveson DA, Grimm J, Lintault L, et al. Restoration of p53 function leads to tumour regression in vivo. Nature 2007; 445:661 - 665
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656 - 660
- Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle 2009; 8:3777 - 3781
- Huang B, Deo D, Xia M, Vassilev LT. Pharmacologic p53 activation blocks cell cycle progression but fails to induce senescence in epithelial cancer cells. Mol Cancer Res 2009; 7:1497 - 1509
- Heminger K, Markey M, Mpagi M, Berberich SJ. Alterations in gene expression and sensitivity to genotoxic stress following HdmX or Hdm2 knockdown in human tumor cells harboring wild-type p53. Aging 2009; 1:89 - 108
- Campisi J. Senescent cells, tumor suppression and organismal aging: Good citizens, bad neighbors. Cell 2005; 120:513 - 522
- Roninson IB. Tumor cell senescence in cancer treatment. Cancer Res 2003; 63:2705 - 2715
- Ferbeyre G, de Stanchina E, Lin AW, Querido E, McCurrach ME, Hannon GJ, et al. Oncogenic ras and p53 cooperate to induce cellular senescence. Mol Cell Biol 2002; 22:3497 - 3508
- Danovi D, Meulmeester E, Pasini D, Migliorini D, Capra M, Frenk R, et al. Amplification of Mdmx (or Mdm4) directly contributes to tumor formation by inhibiting p53 tumor suppressor activity. Mol Cell Biol 2004; 24:5835 - 5843
- Brown JP, Wei W, Sedivy JM. Bypass of senescence after disruption of p21CIP1/WAF1 gene in normal diploid human fibroblasts. Science 1997; 277:831 - 834
- Pantoja C, Serrano M. Murine fibroblasts lacking p21 undergo senescence and are resistant to transformation by oncogenic Ras. Oncogene 1999; 18:4974 - 4982
- Wade M, Wahl GM. Targeting Mdm2 and Mdmx in cancer therapy: Better living through medicinal chemistry?. Mol Cancer Res 2009; 7:1 - 11
- Kortlever RM, Higgins PJ, Bernards R. Plasminogen activator inhibitor-1 is a critical downstream target of p53 in the induction of replicative senescence. Nat Cell Biol 2006; 8:877 - 884
- Carnero A, Beach DH. Absence of p21WAF1 cooperates with c-myc in bypassing Ras-induced senescence and enhances oncogenic cooperation. Oncogene 2004; 23:6006 - 6011
- Li C, Chen L, Chen J. DNA damage induces MDMX nuclear translocation by p53-dependent and -independent mechanisms. Mol Cell Biol 2002; 22:7562 - 7571
- Sharp DA, Kratowicz SA, Sank MJ, George DL. Stabilization of the MDM2 oncoprotein by interaction with the structurally related MDMX protein. J Biol Chem 1999; 274:38189 - 38196
- Reed D, Shen Y, Shelat AA, Arnold LA, Ferreira AM, Zhu F, et al. Identification and characterization of the first small molecule inhibitor of MDMX. J Biol Chem 2010; 285:10786 - 10796
- Kawai H, Lopez-Pajares V, Kim MM, Wiederschain D, Yuan ZM. RING domain-mediated interaction is a requirement for MDM2's E3 ligase activity. Cancer Res 2007; 67:6026 - 6030
- Stad R, Ramos YF, Little NA, Grivell S, Attema J, van De Eb AJ, et al. Hdmx stabilizes Mdm2 and p53. J Biol Chem 2000; 275:28039 - 28044
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 2010; 107:9660 - 9664
- Gilkes DM, Chen L, Chen J. MDMX regulation of p53 response to ribosomal stress. EMBO J 2006; 25:5614 - 5625
- Hu B, Gilkes DM, Chen J. Efficient p53 activation and apoptosis by simultaneous disruption of binding to MDM2 and MDMX. Cancer Res 2007; 67:8810 - 8817
- Markey M, Berberich SJ. Full-length hdmX transcripts decrease following genotoxic stress. Oncogene 2008; 27:6657 - 6666