Abstract
Recently, using a co-culture system, we demonstrated that MCF7 epithelial cancer cells induce oxidative stress in adjacent cancer-associated fibroblasts, resulting in the autophagic/lysosomal degradation of stromal caveolin-1 (Cav-1). However, the detailed signaling mechanism(s) underlying this process remain largely unknown. Here, we show that hypoxia is sufficient to induce the autophagic degradation of Cav-1 in stromal fibroblasts, which is blocked by the lysosomal inhibitor chloroquine. Concomitant with the hypoxia-induced degradation of Cav-1, we see the upregulation of a number of well-established autophagy/mitophagy markers, namely LC3, ATG16L, BNIP3, BNIP3L, HIF-1α and NFκB. In addition, pharmacological activation of HIF-1α drives Cav-1 degradation, while pharmacological inactivation of HIF-1 prevents the downregulation of Cav-1. Similarly, pharmacological inactivation of NFκB-another inducer of autophagy-prevents Cav-1 degradation. Moreover, treatment with an inhibitor of glutathione synthase, namely BSO, which induces oxidative stress via depletion of the reduced glutathione pool, is sufficient to induce the autophagic degradation of Cav-1. Thus, it appears that oxidative stress mediated induction of HIF1- and NFκB-activation in fibroblasts drives the autophagic degradation of Cav-1. In direct support of this hypothesis, we show that MCF7 cancer cells activate HIF-1α- and NFκB-driven luciferase reporters in adjacent cancer-associated fibroblasts, via a paracrine mechanism. Consistent with these findings, acute knock-down of Cav-1 in stromal fibroblasts, using an siRNA approach, is indeed sufficient to induce autophagy, with the upregulation of both lysosomal and mitophagy markers. How does the loss of stromal Cav-1 and the induction of stromal autophagy affect cancer cell survival? Interestingly, we show that a loss of Cav-1 in stromal fibroblasts protects adjacent cancer cells against apoptotic cell death. Thus, autophagic cancer-associated fibroblasts, in addition to providing recycled nutrients for cancer cell metabolism, also play a protective role in preventing the death of adjacent epithelial cancer cells. We demonstrate that cancer-associated fibroblasts upregulate the expression of TIGAR in adjacent epithelial cancer cells, thereby conferring resistance to apoptosis and autophagy. Finally, the mammary fat pads derived from Cav-1 (-/-) null mice show a hypoxia-like response in vivo, with the upregulation of autophagy markers, such as LC3 and BNIP3L. Taken together, our results provide direct support for the "Autophagic Tumor Stroma Model of Cancer Metabolism," and explain the exceptional prognostic value of a loss of stromal Cav-1 in cancer patients. Thus, a loss of stromal fibroblast Cav-1 is a biomarker for chronic hypoxia, oxidative stress and autophagy in the tumor microenvironment, consistent with its ability to predict early tumor recurrence, lymph node metastasis and tamoxifen-resistance in human breast cancers. Our results imply that cancer patients lacking stromal Cav-1 should benefit from HIF-inhibitors, NFκB-inhibitors, anti-oxidant therapies, as well as autophagy/lysosomal inhibitors. These complementary targeted therapies could be administered either individually or in combination, to prevent the onset of autophagy in the tumor stromal compartment, which results in a "lethal" tumor microenvironment.
Introduction
Solid tumors contain areas of hypoxia which trigger important changes, such as increased angiogenesis and metabolic reprogramming. Hypoxia inducible factor 1α (HIF-1α) is the main transcription factor mediating the hypoxic response. HIF-1α promotes transcription of angiogenic factors, such as VEGF and leads to increased glycolysis by inhibition of mitochondrial oxidative phosphorylation.Citation1,Citation2 Under normoxia, HIF-1α is hydroxylated by Prolyl Hydroxylase Domain-containing proteins (PHD) and targeted for degradation by the proteasome.Citation3 Hypoxia, as well as high levels of reactive oxygen species (ROS), inhibit PHD activity,Citation4 leading to HIF-1α stabilization and activation.Citation5,Citation6 ROS are generated mainly in the mitochondria via the electron transport chain,Citation7 when electrons prematurely react with oxygen, generating partially reduced or highly reactive metabolites of oxygen. Inhibition of the mitochondrial respiratory chain leads to increased ROS generation, triggering hypoxia-induced transcription.Citation8–Citation11 Abnormally high levels of ROS induce oxidative stress.
Several studies have now shown that hypoxia and ROS promote the activation of the nuclear factor κB (NFκB) transcription factor. NFκB is a multi-meric inducible transcription factor that plays a role in inflammation, cell survival and proliferation. In unstimulated cells, NFκB subunits (such as p50 and p65) are inhibited and sequestered in the cytoplasm by inhibitor of κB (IκB) proteins. NFκB activation requires the degradation of IκB proteins. This occurs through the activation of the IκB Kinases (IκBK) complex, which targets IκB proteins for degradation by phosphorylation. IκB degradation allows NFκB subunits to translocate to the nucleus, thus promoting transcriptional activity.Citation12,Citation13 Interestingly, as IκBK activity is controlled by O2 sensitive PHD, hypoxia promotes NFκB activation.Citation14
Recent evidence has demonstrated important cross-talk and the inter-dependence of NFκB and HIF-1α signaling. NFκB is a transcriptional activator of HIF-1α and basal NFκB activity is required for HIF-1α accumulation in normoxiaCitation15 and during hypoxia.Citation16 In addition, HIF-1α activation is thought to be nearly coincident with activation of NFκB. For example, IL-1β upregulates the HIF-1α protein under normoxia and activates the HIF-1-responsive gene VEGF via an NFκB-dependent pathway.Citation15 Conversely, HIF-1α accumulation was shown to promote NFκB activity.Citation17,Citation18
Hypoxia and oxidative stress both induce autophagy, a catabolic process whereby cytosolic cargos are entrapped in double-membrane vesicles, called autophagosomes and targeted for degradation by the lysosome. This lysosomal-dependent “self-digestion” process serves either to sustain energy production during nutrient starvation or to eliminate damaged proteins or defective organelles after stress. During oxidative stress, autophagy is particularly important for the removal of damaged mitochondria and of mitochondria producing the most ROS. This occurs via an autophagic process, termed mitophagy. Studies have shown that HIF-1α is a master regulator of the transcriptional program leading to autophagy and mitophagy.Citation19 In addition, it is becoming clear that also NFκB activation plays a key role in inducing autophagy.Citation20–Citation23
The role of autophagy in tumorigenesis is controversial. Studies have shown that autophagy is a pro-survival mechanism that sustains energy generation and maintains protein and organelle quality control. For example, hypoxia-induced autophagy via BNIP3 or BNIP3L was shown to be protective against cell death, thus promoting tumor progression.Citation24 However, several studies have shown that autophagy has tumor suppressing effects. Loss of the autophagy gene, namely Beclin 1 and of other autophagy genes, is observed in human cancers and promotes tumor formation in mice.Citation25,Citation26
The tumor microenvironment has recently gained much attention as a critical determinant of tumor progression and clinical outcome. The tumor microenvironment is composed of several cell-types, including fibroblasts, immune cells, endothelial cells and adipocytes. In particular, cancer associated fibroblasts (CAFs) are myofibroblast-like cells that were shown to induce the formation of a desmoplastic ‘reactive stroma’ and to promote tumor growth.Citation27 Importantly, studies have determined that certain stromal biomarkers are very strong predictors of clinical outcome.
Caveolin-1 (Cav-1) is the principal structural component of caveolae, specialized plasma membrane invaginations involved in the regulation of several cellular processes, including the control of cell signaling. Cav-1 is highly expressed in terminally differentiated stromal cells such as fibroblasts, adipocytes and endothelial cells. Clear evidence has demonstrated that Cav-1 behaves as a tumor suppressor in fibroblasts.Citation28–Citation31 Importantly, a loss of Cav-1 expression was shown to be a hallmark of the aggressive CAF phenotype in breast cancer patients.Citation32 –Citation34
In breast cancer, a loss of stromal Cav-1 expression is a powerful biomarker associated with a poor clinical prognosis. Patients that lack stromal Cav-1 have approximately a 20% 5-year survival rate, as compared to 80% in patients that are positive for stromal Cav-1.Citation33,Citation34 Loss of stromal Cav-1 predicts early tumor recurrence, lymph node metastasis and tamoxifen-resistance in all the most common sub-types of breast cancer, including triple-negative and basal-like breast cancers, as well as progression to invasive disease in ductal carcinoma in situ (DCIS) patients.Citation33,Citation35–Citation37
The mechanism(s) that regulate Cav-1 expression in CAFs are still not well defined. However, the downstream effects by which a loss of stromal Cav-1 promotes aggressive epithelial cancer cell behavior are starting to be elucidated. Of note, a loss of Cav-1 in the stroma was shown to promote (1) myofibroblast conversion, (2) a glycolytic switch in CAFs and (3) oxidative stress. Gene profiling and proteomic analysis of Cav-1 (−/−) null stromal cells shows the upregulation of myofibroblast markers (vimentin, calponin2, tropomyosin, gelsolin and prolyl 4-hydroxylase alpha), glycolytic enzymes and anti-oxidant enzymes.Citation38 Further analysis of the transcriptional profile of Cav-1 (−/−) mesenchymal cells demonstrates that a loss of Cav-1 induces oxidative stress, leading to aerobic glycolysis and inflammation in the tumor microenvironment stroma. On the basis of these data, we have proposed a novel model for explaining tumor metabolism, termed the “reverse Warburg effect,” whereby cancer cells reprogram CAFs to undergo aerobic glycolysis and to secrete energy-rich nutrients (such as pyruvate/lactate) that directly feed into mitochondrial oxidative metabolism in cancer cells. Thus, we believe that loss of Cav-1 is necessary to induce metabolic coupling between CAFs and epithelial cancer cells, resulting in the formation of a true host-parasite relationship.
We have previously employed a co-culture model of human fibroblasts and MCF7 breast cancer cells to recapitulate several features of the tumor microenvironment and the heterotypic signaling between cancer cells and CAFs. We demonstrated that MCF7 cancer cells drive the conversion of fibroblasts to CAFs, by inducing oxidative stress in adjacent fibroblasts, leading to the autophagic degradation of Cav-1.Citation39,Citation40 A loss of Cav-1 triggers HIF-1α stabilization in CAFs, with increased aerobic glycolysis and enhanced autophagy. In support of this notion, we have shown that fibroblasts (co-cultured with cancer cells) accumulate vesicular structures that we have identified as autophagosomes, by immuno-staining with the autophagy marker LC3.
The aim of the current study is to dissect the detailed signaling mechanism(s) underlying the autophagic/lysosomal degradation of stromal Cav-1 and to identify the molecular drivers that are sufficient to induce Cav-1 downregulation. As an experimental system, we employed the co-culture model of human hTERT-fibroblasts and MCF7 breast cancer epithelial cells. Complimentary experiments were also performed via the acute knockdown of Cav-1 in fibroblasts, using an siRNA gene silencing approach.
Here, we show that a loss of Cav-1 can be induced by three distinct pro-autophagic stimuli: (1) hypoxia, (2) hypoxia-mimetic drugs and (3) oxidative stress. Moreover, we demonstrate that a loss of Cav-1 occurs concomitantly with the upregulation of a number of well-established autophagy/mitophagy markers, namely LC3, ATG16L, BNIP3 and BNIP3L. Activation of the two pro-autophagic transcription factors, namely HIF-1α and NFκB, is sufficient to induce a loss of Cav-1 in fibroblasts, suggesting that the induction of HIF-1- and NFκB-activation drives the autophagic degradation of Cav-1. Functionally, we show that a loss of Cav-1 in fibroblasts protects cancer cells from apoptosis. Finally, we present evidence that the upregulation of a newly discovered protein, TIGAR (TP53-induced glycolysis and apoptosis regulator), provides a mechanism by which cancer cells are protected against apoptosis.
Results
In co-culture, fibroblasts display numerous lysosomes/phagosomes and MCF7 cells exhibit abundant mitochondria.
We have previously established a co-culture model of human fibroblasts and MCF7 cancer cells and we demonstrated that cancer cells induce a loss of Cav-1 in adjacent fibroblasts. Importantly, we have shown that chloroquine, a lysosomal-autophagy inhibitor, is sufficient to restore Cav-1 expression in co-cultured fibroblasts.Citation40 In addition, we have shown that co-cultured fibroblasts cluster vesicular structures that we have identified as autophagosomes, by immunostaining with the autophagy marker LC3.Citation39
Based on these findings, we attempted to provide additional morphological evidence to demonstrate that co-cultured fibroblasts undergo autophagy. Electron microscopy (EM) is the gold standard for identifying morphological structures, such as lysosomes and autophagosomes. Thus, co-cultures of hTERT-fibroblasts and MCF7 cells were analyzed by EM. Representative EM images are shown in and B and demonstrate that co-cultured fibroblasts display numerous lysosomes (dark electron-dense vesicles) and autophagosomes (vesicles surrounded by a double membrane). Interestingly, co-cultured MCF7 cells exhibit the presence of numerous mitochondria (), consistent with the idea that cancer cells do not rely solely upon glycolysis for energy production.
Hypoxia-induced autophagy leads to Cav-1 downregulation in fibroblasts.
As hypoxia is well known to promote autophagy, and as cancer cells induce a loss of Cav-1 in adjacent fibroblasts, we then hypothesized that hypoxia-induced autophagy may further exacerbate Cav-1 downregulation in co-cultured fibroblasts. To this end, hTERT-fibroblast-MCF7 co-cultures were incubated in hypoxia (0.5% O2) or normoxia (21% O2) for three days. We focused on day 3, because at this time point we do not usually observe significant Cav-1 downregulation in co-cultured fibroblasts. Interestingly, at day 3, hypoxia greatly downregulates Cav-1 expression in co-cultured fibroblasts, as compared to normoxia (). During hypoxia, Cav-1 becomes localized to intracellular vesicles, consistent with autophagic/lysosomal degradation.
To further substantiate these findings, homotypic cultures of hTERT-fibroblasts were subjected to hypoxia (0.5% O2) or normoxia (21% O2) for three days. Then, the cells were fixed and stained with anti-Cav-1 antibodies. shows that Cav-1 expression is greatly downregulated in fibroblasts in hypoxia, as compared to normoxia.
It is well established that hypoxia is sufficient to induce autophagy. We first attempted to evaluate if a loss of Cav-1 correlates with the expression of autophagic markers. For this purpose, hTERT-fibroblasts were subjected to hypoxia or normoxia and stained with antibodies directed against either Cav-1 or markers of autophagy (LC3A/B and ATG16L) and mitophagy (BNIP3 and BNIP3L). shows that the hypoxia-induced expression of both autophagy and mitophagy markers directly correlates with Cav-1 downregulation in fibroblasts.
Then, we examined whether hypoxia-induced Cav-1 down-regulation is mediated via an autophagic/lysosomal mechanism. To this end, Cav-1 immuno-staining was performed on fibroblasts subjected to hypoxia in the presence of chloroquine or vehicle alone. By raising the intra-lysosomal pH, chloroquine blocks autophagic protein degradation. shows that treatment with chloroquine inhibits the hypoxia-induced degradation of Cav-1, clearly indicating that a loss of Cav-1 during hypoxia is mediated by an autophagic-lysosomal mechanism.
Oxidative stress-induced autophagy correlates with Cav-1 downregulation.
We have previously shown that a loss of Cav-1 could be used as a marker of oxidative stress.Citation39 Thus, we attempted to directly evaluate if oxidative stress-induced autophagy mediates a loss of Cav-1 in fibroblasts. shows that treatment of hTERT-fibroblasts with the pro-oxidant buthionine sulfoximine (BSO) induces the accumulation of the active autophagosomal membrane-bound form of LC3B (LC3B-II). Importantly, we observe Cav-1 downregulation, concomitant with the accumulation of active LC3B-II.
Activation of pro-autophagic HIF-1α is sufficient to induce Cav-1 downregulation.
Next, we evaluated if activation of HIF-1α, a master regulator of autophagy, is sufficient to trigger Cav-1 downregulation. Thus, we first performed Western blot analysis on hTERT-fibroblasts subjected to hypoxia and normoxia. shows that HIF-1α accumulation precisely correlates with decreased Cav-1 levels.
Is the hypoxia-induced downregulation of Cav-1 reversible? To address this issue, fibroblast-MCF7 co-cultures were incubated in hypoxia or normoxia for three days. A subset of plates in hypoxia were then re-oxygenated for an additional 24 hours. Finally, the cells were immuno-stained with antibodies directed against Cav-1 and HIF-1α. As expected, shows that hypoxia downregulates Cav-1 expression in co-cultured fibroblasts (top panels), compared to normoxia (bottom panels) and re-oxygenation restores Cav-1 levels (middle panels, hypoxia + normoxia). These results indicate that Cav-1 downregulation is reversible and is likely independent of epigenetic mechanisms, such as DNA-methylation. Interestingly, shows also that HIF-1α is highly expressed in co-cultured fibroblasts in hypoxia (upper panels), but is undetectable in fibroblasts upon re-oxygenation (middle panels). These results demonstrate a tight inverse relationship between Cav-1 and HIF-1α levels in fibroblasts. During hypoxia, fibroblasts that have low Cav-1 levels display high HIF-1α activation. Re-oxygenation restores Cav-1 levels, while downregulating HIF-1α.
Regarding the epithelial cancer cells, HIF-1α is detected in co-cultured MCF7 cells at a high level during hypoxia and at low levels in normoxia (). Interestingly, however, HIF-1α levels are highest in co-cultured MCF7 cells after re-oxygenation (middle panels). The persistence of HIF-1α activation in MCF7 cells after re-oxygenation is consistent with the idea that fibroblasts and MCF7 cells are metabolically coupled during hypoxia, with Cav-1-low fibroblasts secreting energy-rich nutrients (such as lactate) that can directly feed tumor cells. However, during re-oxygenation, restoration of Cav-1 levels leads to the metabolic uncoupling of fibroblasts and MCF7 cells, and as a consequence, MCF7 cells experience a more severe “hypoxic stress” under normoxic conditions.
To directly dissect the ability of HIF-1α to regulate Cav-1 expression, we next employed conditions that pharmacologically activate or inhibit HIF-1α. First, fibroblast-MCF7 cell co-cultures were treated with three PHD inhibitors, namely DMOG, 2,4 DPD and 1,4 DPCA. PHD inhibitors block the proteasomal degradation of HIF-1α, inducing constitutive HIF-1α activation. shows that treatment of co-cultures with all three PHD inhibitors decreases Cav-1 expression in fibroblasts, as compared with vehicle alone controls. Similarly, Western blot analysis shows that DMOG treatment downregulates Cav-1 levels in homotypic cultures of hTERT-fibroblasts ().
Conversely, hTERT-fibroblasts in hypoxia were treated with the HIF-1α inhibitor, echinomycin, or with vehicle alone (DMSO) for 24 hours. By inhibition of HIF-1α DNA-binding activity, echinomycin blocks HIF-1α transcriptional activity and its downstream effects.Citation41 shows that treatment with echinomycin blocks the hypoxia-induced downregulation of Cav-1. Similarly, western blot analysis of hypoxic fibroblasts indicates that echinomycin treatment for 48 hours rescues Cav-1 expression levels (), showing that loss of Cav-1 is mediated by HIF-1α activation.
Activation of the pro-autophagic molecule NFκB leads to Cav-1 downregulation.
We also evaluated whether another autophagy inducer, namely NF.B, can downregulate Cav-1 protein levels. First, we assessed if an autophagic stimulus, i.e., hypoxia, is sufficient to lead to NFκB activation in fibroblasts. shows that hypoxia induces the accumulation of the phosphorylated/active form of NFκB in the nuclei of hTERT-fibroblasts.
As we have shown that co-cultured cancer cells induce Cav-1 downregulation in adjacent fibroblasts, we also determined if cancer cells can promote NFκB activation in adjacent fibroblasts, leading to the autophagic-degradation of Cav-1. To this end, fibroblast-MCF7 cell co-cultures were immuno-stained with antibodies directed against the phosphorylated form of NFκB. Homotypic cultures of fibroblasts and MCF7 cells were processed and evaluated in parallel. Interestingly, nuclear expression of phospho-NFκB was detected in co-cultured fibroblasts, but not in homotypic control cells (), demonstrating that cancer cells induce NFκB activation in adjacent fibroblasts.
Is NFκB inhibition sufficient to restore Cav-1 expression in fibroblasts? To block the NFκB pathway, fibroblast-MCF7 cell co-cultures were treated with PS1145, an IκBK inhibitor, or vehicle alone for 24 hours. IκBK inhibition blocks IκB degradation, leading to NFκB cytoplasmic sequestration. Immuno-staining with anti-phospho-NFκB antibodies directly demonstrates NFκB inhibition in co-cultured fibroblasts (). Interestingly, shows that pre-treatment with PS1145 restores Cav-1 expression in co-cultured fibroblasts, compared to vehicle alone controls, clearly indicating that NFκB activation drives Cav-1 downregulation in CAFs.
Loss of Cav-1 is sufficient to induce autophagy.
Is loss of Cav-1 sufficient to trigger autophagy in fibroblasts? To address this issue, we evaluated if acute knockdown of Cav-1 is sufficient to promote the activation of the autophagy inducer NFκB. To this end, phospho-NFκB staining was performed on hTERT-fibroblasts treated with Cav-1 siRNA or control siRNA. shows the nuclear localization of phospho-NFκB in Cav-1 siRNA treated cells, indicating that acute loss of Cav-1 drives NFκB activation in fibroblasts. Taken together, our data indicate that a loss of Cav-1 and NFκB activation are under a positive “feed-forward” control mechanism, with a loss of Cav-1 triggering NFκB activation and NFκB activation further enhancing Cav-1 downregulation.
These results suggest that acute loss of Cav-1 may be sufficient to activate the autophagy and/or mitophagy program in fibroblasts. To test this hypothesis directly, western blot analysis was performed on hTERT-fibroblasts treated with Cav-1 siRNA or control siRNA, using antibodies directed against a panel of autophagy markers. shows that acute Cav-1 knockdown in fibroblasts drives the increased expression of six autophagy markers, including Cathepsin B (active form), LAMP-1, LC3B, Beclin 1, ATG16L and BNIP3.
To independently validate these results, hTERT-fibroblasts treated with Cav-1 siRNA or control siRNA were also immunostained with a subset of autophagy/mitophagy markers. shows that Beclin 1, BNIP3 and BNIP3L are greatly increased in Cav-1 knockdown cells, indicating that an acute loss of Cav-1 is sufficient to promote autophagy. Taken together, our current findings indicate that oxidative stress and hypoxia induce the autophagy-mediated loss of Cav-1 in fibroblasts, and that loss of Cav-1 further promotes autophagy/mitophagy in fibroblasts, via a feed-forward mechanism.
Cancer cells promote pro-autophagic signaling pathways in adjacent fibroblasts.
Do cancer cells trigger the activation of pro-autophagic HIF-1α and NFκB signaling pathways in adjacent fibroblasts? The activation of both HIF-1α and NFκB is transient and time-dependent. In order to detect NFκB and HIF-1α activation in co-cultured fibroblasts, we employed a luciferase-reporter approach. NIH3T3 cells were engineered to express a luciferase reporter gene either under the control of a NFκB response element (NFκB-Luc cells) or under the control of a HIF-1α response element (HIF-Luc cells). These fibroblasts were then co-cultured with MCF7 cells for up to 5 days. Luciferase activity was measured at day 0, 3 and 5. Homotypic cultures of NFκB-Luc cells and HIF-Luc cells were evaluated in parallel.
(left panel) shows that luciferase activity is increased by 9.4- and 2.4-fold at day 0 and day 3, respectively, in NFκB-Luc co-cultured fibroblasts, as compared to the same fibroblasts cultured alone. This suggests that the relative increase in NFκB activation is highest early during co-culture. By day 5, NFκB activation has declined in co-cultured fibroblasts. (In this experiment, it is important to note that the day 0 time point represents 8 hours of co-culture; see Materials and Methods).
Conversely, (right panel) shows that luciferase activity is increased by 4-fold at day 5 in HIF-Luc co-cultured fibroblasts, as compared to fibroblasts alone, indicating that HIF-1α activation occurs at a time point where NFκB activation has subsided. HIF1 activation (by ∼2-fold) was also observed on day 4 of co-culture (see Suppl. Fig. 1).
In summary, cancer cells induce NFκB and HIF-1α activation in adjacent fibroblasts, with NFκB activation occurring at an early time point, and decreasing just before the peak of HIF-1α activation.
Loss of Cav-1 in stromal fibroblasts protects adjacent cancer cells against apoptosis.
What are the functional consequences of the autophagy-mediated loss of Cav-1 in fibroblasts? One hypothesis is that autophagy in fibroblasts generates nutrients (such as free amino acids and nucleotides) that can be transferred to cancer cells to sustain their high-energy demand. We have previously shown that cancer-associated fibroblasts do not promote cancer cell proliferation, but rather protect cancer cells from apoptosis.Citation39 Thus, a loss of stromal Cav-1 may be a mechanism to further protect cancer cells against apoptosis. To test this hypothesis, MCF7 cells were co-cultured with hTERT-fibroblasts carrying a GFP (+) Cav-1 shRNA vector (KD) or a GFP (+) control shRNA vector (CTR). Then, the cell populations were subjected to annexin-V staining and analyzed by FACS. Corresponding homotypic cultures were processed in parallel. Thus, the GFP (+) and GFP (−) cells represent hTERT-fibroblasts and MCF7 cells, respectively.
Consistent with our previous data, shows that MCF7 cells co-cultured with CTR fibroblasts demonstrate a 3-fold reduction in apoptosis (a 14% apoptotic rate for MCF7 cells alone, versus a 4.5% apoptotic rate for MCF7 cells co-cultured with CTR fibroblasts). Importantly, MCF7 cells maintained in co-culture with Cav-1 KD fibroblasts display an 8.5-fold reduction in apoptosis (a 14% apoptotic rate for MCF7 cells alone, versus a 1.7% apoptotic rate for MCF7 cells co-cultured with Cav-1 KD fibroblasts). These data clearly indicate that a loss of Cav-1 in fibroblasts greatly protects cancer cells from apoptosis.
Does Cav-1 KD also protect fibroblasts against apoptosis? shows that in mono-culture Cav-1 KD fibroblasts demonstrate a 2.7-fold reduction in apoptosis as compared to CTR fibroblasts (a 0.8% apoptotic rate for CTR cells versus 0.3% of Cav-1 KD cells). Interestingly, co-culture with MCF7 cells induces a 2.4-fold increase in apoptosis in CTR fibroblasts (a 1.9% apoptotic rate for co-cultured CTR fibroblasts versus 0.8% for CTR cells alone). However, Cav-1 knockdown protects co-cultured fibroblasts from apoptosis. We observe that co-cultured Cav-1 KD fibroblasts exhibit a 2-fold decrease in apoptosis as compared to co-cultured CTR fibroblasts (a 1.9% apoptotic rate for co-cultured CTR fibroblasts versus 0.9% for co-cultured Cav-1 KD fibroblasts). Thus, our results directly show that a loss of Cav-1 in fibroblasts protects both cancer cells and co-cultured fibroblasts from apoptosis.
Fibroblasts induce the upregulation of the apoptosis and autophagy inhibitor TIGAR in cancer cells.
To begin to understand the mechanism(s) by which cancer cells are protected from apoptosis, we next tested the hypothesis that fibroblasts induce the upregulation of TIGAR (TP53-induced glycolysis and apoptosis regulator) in epithelial cancer cells. TIGAR protects cells from oxidative stress by simultaneously inhibiting 3 inter-related cellular processes, namely (1) aerobic glycolysis, (2) apoptosis and (3) autophagy.Citation42,Citation43
Thus, we hypothesized that TIGAR expression would be increased in MCF7 cells in co-culture, thereby protecting them from apoptosis. To this end, fibroblast-MCF7 cell co-cultures and the corresponding mono-cultures were immuno-stained with anti-TIGAR antibodies. Interestingly, shows that TIGAR is dramatically increased in co-cultured MCF7 cells, as compared to MFC7 cells cultured alone, suggesting that TIGAR upregulation may be a key mechanism by which cancer cells protect themselves from apoptosis. Note that TIGAR was not increased in adjacent fibroblasts.
Loss of Cav-1 induces a pseudo-hypoxic state in vivo.
Analysis of the transcriptional profile of Cav-1 (−/−) mesenchymal cells has shown that a loss of Cav-1 induces oxidative stress.Citation44 Similarly, acute Cav-1 knockdown is sufficient to promote ROS generation in fibroblasts.Citation39 However, it remains unknown if a loss of Cav-1 is sufficient to induce hypoxic stress in vivo. To this end, the hypoxia marker pimonidazole was injected into the tail vein of WT and Cav-1 KO mice. After one hour, mice were sacrificed and the mammary glands and lungs were analyzed by immunohistochemistry using a rabbit anti-serum to pimonidazole. shows that mammary fat pads and lung parenchyma derived from Cav-1 (−/−) mice display strong pimonidazole-positive staining, as compared to WT controls. These data strongly suggest that Cav-1 (−/−) mice undergo “pseudo-hypoxic stress” at steady-state, and indicates that a loss of Cav-1 is sufficient to drive hypoxic signaling in vivo. In this regard, is important to note that Cav-1 (−/−) null mice do not show any defects in proper oxygenation (See Table 1 in ref. Citation45).
Cav-1 (−/−) mammary fat pads display increased autophagy.
As Cav-1 (−/−) mice undergo pseudo-hypoxia at steady-state, we evaluated if Cav-1 gene deletion is sufficient to promote autophagy in vivo. To this end, paraffin-embedded sections from WT and Cav-1 (−/−) mammary glands were immuno-stained with two markers of autophagy, namely LC3A/B and BNIP3L. shows that both LC3A/B and BNIP3L are increased in Cav-1 (−/−) mammary fat pads. Interestingly, Cav-1 (−/−) adipocytes show a large increase in LC3-positive vesicles.
Human breast cancers lacking stromal Cav-1 display increased stromal BNIP3L.
We have shown above that a loss of Cav-1 promotes autophagy and that Cav-1 is degraded via an autophagic mechanism. To evaluate the translational significance of our findings, we evaluated if a loss of stromal Cav-1 in human breast cancer correlates with increased autophagy. To this end, we selected a number of human breast cancer samples that lack Cav-1 in the stroma to perform immuno-staining with the autophagy/mitophagy marker BNIP3L.
shows that BNIP3L is highly expressed in the stromal compartment of human breast cancers that lack Cav-1. These results directly support the “autophagic tumor stroma model of cancer metabolism.”Citation46 As a loss of Cav-1 is a powerful predictor of poor clinical outcome in breast cancers,Citation33,Citation35 our findings indicate that in human breast cancer a loss of Cav-1 promotes autophagy/mitophagy in the stroma, to support the growth and aggressive behavior of adjacent cancer cells.
Discussion
We have previously shown using a co-culture model, that breast cancer cells induce oxidative stress in adjacent cancer associated fibroblasts, resulting in the autophagic/lysosomal degradation of stromal Cav-1.Citation39,Citation40 However, the detailed signaling mechanism(s) underlying this process remain largely unknown. As loss of stromal Cav-1 is a powerful predictor of clinical outcome in human breast cancers,Citation33–Citation37 a clear understanding of the signaling mechanism(s) leading to the degradation of Cav-1 is of foremost importance.
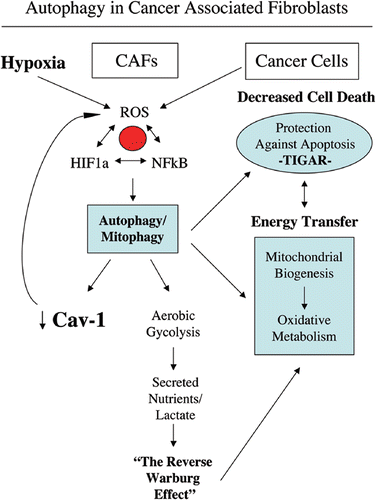
Our data suggest a model whereby cancer cells trigger oxidative stress, and activate two pro-autophagic drivers, namely HIF-1α and NFκB, in adjacent stromal fibroblasts (). As a result, CAFs undergo autophagy and mitophagy, leading to loss of Cav-1 and metabolic re-programming. A loss of stromal Cav-1 aggravates oxidative stress and further promotes autophagy and mitophagy. Stromal autophagy generates building blocks (such as recycled free amino acids, fatty acids and nucleotides) that can be directly utilized by cancer cells to sustain growth and maintain cell viability. At the same time, HIF-1α activation and consequent mitophagy in CAFs induces mitochondrial dysfunction and enhances aerobic glycolysis, leading to the secretion of high-energy nutrients (such as lactate and pyruvate) that can directly feed oxidative mitochondrial metabolism in cancer cells. We also provide evidence that a loss of Cav-1 in stromal cells protects cancer cells from apoptosis, at least in part via TIGAR upregulation. Thus, our “working model” indicates that cancer cells exploit CAFs to satisfy their increased energy demand by forcing stromal cells to undergo a unilateral and vectorial energy transfer to sustain cancer cell growth. As such, catabolism in the tumor stroma promotes the anabolic growth of adjacent cancer cells, by providing a steady stream of recycled nutrients, independently of angiogenesis.
We show here that three pro-autophagic stimuli are sufficient to induce a loss of Cav-1 in fibroblasts: (1) hypoxia, (2) hypoxia-mimetic drugs and (3) oxidative stress. To our knowledge, this is the first demonstration that hypoxia-induced autophagy leads to Cav-1 downregulation. A previous report had suggested that chronic myocardial hypoxia may decrease the expression of caveolin-3, the muscle-specific isoform of the caveolin gene family, as a way to increase nitric oxide production and protect against ischemia.Citation47 Importantly, we show here that hypoxia-induced Cav-1 downregulation occurs by an autophagic mechanism and serves to protect adjacent cancer cells against apoptosis.
Concomitant with Cav-1 downregulation, we observe the upregulation of numerous autophagy and mitophagy markers. Treatment with the autophagic/lysosomal inhibitor chloroquine reverts the hypoxia-induced downregulation of Cav-1. We also show that activation of the autophagic molecular driver, HIF-1α, is sufficient to induce a loss of Cav-1 in fibroblasts. Interestingly, pharmacological activation of HIF-1α induces Cav-1 downregulation, whereas pharmacological inhibition of HIF-1α abrogates the hypoxia-induced degradation of Cav-1. Similarly, pharmacological inactivation of NFκB, another inducer of autophagy, prevents Cav-1 degradation. Moreover, treatment with an inducer of oxidative stress, namely BSO, is sufficient to induce the autophagic degradation of Cav-1. Thus, it appears that oxidative stress mediated via the induction of HIF1- and NFκB-activation drives the autophagic degradation of Cav-1. In direct support of this hypothesis, we employed HIF-1α- and NFκB-driven luciferase reporters to monitor the ability of cancer cells to promote proautophagic signaling. Using this approach, we show that epithelial cancer cells promote the activation of HIF-1α and NFκB in adjacent cancer-associated fibroblasts, via a paracrine mechanism. Thus, a loss of stromal Cav-1 in breast cancers is a biomarker of hypoxia, oxidative stress and autophagy in the tumor stromal microenvironment.
Little is known about HIF-1α activation in the tumor stromal compartment. It was recently shown that oxidative stress promotes myofibroblast differentiation through accumulation of HIF-1α and the chemokine CXCL12. Importantly, oxidative stress was shown to increase the migration of stromal fibroblasts, and to enhance tumor dissemination.Citation48 We have also previously demonstrated that acute loss of Cav-1 in stromal fibroblasts is sufficient to drive HIF-1α accumulation, leading to mitochondrial dysfunction and aerobic glycolysis.Citation39 Here, we show that HIF-1α activation in the stroma drives the autophagic degradation of Cav-1, providing a positive “feed-forward” control loop between HIF-1α activation and Cav-1 downregulation.
Activation of NFκB signaling in stromal macrophages is known to promote metastasis,Citation49,Citation50 however, the role of NFκB activity in CAFs is just starting to be unraveled. Previous studies have suggested that NFκB activation in CAFs may promote tumor growth. For example, stromal fibroblasts isolated from colorectal liver metastases display the upregulation of IL-8, a chemokine important for invasion and angiogenesis, via NFκB activation.Citation51 In addition, transcriptome analysis has revealed that cancer-associated fibroblasts from breast, pancreas and skin cancers exhibit a pro-inflammatory gene signature compared to normal fibroblasts. Importantly, these inflammatory fibroblasts enhance tumor growth in an NFκB-dependent mechanism.Citation52 We have previously shown that Cav-1 gene deletion in stromal cells drives the transcriptional activation of NFκB and of NFκB target genes.Citation44 Importantly, we show here that cancer cells promote NF.B activation in adjacent fibroblasts. In addition, we provide evidence that pharmacological inactivation of NFκB prevents Cav-1 degradation in CAFs, and that acute loss of Cav-1 is sufficient to promote NFκB activation and autophagy, clearly indicating that in stromal cells, a loss of Cav-1 and NFκB activation can reciprocally promote each other.
Interestingly, our data indicate that acute loss of Cav-1 is sufficient to potently induce the expression of several autophagic and mitophagic markers. In addition, we show that in vivo, genetic ablation of Cav-1 in the mammary gland induces a pseudohypoxic state with increased autophagy. Our findings are consistent with recent studies suggesting that a loss of Cav-1 may promote autophagy. For example, transmission electron microscopy on murine myocardium has demonstrated increased assembly of autophagosomes in Cav-1 (−/−) null cardiac myocytes, as compared to WT controls.Citation53 In addition, adipocytes from Cav-1 (−/−) null mice show increased LC3 staining and increased autophagic vacuoles.Citation54
We show here that acute loss of Cav-1 is sufficient to trigger an autophagic response in fibroblasts. More importantly, our current data demonstrate that induction of autophagy via a Cav-1 deficiency may have important translational implications. We have recently proposed the “autophagic tumor stroma model of cancer metabolism,” whereby autophagy/mitophagy in the tumor stroma provides cancer cells with essential chemical building blocks (such as amino acids and nucleotides) to drive tumor progression and metastasis.Citation46 Thus, a loss of stromal Cav-1 drives tumor progression by increasing “catabolism/autophagy” in the tumor microenvironment.
In direct support of our hypothesis, several studies have shown that overexpression of autophagic markers in the stroma, but not in tumor cells, correlates with poor clinical outcome in human cancers. For example, high stromal expression of ATG16L is associated with lympho-vascular invasion and lymph node metastasis in oral squamous cell carcinoma.Citation55 In addition, stromal cathepsin K expression was found to be significantly higher in invasive breast cancers, compared with in situ carcinomas and in one patient set, it correlated with higher tumor stage.Citation56 Similarly, in invasive breast ductal carcinoma, cathepsin D staining of tumor cells is associated with a lower nuclear grade and well-differentiated histology, whereas cathepsin D staining of stromal cells correlates with increased tumor size, poorly differentiated histology and shorter disease-free and overall survivals.Citation57 Interestingly, cathepsin B was shown to be necessary for extracellular matrix degradation in v-Src transformed fibroblasts,Citation58 and to drive proliferation and liver fibrosis in hepatic stellate cells.Citation59 These data clearly suggest that the induction of an autophagic program in the tumor microenvironment correlates with poor clinical outcome and promotes aggressive cancer cell behavior.
What are the functional consequences of a loss of stromal Cav-1 and the induction of autophagy in the stromal compartment? Interestingly, we provide evidence that a loss of Cav-1 in stromal fibroblasts protects adjacent cancer cells against apoptosis. Thus, autophagic cancer-associated fibroblasts, in addition to providing recycled nutrients to “feed” cancer cell metabolism, also play a protective role in preventing the death of adjacent epithelial cancer cells. Other studies have suggested that CAFs may promote tumor growth through decreased apoptosis. For example, conditioned media derived from CAFs expressing high levels of the GPI-anchored protein CD90 (Thy-1) was shown to protect prostate cancer cells from oxidative stress-induced apoptosis.Citation60 Similarly, TGFβ1 treatment of prostate stromal cells was demonstrated to promote myofibroblast differentiation and to decrease apoptotic rates of co-cultured prostate cancer cells.Citation61 We show here that loss of Cav-1 in stromal fibroblasts protects breast cancer cells from apoptosis. As Cav-1 is a potent inhibitor of TGFβ signaling, and we have previously shown that Cav-1 knockdown is sufficient to promote constitutive activation of TGFβ pathway,Citation40,Citation62 we speculate that TGFβ activation may play a key role in the loss of stromal Cav-1 and its enhancement of cancer cell survival.
Also, we provide evidence that increased expression of TIGAR may be one of the mechanism(s) by which cancer cells are protected against apoptosis. Co-culture with fibroblasts induces the compartment-specific expression of TIGAR in adjacent epithelial cancer cells. TIGAR is a newly discovered protein that protect cells against oxidative stress,Citation43 and inhibits three related cellular processes, namely aerobic glycolysis, apoptosis and autophagy.Citation42 Thus, TIGAR expression would render anabolic epithelial cancer cells resistant to the induction of aerobic glycolysis, apoptosis and autophagy, thereby facilitating mitochondrial biogenesis in cancer cells and promoting the transfer of nutrients from autophagic cancer-associated fibroblasts. These properties make TIGAR an ideal candidate to explain the “reverse Warburg effect,”Citation63 and the “autophagic tumor stroma model of cancer metabolism.”Citation46
In summary, we believe that during tumor formation, cancer cells and adjacent fibroblasts are metabolically coupled. Cancer cells induce a ROS-dependent loss of Cav-1 in adjacent fibroblasts via autophagic/lysosomal degradation. A loss of stromal Cav-1 then (1) promotes aerobic glycolysis and autophagy, thus producing nutrients (such as lactate and pyruvate) and chemical building blocks (such as amino acids, fatty acids and nucleotides) to “feed” adjacent cancer cells undergoing oxidative mitochondrial metabolism; and (2) protects cancer cells from apoptosis. Thus, upregulation of TIGAR in cancer cells could serve as a mechanism to promote cancer cell survival and prevent ROS generation, while inhibiting the two key metabolic processes, namely aerobic glycolysis and autophagy, in epithelial cancer cells that we show are activated in the tumor stroma. This model allows for the unilateral and vectorial transfer of nutrients from autophagy-prone catabolic stromal cells to autophagy-resistant anabolic cancer cells. Further studies will be required to fully address the role of stromal Cav-1 in modulating TIGAR expression in epithelial cancer cells.
Materials and Methods
Materials.
Antibodies were as follows: Cav-1 (sc-894, Santa Cruz Biotech for immunofluorescence; 610407, BD Biosciences for WB); cytokeratin 8/18 (20R-CP004, Fitzgerald Industries International); HIF-1α (NB100-123, Novus Biologicals for IF; 610959, BD Biosciences for WB); phospho-serine 276 NFκB p65 subunit (3037, Cell Signaling); Cathepsin B (sc-13985, Santa Cruz); LAMP-1 (sc-17768, Santa Cruz); LC3B (ab48394, Abcam); Beclin 1 (NBP1-00085, Novus for WB; 2026-1, Epitomics for IF); LC3A/B (ab58610, Abcam); ATG16L (AM18176B for IF, AM18176D for WB, Abgent); BNIP3 (ab10433, Abcam); BNIP3L (AP1320A, Abgent); TIGAR, (ab37910, Abcam); rabbit antiserum to pimonidazole (PAb2627, Hydroxyprobe Gemini Kit, Hydroxyprobe, Inc., Burlington, MA); β-tubulin and β-actin were from Sigma; secondary antibodies for immunofluorescence were Alexa green 488 nm and Alexa Orange-Red 546 nm (Invitrogen). Other reagents were as follows. PS1145 and buthionine sulfoxide (BSO) were from Sigma. 4,6-diamidino-2-phenylindole (DAPI) (D3571), Prolong Gold Antifade mounting reagent (P36930), Slow-Fade Antifade reagent (S2828) were from Invitrogen. 2,4 DPD (2,4-Diethylpyridine dicarboxylate), 1,4 DPCA (1,4-dihydrophenonthrolin-4-one-3-Carboxylic acid) and DMOG (Dimethyloxallyl Glycine) were from Cayman Chemicals. Echinomycin was from Enzo Life Sciences.
Cell cultures.
Human skin fibroblasts immortalized with telomerase reverse transcriptase protein (hTERT-BJ1) were originally purchased from Clontech, Inc. The breast cancer cell line MCF7 was purchased from ATCC. NIH3T3-HIF-Luc reporter cells and NIH3T3-NFκB-Luc reporter cells were purchased from Panomics. All cells were maintained in DMEM with 10% fetal bovine serum (FBS) and penicillin 100 units/mL-Streptomycin 100 µg/mL. Cells were maintained at 37°C in a humidified atmosphere containing 5% CO2. Hypoxia experiments were performed using a hypoxia chamber with 0.5% O2.
Co-culture of MCF7 cells and fibroblasts.
hTERT-fibroblasts and MCF7 cells were plated on glass coverslips in 12-well plates in 1-ml of complete media. MCF7 cells were plated within 2 hours of fibroblast plating. The total number of cells per well was 1 × 105 cells. Experiments were performed at a 5:1 ratio of fibroblasts-to-MCF7 cells. As controls, mono-cultures of fibroblasts and MCF7 cells were seeded using the same number of cells as in the corresponding co-cultures. The day after, media was changed to DMEM with 10% NuSerum (a low protein alternative to FBS, BD Biosciences) and Pen-Strep.
Confocal microscopy.
Images were collected with a Zeiss LSM510 meta confocal system using a 405 nm Diode excitation laser with a band pass filter of 420–480 nm, a 488 nm Argon excitation laser with a band pass filter of 505–550 nm and a 543 nm HeNe excitation laser with a 561–604 nm filter. Images were acquired with a 40x or 60x objective, as stated in the Figure legends.
Immunocytochemistry.
Immunocytochemistry was performed as previously described.Citation64 All steps were performed at room temperature. Briefly, after 30 minutes fixation in 2% paraformaldehyde, cells were permeabilized for 10 minutes with immunofluorescence (IF) buffer (PBS, 0.2% BSA, 0.1% TritonX-100). Then, cells were incubated for 10 minutes with NH4Cl in PBS to quench free aldehyde groups. Primary antibodies were incubated in IF buffer for 1 hour. After washing with IF buffer (3x, 10 minutes each), cells were incubated for 30 minutes with fluorocrome-conjugated secondary antibodies diluted in IF buffer. Finally, slides were washed with IF buffer (3x, 10 minutes each), incubated with the DAPI nuclear stain and mounted.
Electron microscopy.
For electron microscopy evaluation, co-cultures were fixed with 2.5% glutaraldehyde in 0.1 M sodium cacodylate buffer, pH 7.4 for 30 minutes at room temperature. Then, cells were post-fixed with 1% osmium tetroxide in 0.1 M sodium cacodylate buffer, pH 7.4 for 1 hour, contrasted with 1% tannic acid in 0.05 M sodium cacodylate, followed by dehydration through graded alcohols and propylene oxide. Infiltration was performed in graded epon solutions before final embedding in EMbed 812 (Electron Microscopy Sciences, Hatfield, PA). Thin sections were cut on an UltraCut E ultramicrotome and stained with uranyl acetate and lead as previously described.Citation65,Citation66 Images were collected with an AMT XR41-B 4 megapixel camera on a Hitachi H-7000 electron microscope.
Western blotting.
Cells were harvested in lysis buffer (10 mM Tris-HCl pH 7.5, 150 mM NaCl, 1% Triton X-100, 60 mM octylglucoside), containing protease and phosphatase inhibitors (Roche Applied Science). Samples were incubated for 40 min at 4°C in a rotating platform and centrifuged 10 min at 13,000x g at 4°C. Protein concentrations were determined using the BCA reagent (Pierce). To detect HIF-1α, cells were scraped in urea lysis buffer (6.7 M urea, 10% glycerol, 1% SDS, 10 mM Tris-HCl pH 6.6, 1% Triton X-100, protease inhibitors), homogenized for 5 seconds and incubated on ice for 10 minutes. After centrifugation, protein concentration was determined using the Bradford assay (BioRad). 30 µg of proteins were loaded and separated by SDS-PAGE and transferred to a 0.2 µm nitrocellulose membrane (Fisher Scientific). After blocking for 30 min in TBST (10 mM Tris-HCl pH 8.0, 150 mM NaCl, 0.05% Tween-20) with 5% nonfat dry milk, membranes were incubated with the primary antibody for 1 hour, washed and incubated for 30 minutes with horseradish peroxidase-conjugated secondary antibodies. The membranes were washed and incubated with enhanced chemiluminescence substrate (Thermo Scientific).
Cav-1 knockdown.
siRNA-mediated Cav-1 knockdown was performed using the HiPerFect transfection reagent (Qiagen) as per manufacturer instructions, with minor modifications. Briefly, 1 × 105 cells were seeded in 12 well plates in 350 µL of complete media and were transfected within 30 minutes of plating. 100 ng of Cav-1 siRNA (SI00299635, Qiagen) or 100 ng control siRNA (1022076, Qiagen) were mixed with 100 µL of serum free media and 6 µL of HiPerFect reagent. After vortexing for 30 seconds, the mixture was incubated for 15 minutes at room temperature and then added drop-wise to the freshly seeded cells. Three hours post-transfection, 800 µL of complete media was added to the cells. Cav-1 knockdown was monitored 36–48 h post-transfection.
PHD inhibitors.
DMOG, 2,4 DPD and 1,4 DPCA were incubated with cells in DMEM 10% NuSerum for 48 hours. DMOG and 1,4 DPCA were dissolved in DMSO and used at a final concentration of 500 and 4 µM, respectively. 2,4 DPD was re-suspended in ethanol and added at a final concentration of 10 µM. All control experiments were carried out with the corresponding vehicle alone.
Luciferase activity.
NIH3T3 fibroblasts stably transfected with an NFκB luciferase reporter (RC0015, Panomics) or a HIF-1α luciferase reporter (RC0017, Panomics) were seeded in complete media (DMEM with 10% FBS) in 24 well plates. For co-culture, 5 × 104 cells were seeded at a fibroblast-to-MCF7 cell ratio of 5:1. As controls, fibroblasts were plated in mono-culture using the same cell numbers, as in the corresponding co-cultures. After cells attached, i.e., about 8 hours later, the media was changed to DMEM with 10% NuSerum and that was considered day 0. Luciferase activity was measured at day 0, day 3 and day 5, as previously described.Citation67 In brief, cell lysis buffer was added to each well. Then, ATP mix was added to the lysates to serve as the substrate for the chemiluminescent oxidation-reduction reaction which leads to light emission and oxy-luciferin generation. A luminometer was used to measure light emitted at 560 nm.
Animal studies.
Animals were housed and maintained in a pathogen-free environment/barrier facility at the Kimmel Cancer Center at Thomas Jefferson University under National Institutes of Health (NIH) guidelines. Mice were kept on a 12 hour light/dark cycle with ad libitum access to chow and water. Approval for all animal protocols used for this study was reviewed and approved by the Institutional Animal Care and Use Committee (IACUC). To detect hypoxia in vivo, pimonidazole HCl (1.5 mg/mouse, Hydroxyprobe, Inc.) was injected in the tail vein of WT and Cav-1 KO mice (in the FVB/N genetic background). After 1 hour, mice were sacrificed and lungs and mammary glands were harvested and fixed with 10% formalin.
Immunohistochemistry.
Paraffin-embedded sections of lung and mammary gland were immuno-stained as previously described.Citation68 Briefly, sections were deparaffinized, rehydrated and washed in PBS. Antigen retrieval was performed in 10 mM sodium citrate, pH 6.0 for 10 min using a pressure cooker. Then, sections were blocked with 3% hydrogen peroxide for 10 minutes followed by an incubation using a avidin-biotin blocking kit (Dako) to block endogenous biotin. After incubation with 10% goat serum for 1 hour, sections were incubated with primary antibodies overnight at 4°C (except for pimonidazole staining on the mammary glands, in which the antibody was incubated for 1 hour at room temperature). Antibody binding was detected using a biotinylated secondary (Vector Labs, Burlingame, CA) followed by strepavidin-HRP (Dako, Carpinteria, CA). Immunoreactivity was revealed using 3, 3′ diaminobenzidine.
Annexin-V apoptosis detection.
Apoptosis was quantified by flow cytometry using the Annexin-V-Cy5 apoptosis detection kit, as the per manufacturer's instructions (Abcam). Briefly, MCF7 cells were plated in 12 well plates with GFP (+) hTERT-fibroblasts carryng either a control shRNA (CTR) vector or a Cav-1 shRNA (KD) vector. Homotypic cultures were plated in parallel. The day after, media was changed to DMEM with 10% NuSerum. After 48 hours, cells were collected by centrifugation and re-suspended in 500 µL of Annexin-V Binding Buffer. Then, the annexin V-Cy5 conjugate (5 µL) was added and incubated in the dark at room temperature for 5 minutes. Cells were then analyzed by flow cytometry using a GFP signal detector with excitation wavelength of 488 nm and emission of 530 nm (to detect fibroblasts) and a Cy5 signal detector with excitation wavelength of 649 nm and emission of 670 nm (to detect apoptotic cells).
Generation of stable Cav-1 sh-RNA knockdown cells.
hTERT-BJ1 human fibroblasts were retrovirally-transduced to express GFP and either a short-hair-pin RNA against Cav-1 or a scrambled control RNA under the control of the cytomegalovirus (CMV) promoter. Successfully infected cells were selected via FACS analysis. We validated the loss of Cav-1 expression in fibroblasts harboring the Cav-1 sh-RNA by western blot analysis; Cav-1 sh-RNA fibroblasts showed a >95% reduction in Cav-1 protein levels, as compared with fibroblasts harboring the control scrambled sh-RNA vector (data not shown).
Abbreviations
| BSO | = | buthionine sulfoximine |
| CAFs | = | cancer associated fibroblasts |
| Cav-1 | = | caveolin-1 |
| DCIS | = | ductal carcinoma in situ |
| EM | = | electron microscopy |
| K8/18 | = | cytokeratin 8/18 |
| HIF-1α | = | hypoxia inducible factor 1α |
| IκB | = | inhibitor of κB |
| IκBK | = | IκB Kinase |
| NFκB | = | nuclear factor κB |
| ROS | = | reactive oxygen species |
| PHDs | = | prolyl hydroxylase domain-containing proteins |
| hTERT | = | human telomerase reverse transcriptase |
| TIGAR | = | TP53-induced glycolysis and apoptosis regulator |
Figures and Tables
Figure 1 Electron microscopy shows that fibroblasts in co-culture display increased autophagy and MCF7 cells exhibit abundant mitochondria. To monitor the status of autophagy, hTERT-fibroblasts and MCF7 cells were co-cultured for three days, fixed and evaluated by electron microscopy. (A and B) Co-cultured fibroblasts display numerous lysosomes and autophagosomes. (A) The EM image represents a fibroblast with numerous lysosomes (arrows) and autophagosomes (boxed area). (B) The higher magnification of the boxed area depicts an autophagosome. Note that the autophagosome shows (i) a double-membrane (arrowhead) and (ii) organelles within that appear to be degraded mitochondria (arrows). Bars = 1 µm for (A); = 0.2 µm for (B). (C) Co-cultured MCF7 cells exhibit the presence of many mitochondria. Note that co-cultured MCF7 cells exhibit the presence of many mitochondria (arrows) and of intermediate keratin filaments (demonstrating their epithelial origin). N, nucleus.
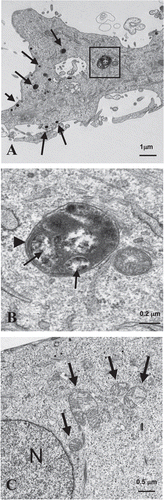
Figure 2 Hypoxia induces Cav-1 downregulation in co-cultured fibroblasts. hTERT-fibroblast-MCF7 co-cultures were placed in hypoxia (0.5% O2) or normoxia (21% O2) for 3 days. Then, cells were fixed and immuno-stained with anti-Cav-1 (Red) and anti-K8-18 (Green) antibodies. Cav-1 staining (red only) is shown on the left to better appreciate hypoxia-induced Cav-1 downregulation. Note that during hypoxia, Cav-1 is localized to intracellular vesicles. The boxed area is shown enlarged on the left to illustrate that Cav-1 accumulates in intracellular vesicles, consistent with an autophagic/lysosomal degradation mechanism. Original magnification, 40x.
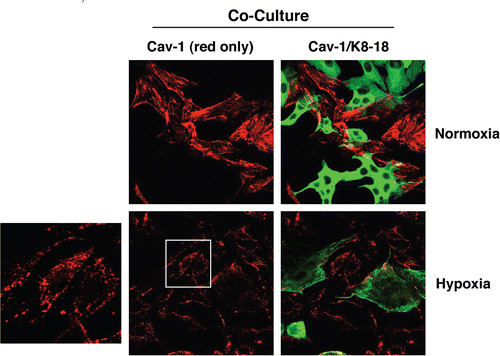
Figure 3 Hypoxia-induced autophagy drives Cav-1 degradation in fibroblasts: Rescue with chloroquine, an autophagy inhibitor. (A) Hypoxia decreases Cav-1 levels in homotypic fibroblasts. Homotypic cultures of hTERT-fibroblasts were placed in hypoxia (0.5% O2) or normoxia (21% O2) for three days. Then, cells were fixed and stained with anti-Cav-1 (red) antibodies and DAPI nuclear stain (blue). Cav-1 staining (red only) is shown at left to better appreciate the hypoxia-induced Cav-1 degradation. Original magnification, 40x. (B) Hypoxia-induced Cav-1 downregulation correlates with the expression of autophagic markers. hTERT-fibroblasts were subjected to hypoxia (0.5% O2, lower panels) or normoxia (21% O2, upper panels) for 48 hours. Then, the cells were fixed and stained with antibodies against either Cav-1 (green) or the indicated autophagic markers (red). DAPI was used to stain nuclei (blue). Note that hypoxia induces Cav-1 downregulation, while promoting autophagy. Original magnification, 80x. (C) The autophagy inhibitor chloroquine rescues the hypoxia-induced downregulation of Cav-1. To block hypoxia-induced Cav-1 degradation, hTERT-fibroblasts were subjected to hypoxia in the presence of the autophagic inhibitor chloroquine (25 µM) or vehicle alone control (H2O) for 24 hours. Then, cells were fixed and stained with anti-Cav-1 (green) antibodies and DAPI nuclear stain (blue). Note that chloroquine treatment prevents the hypoxia-induced degradation of Cav-1, as compared with control cells. Original magnification, 60x.
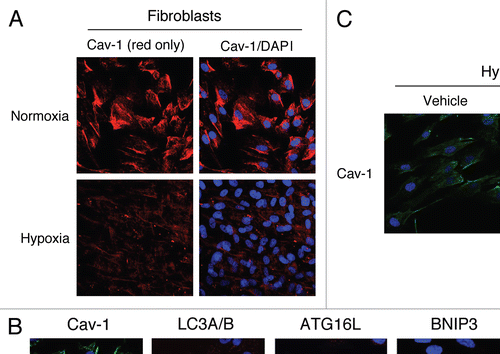
Figure 4 Oxidative stress-induced autophagy correlates with Cav-1 downregulation. BSO treatment downregulates Cav-1 while promoting autophagy. To induce oxidative stress, hTERT-fibroblasts were treated with increasing concentration of BSO (1 µM and 1 mM) for 48 hours. Cell lysates were then analyzed by Western blot analysis using anti-Cav-1 and LC3B antibodies. Note that Cav-1 levels are greatly decreased upon treatment with 1 mM of the pro-oxidant BSO. At the same concentration of BSO, the accumulation of the active LC3B-II form is highest. β-actin was used as a control to assess equal protein loading.
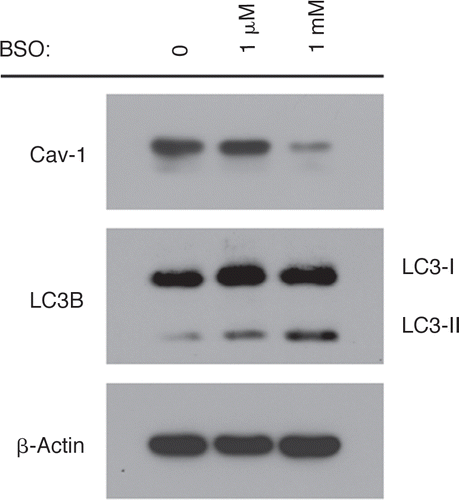
Figure 5 Activation of HIF-1α correlates with Cav-1 downregulation in fibroblasts. (A) Hypoxia-induced HIF-1α activation correlates with decreased Cav-1 levels in fibroblasts. Homotypic cultures of hTERT-fibroblasts were placed in hypoxia (0.5% O2) or normoxia (21% O2) for 3 days. Cells were lysed and analyzed by western blot analysis using anti-Cav-1 and HIF-1α antibodies. Note that HIF-1α accumulation correlates with decreased Cav-1 levels during hypoxia. β-tubulin was used as a control to assess equal protein loading. (B) Re-oxygenation restores Cav-1 levels. Co-cultures of hTERT-fibroblasts and MCF7 cells were placed either in normoxia or in hypoxia or in hypoxia followed by normoxia (hypoxia + normoxia). Then, the cells were fixed and immuno-stained with anti-Cav-1 (red) and anti-HIF-1α (green) antibodies. The left panels show the red channel only to better appreciate Cav-1 staining. Upon hypoxia, fibroblasts display HIF-1α nuclear accumulation and decreased Cav-1 expression. The boxed area is shown enlarged on the left to better illustrate the distribution of Cav-1 within intracellular vesicles, consistent with the idea that Cav-1 is degraded by an autophagic/lysosomal mechanism. Interestingly, re-oxygenation (hypoxia + normoxia, middle panels) restores Cav-1 expression, while inducing HIF-1α degradation in fibroblasts. However, HIF-1α is further activated and maintained in MCF7 cells after re-oxygenation (hypoxia + normoxia, middle panels). Original magnification, 40x.
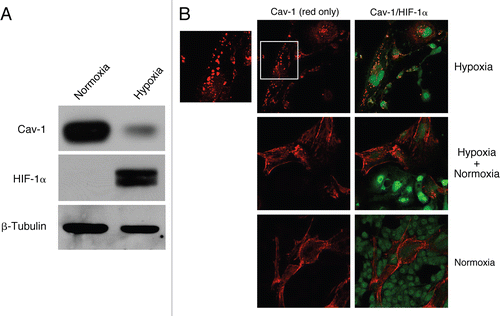
Figure 6 Pharmacological activation of HIF-1α downregulates Cav-1 levels. (A) Immunofluorescence. to pharmacologically activate HIF-1α, co-cultures of hTERT-fibroblasts and MCF7 cells were treated with PHD inhibitors, such as DMOG, 2,4 DPD and 1,4 DPCA. Cells were then fixed and immuno-stained with anti-Cav-1 (red) and anti-K8-18 (green) antibodies. Nuclei were stained with DAPI (blue). Cav-1 staining (red channel only) is shown on the left to better appreciate the decreased Cav-1 levels, after treatment with the PHD inhibitors. Importantly, images were acquired using identical exposure settings. Original magnification, 40x. (B) Western blot. Homotypic cultures of hTERT-fibroblasts were treated with the PHD inhibitor DMOG (500 µM) or vehicle control (DMSO) for 24 hours. Cell lysates were analyzed by western blot analysis using anti-Cav-1 antibodies. Note that Cav-1 levels are greatly decreased upon treatment with the HIF-1α inducer DMOG. β-actin was used as a control for equal protein loading.
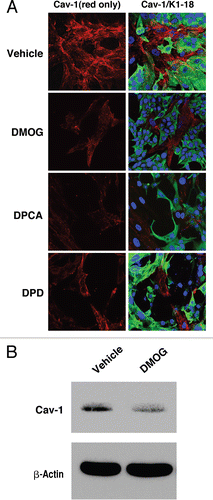
Figure 7 Pharmacological inhibition of HIF-1α rescues the hypoxia-induced downregulation of Cav-1. (A) Immunofluorescence. hTERT-fibroblasts were subjected to hypoxia (0.5% O2) for 24 hours in the presence of the HIF-1α inhibitor echinomycin (10 ng/ml) or vehicle alone (DMSO). Then, the cells were fixed and stained with anti-Cav-1 (green) antibodies and DAPI nuclear stain (blue). Note that treatment with the HIF-1α inhibitor rescues the hypoxia-induced downregulation of Cav-1, as compared to vehicle alone treated cells. Original magnification, 60x. (B) Western blot. hTERT-fibroblasts were subjected to hypoxia (0.5% O2) for 48 hours in the presence of echinomycin (2 ng/ml) or vehicle alone (DMSO). Cell lysates were analyzed by western blot analysis using anti-Cav-1 antibodies. Note that Cav-1 levels are greatly increased upon treatment with the HIF-1α inhibitor. β-actin was used as an equal loading control.
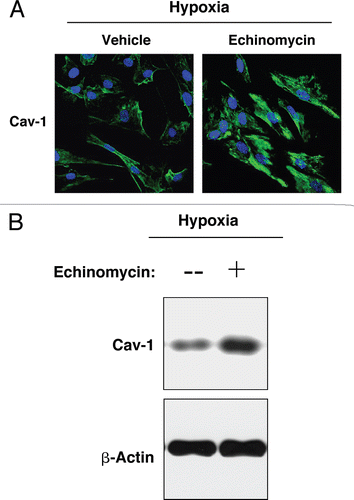
Figure 8 Activation of the pro-autophagic NFκB pathway induces Cav-1 downregulation. (A) NFκB is activated during hypoxia. hTERT-fibroblasts were subjected to hypoxia (0.5% O2) or normoxia for 48 hours. Then, the cells were fixed and stained with phospho-NFκB (phospho-p65 at Ser 276, red) antibodies and DAPI nuclear stain (blue). Note that hypoxia promotes the activation of the pro-autophagic NFκB pathway. Original magnification, 40x. (B) NFκB signaling is activated in co-cultured fibroblasts. Day 5 hTERT-fibroblast-MCF7 cell co-cultures were fixed and immuno-stained with antibodies against phospho-NFκB (phospho-p65 at Ser 276, red) and anti-K8-18 (green). As control, homotypic cultures of fibroblasts and MCF7 cells were fixed and stained in parallel. Note that phospho-NFκB is exclusively detected in co-cultured fibroblasts nuclei (white arrows). Importantly, images were acquired using identical exposure settings. Original magnification, 40x. (C and D) Treatment with the NFκB inhibitor PS115 restores Cav-1 expression in co-culture. Day 5 hTERT-fibroblast-MCF7 co-cultures were treated with 10 µM PS1145 or vehicle alone (DMSO) for 24 hours. (C) Cells were fixed and immuno-stained with anti-phospho-NFκB antibodies (red). Nuclei were stained with DAPI (blue). As expected, the accumulation of phospho-NFκB in co-cultured cells (white arrows) is abolished by treatment with the NFκB inhibitor. Original magnification, 40x. (D) Cells were fixed and immunostained with anti-Cav-1 (red) and anti-K8-18 (green) antibodies. Nuclei were stained with DAPI (blue). Note that treatment with the NFκB inhibitor PS1145 restores Cav-1 expression in co-cultured fibroblasts. The red only image is shown on the left to better appreciate Cav-1 staining. Original magnification, 40x.
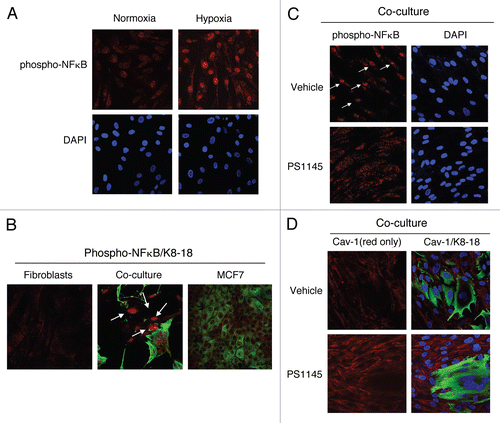
Figure 9 Cav-1 knockdown is sufficient to promote autophagy/mitophagy. (A) Acute Cav-1 downregulation activates the autophagy inducer NFκB. hTERT-fibroblasts treated with Cav-1 siRNA (right) or control siRNA (left) were fixed and immuno-stained with antibodies against phospho-NFκB (phospho-p65 at Ser 276, green). DAPI was used to visualize nuclei (blue). Note that Cav-1 knockdown is sufficient to induce phospho-NFκB nuclear localization and activation. White arrows point at the nuclear localization of phospho-NFκB in Cav-1 siRNA treated cells. Importantly, images were acquired using identical exposure settings. Original magnification, 40x. (B and C) Acute loss of Cav-1 increases the expression of autophagic markers. hTERT-fibroblasts were treated with Cav-1 siRNA or control (CTR) siRNA. (B) Western blot analysis. Cells were analyzed by western blot analysis using antibodies against the indicated autophagic markers. β-tubulin was used as equal loading control. (C) Immunofluorescence. Cells were fixed and immuno-stained with antibodies against beclin 1, BNIP3 and BNIP3L. DAPI was used to visualize nuclei (blue). Importantly, paired images were acquired using identical exposure settings. Original magnification, 40x. Note that acute Cav-1 knockdown is sufficient to greatly increase the expression levels of all the autophagy/mitophagy markers we examined.
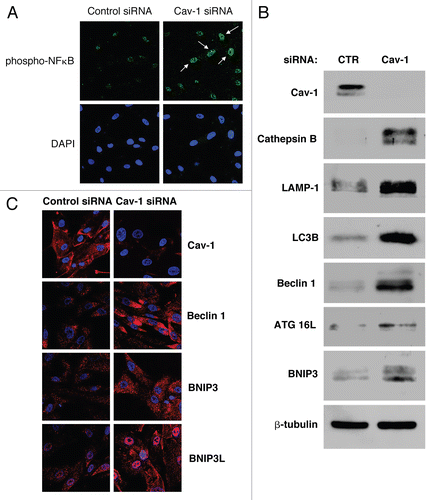
Figure 10 Cancer cells promote the activation of the pro-autophagic NFκB and HIF-1α pathways in adjacent fibroblasts. NIH3T3-HIF-Luc or NIH3T3-NFκB-Luc reporter fibroblasts were plated with MCF7 cells. The next day, the media was changed and that was considered day 0. Cells were then cultured for up to 5 days. As a control, homotypic cultures of fibroblasts were established and processed in parallel. Luciferase activity was measured at day 0, 3 and 5. The graph on the left shows data generated using NIH3t3-NFκB-Luc cells, whereas the graph on the right graph represents data from the use of NIH3T3-HIF-Luc cells. Note that luciferase activity is increased by 9-fold and 2-fold respectively at day 0 and 3 in NFκB-Luc co-cultured fibroblasts, compared to homotypic cultures. These results suggest that the NFκB pathway is potently activated as an early event during co-culture. Conversely, luciferase activity is increased by ∼4-fold at day 5 in NIH3T3-HIF-Luc co-cultured cells, compared to homotypic cultures, suggesting that HIF-1α activation occurs at a later time-point. *p = 0.000004 for day 0 NFκB-Luc, *p = 0.006 for day 3 NFκB-Luc, *p = 0.0003 for day 5 NFκB-Luc, *p = 0.003 for day 5 HIF-Luc (Student's t-test).
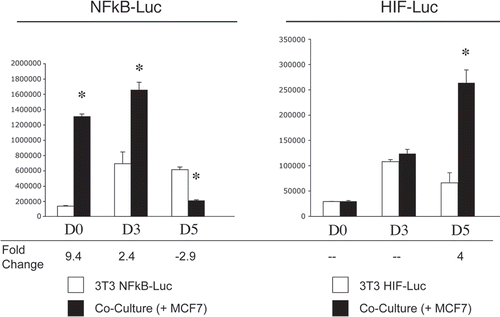
Figure 11 Loss of Cav-1 in fibroblasts protects adjacent MCF7 cells against apoptosis. MCF7 cells were co-cultured for 72 hours with hTERT-fibroblasts carrying either a GFP (+) control shRNA (CTR) vector or a GFP (+) Cav-1 shRNA (KD) vector. Corresponding homotypic cultures were established in parallel. Then, the cells were subjected to annexin-V staining and analyzed by FACS. Thus, the GFP (+) and GFP (−) cells represent hTERT-fibroblasts and MCF7 cells, respectively. (A) MCF7 cell apoptotic rate: Cav-1 knockdown fibroblasts protect MCF7 cells against apoptosis. MCF7 cells co-cultured with CTR-fibroblasts show an ∼3-fold reduction in apoptosis, as compared to MCF7 cells cultured alone. However, co-cultures with KD fibroblasts provide MCF7 cells with a greater protection against apoptosis (8.5-fold decrease in apoptotic rate compared to MCF7 cell mono-cultures). The upper graph represents the percentage of annexin-V (+) cells. The lower graph represents the fold change versus MCF7 cells cultured in the absence of fibroblasts. *p ≤ 0.0002, **p ≤ 0.0000006 versus MCF7 cells cultured alone. *p ≤ 0.02 versus Cav-1 KD (Student's t-test). (B) Fibroblast apoptotic rate: Cav-1 knockdown protects fibroblasts against apoptosis. In homotypic cultures, KD fibroblasts are protected by 2.7-fold against apoptosis as compared to CTR fibroblasts. In addition, CTR fibroblasts co-cultured with MCF7 cells display a 2.4-fold increase in apoptosis, compared to CTR mono-cultures. Interestingly, co-cultured KD fibroblasts exhibit a 2-fold decrease in apoptosis compared to co-cultured CTR fibroblasts. These results suggest that a loss of Cav-1 protects fibroblasts in co-culture against apoptosis. *p = 0.03 versus the other three experimental conditions (Student's t-test).
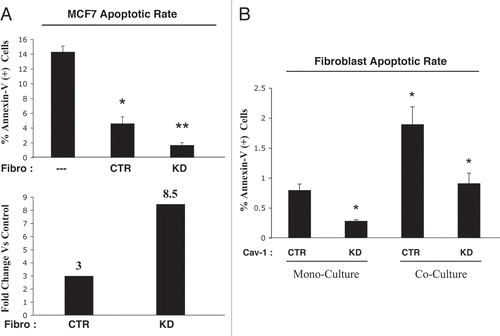
Figure 12 Co-cultured MCF7 cells display the upregulation of the autophagy and apoptosis inhibitor TIGAR. Day 5 hTERT-fibroblast-MCF7 co-cultures were fixed and immuno-stained with antibodies against TIGAR (red) and anti-K8-18 (green). DAPI was used to visualize nuclei (blue). As a control, homotypic cultures of fibroblasts and MCF7 cells were fixed and stained in parallel. Note that TIGAR is greatly upregulated in co-cultured MCF7 cells as compared to MCF7 cells mono-cultures. Importantly, images were acquired using identical exposure settings. Original magnification, 40x.
Figure 13 Cav-1 gene deletion in mice induces a pseudo-hypoxic phenotype. To monitor hypoxia signaling in vivo, the hypoxia marker pimonidazole was injected in the tail vein of WT and Cav-1 (−/−) null mice. One hour post-injection, mice were sacrificed and the mammary glands and lungs were collected. Paraffin-embedded sections of mammary gland and lung were immuno-stained using a rabbit antiserum to pimonidazole. Slides were counter-stained with hematoxylin. Note that both the mammary fat pads and the lung parenchyma of Cav-1 (−/−) null mice display strong anti-pimonidazole staining. These data suggest that Cav-1 (−/−) mice undergo pseudo-hypoxic stress at steady-state. Original magnification, 40x.
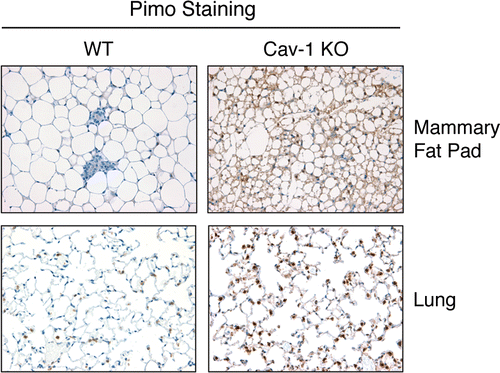
Figure 14 Cav-1 (−/−) null mammary glands exhibit increased autophagy. To monitor autophagy in vivo, paraffin-embedded sections of mammary glands from 5 month old WT and Cav-1 (−/−) null mice were immuno-stained with two markers of autophagy, LC3A/B (A) and BNIP3L (B). Slides were counter-stained with hematoxylin. Note that both LC3A/B and BNIP3L are greatly increased in Cav-1 (−/−) mammary fat pads. The boxed area of LC3A/B staining is shown at higher magnification to better appreciate the LC3 positive vesicles in Cav-1 (−/−) adipocytes. Original magnification, 60x.
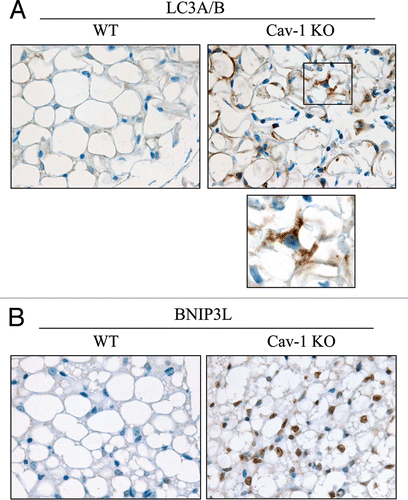
Figure 15 BNIP3L is highly increased in the stroma of human breast cancers that lack stromal Cav-1. Paraffin-embedded sections of human breast cancer samples lacking stromal Cav-1 were immuno-stained with antibodies directed against BNIP3L (ab59908; Abcam). Slides were then counter-stained with hematoxylin. Note that BNIP3L is highly expressed in the stromal compartment of human breast cancers that lack stromal Cav-1. The boxed area shown at higher magnification reveals punctate staining, consistent with mitochondrial and/or lysosomal localization. Original magnification, 40x and 60x, as indicated.
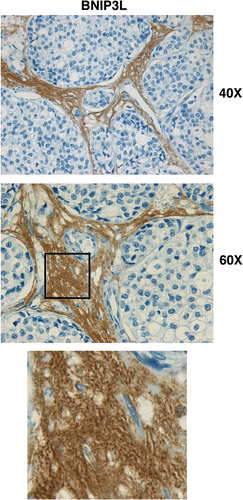
Figure 16 Autophagy in cancer associated fibroblasts (CAFs) fuels tumor cell survival. Here, we present a new model in which cancer cells trigger oxidative stress and activate two pro-autophagic drivers, namely HIF-1α and NFκB, in adjacent fibroblasts. Thus, CAFs undergo autophagy and mitophagy, leading to a loss of Cav-1 and metabolic re-programming. A loss of stromal Cav-1 aggravates oxidative stress and further promotes autophagy and mitophagy. Stromal autophagy generates building blocks (such as recycled free amino acids, fatty acids and nucleotides) that can be directly utilized by cancer cells to sustain growth and maintain cell viability. HIF-1α activation and consequent mitophagy in CAFs induces mitochondrial dys-function and enhances aerobic glycolysis, leading to the secretion of high-energy nutrients (such as lactate and pyruvate) that can directly feed mitochondrial biogenesis and oxidative mitochondrial metabolism in cancer cells. Loss of Cav-1 in stromal cells protects cancer cells from apoptosis, at least in part via TIGAR upregulation. In this model, TIGAR protects epithelial cancer cells from oxidative stress by simultaneously inhibiting three inter-related cellular processes, namely (1) aerobic glycolysis, (2) apoptosis and (3) autophagy. Thus, epithelial cancer cells exploit CAFs to satisfy their increased energy demand by forcing these stromal cells to undergo a unilateral and vectorial energy transfer (via compartment-specific autophagy) to sustain epithelial cancer cell growth. Transfer of nutrients from autophagy-prone catabolic stromal cells to autophagy-resistant anabolic cancer cells promotes epithelial cancer cell survival, thereby enhancing tumor growth.
Additional material
Download Zip (74.3 KB)Acknowledgements
F.S. and her laboratory were supported by grants from the W.W. Smith Charitable Trust, the Breast Cancer Alliance (BCA) and a Research Scholar Grant from the American Cancer Society (ACS). M.P.L. was supported by grants from the NIH/NCI (R01-CA-080250; R01-CA-098779; R01-CA-120876; R01-AR-055660) and the Susan G. Komen Breast Cancer Foundation. A.K.W. was supported by a Young Investigator Award from Breast Cancer Alliance, Inc., and a Susan G. Komen Career Catalyst Grant. R.G.P. was supported by grants from the NIH/NCI (R01-CA-70896, R01-CA-75503, R01-CA-86072 and R01-CA-107382) and the Dr. Ralph and Marian C. Falk Medical Research Trust. The Kimmel Cancer Center was supported by the NIH/NCI Cancer Center Core grant P30-CA-56036 (to R.G.P.). Funds were also contributed by the Margaret Q. Landenberger Research Foundation (to M.P.L.). This project is funded, in part, under a grant with the Pennsylvania Department of Health (to M.P.L. and F.S.). The Department specifically disclaims responsibility for any analyses, interpretations or conclusions. This work was also supported, in part, by a Centre grant in Manchester from Breakthrough Breast Cancer in the UK (to A.H.) and an Advanced ERC Grant from the European Research Council.
References
- Papandreou I, Cairns RA, Fontana L, Lim AL, Denko NC. HIF-1 mediates adaptation to hypoxia by actively downregulating mitochondrial oxygen consumption. Cell Metab 2006; 3:187 - 197
- Kim JW, Tchernyshyov I, Semenza GL, Dang CV. HIF-1-mediated expression of pyruvate dehydrogenase kinase: a metabolic switch required for cellular adaptation to hypoxia. Cell Metab 2006; 3:177 - 185
- Salceda S, Caro J. Hypoxia-inducible factor 1alpha (HIF-1alpha) protein is rapidly degraded by the ubiquitin-proteasome system under normoxic conditions. Its stabilization by hypoxia depends on redox-induced changes. J Biol Chem 1997; 272:22642 - 22647
- Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, et al. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 2005; 1:401 - 408
- Schofield CJ, Ratcliffe PJ. Signalling hypoxia by HIF hydroxylases. Biochem Biophys Res Commun 2005; 338:617 - 626
- Valko M, Rhodes CJ, Moncol J, Izakovic M, Mazur M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem Biol Interact 2006; 160:1 - 40
- Haynes CM, Titus EA, Cooper AA. Degradation of misfolded proteins prevents ER-derived oxidative stress and cell death. Mol Cell 2004; 15:767 - 776
- Mills E, Jobsis FF. Mitochondrial respiratory chain of carotid body and chemoreceptor response to changes in oxygen tension. J Neurophysiol 1972; 35:405 - 428
- Wyatt CN, Buckler KJ. The effect of mitochondrial inhibitors on membrane currents in isolated neonatal rat carotid body type I cells. J Physiol 2004; 556:175 - 191
- Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 1998; 95:11715 - 11720
- Chandel NS, McClintock DS, Feliciano CE, Wood TM, Melendez JA, Rodriguez AM, et al. Reactive oxygen species generated at mitochondrial complex III stabilize hypoxia-inducible factor-1alpha during hypoxia: a mechanism of O2 sensing. J Biol Chem 2000; 275:25130 - 25138
- Perkins ND, Gilmore TD. Good cop, bad cop: the different faces of NFkappaB. Cell Death Differ 2006; 13:759 - 772
- Perkins ND. Post-translational modifications regulating the activity and function of the nuclear factor kappa B pathway. Oncogene 2006; 25:6717 - 6730
- Cummins EP, Berra E, Comerford KM, Ginouves A, Fitzgerald KT, Seeballuck F, et al. Prolyl hydroxylase-1 negatively regulates IkappaB kinase-beta, giving insight into hypoxia-induced NFkappaB activity. Proc Natl Acad Sci USA 2006; 103:18154 - 18159
- Jung Y, Isaacs JS, Lee S, Trepel J, Liu ZG, Neckers L. Hypoxia-inducible factor induction by tumour necrosis factor in normoxic cells requires receptor-interacting protein-dependent nuclear factor kappaB activation. Biochem J 2003; 370:1011 - 1017
- Rius J, Guma M, Schachtrup C, Akassoglou K, Zinkernagel AS, Nizet V, et al. NFkappaB links innate immunity to the hypoxic response through transcriptional regulation of HIF-1alpha. Nature 2008; 453:807 - 811
- Pantuck AJ, An J, Liu H, Rettig MB. NFkappaB-dependent plasticity of the epithelial to mesenchymal transition induced by Von Hippel-Lindau inactivation in renal cell carcinomas. Cancer Res 2010; 70:752 - 761
- An J, Rettig MB. Mechanism of von Hippel-Lindau protein-mediated suppression of nuclear factor kappa B activity. Mol Cell Biol 2005; 25:7546 - 7556
- Zhang H, Bosch-Marce M, Shimoda LA, Tan YS, Baek JH, Wesley JB, et al. Mitochondrial autophagy is an HIF-1-dependent adaptive metabolic response to hypoxia. J Biol Chem 2008; 283:10892 - 10903
- Criollo A, Senovilla L, Authier H, Maiuri MC, Morselli E, Vitale I, et al. The IKK complex contributes to the induction of autophagy. EMBO J 2010; 29:619 - 631
- Copetti T, Bertoli C, Dalla E, Demarchi F, Schneider C. p65/RelA modulates BECN1 transcription and autophagy. Mol Cell Biol 2009; 29:2594 - 2608
- Copetti T, Demarchi F, Schneider C. p65/RelA binds and activates the beclin 1 promoter. Autophagy 2009; 5:858 - 859
- Nivon M, Richet E, Codogno P, Arrigo AP, Kretz-Remy C. Autophagy activation by NFkappaB is essential for cell survival after heat shock. Autophagy 2009; 5:766 - 783
- Bellot G, Garcia-Medina R, Gounon P, Chiche J, Roux D, Pouyssegur J, et al. Hypoxia-induced autophagy is mediated through hypoxia-inducible factor induction of BNIP3 and BNIP3L via their BH3 domains. Mol Cell Biol 2009; 29:2570 - 2581
- Liang XH, Jackson S, Seaman M, Brown K, Kempkes B, Hibshoosh H, et al. Induction of autophagy and inhibition of tumorigenesis by beclin 1. Nature 1999; 402:672 - 676
- Levine B, Kroemer G. Autophagy in the pathogenesis of disease. Cell 2008; 132:27 - 42
- Kalluri R, Zeisberg M. Fibroblasts in cancer. Nat Rev Cancer 2006; 6:392 - 401
- Koleske AJ, Baltimore D, Lisanti MP. Reduction of caveolin and caveolae in oncogenically transformed cells. Proc Natl Acad Sci USA 1995; 92:1381 - 1385
- Engelman JA, Wykoff CC, Yasuhara S, Song KS, Okamoto T, Lisanti MP. Recombinant expression of caveolin-1 in oncogenically transformed cells abrogates anchorage-independent growth. J Biol Chem 1997; 272:16374 - 16381
- Lee SW, Reimer CL, Oh P, Campbell DB, Schnitzer JE. Tumor cell growth inhibition by caveolin re-expression in human breast cancer cells. Oncogene 1998; 16:1391 - 1397
- Galbiati F, Volonte D, Engelman JA, Watanabe G, Burk R, Pestell RG, et al. Targeted downregulation of caveolin-1 is sufficient to drive cell transformation and hyperactivate the p42/44 MAP kinase cascade. EMBO J 1998; 17:6633 - 6648
- Mercier I, Casimiro MC, Wang C, Rosenberg AL, Quong J, Minkeu A, et al. Human breast cancer-associated fibroblasts (CAFs) show caveolin-1 downregulation and RB tumor suppressor functional inactivation: Implications for the response to hormonal therapy. Cancer Biol Ther 2008; 7:1212 - 1225
- Witkiewicz AK, Dasgupta A, Sotgia F, Mercier I, Pestell RG, Sabel M, et al. An absence of stromal caveolin-1 expression predicts early tumor recurrence and poor clinical outcome in human breast cancers. Am J Pathol 2009; 174:2023 - 2034
- Sloan EK, Ciocca DR, Pouliot N, Natoli A, Restall C, Henderson MA, et al. Stromal cell expression of caveolin-1 predicts outcome in breast cancer. Am J Pathol 2009; 174:2035 - 2043
- Witkiewicz AK, Dasgupta A, Sammons S, Er O, Potoczek MB, Guiles F, et al. Loss of stromal caveolin-1 expression predicts poor clinical outcome in triple negative and basal-like breast cancers. Cancer Biol Ther 2010; 10:135 - 143
- Witkiewicz AK, Dasgupta A, Nguyen KH, Liu C, Kovatich AJ, Schwartz GF, et al. Stromal caveolin-1 levels predict early DCIS progression to invasive breast cancer. Cancer Biol Ther 2009; 8:1071 - 1079
- Witkiewicz AK, Casimiro MC, Dasgupta A, Mercier I, Wang C, Bonuccelli G, et al. Towards a new “stromal-based” classification system for human breast cancer prognosis and therapy. Cell Cycle 2009; 8:1654 - 1658
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: Aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001
- Martinez-Outschoorn UE, Balliet RM, Rivadeneira DB, Chiavarina B, Pavlides S, Wang C, et al. Oxidative stress in cancer associated fibroblasts drives tumor-stroma co-evolution: A new paradigm for understanding tumor metabolism, the field effect and genomic instability in cancer cells. Cell Cycle 2010; 9:3256 - 3276
- Martinez-Outschoorn UE, Pavlides S, Whitaker-Menezes D, Daumer KM, Milliman JN, Chiavarina B, et al. Tumor cells induce the cancer associated fibroblast phenotype via Caveolin-1 degradation: Implications for breast cancer and DCIS therapy with autophagy inhibitors. Cell Cycle 2010; 9:2423 - 2433
- Kong D, Park EJ, Stephen AG, Calvani M, Cardellina JH, Monks A, et al. Echinomycin, a small-molecule inhibitor of hypoxia-inducible factor-1 DNA-binding activity. Cancer Res 2005; 65:9047 - 9055
- Bensaad K, Cheung EC, Vousden KH. Modulation of intracellular ROS levels by TIGAR controls autophagy. EMBO J 2009; 28:3015 - 3026
- Bensaad K, Tsuruta A, Selak MA, Vidal MN, Nakano K, Bartrons R, et al. TIGAR, a p53-inducible regulator of glycolysis and apoptosis. Cell 2006; 126:107 - 120
- Pavlides S, Tsirigos A, Vera I, Flomenberg N, Frank PG, Casimiro MC, et al. Loss of stromal Caveolin-1 leads to oxidative stress, mimics hypoxia and drives inflammation in the tumor microenvironment, conferring the “Reverse Warburg Effect”: A transcriptional informatics analysis with validation. Cell Cycle 2010; 9:2201 - 2219
- Razani B, Combs TP, Wang XB, Frank PG, Park DS, Russell RG, et al. Caveolin-1-deficient mice are lean, resistant to diet-induced obesity and show hypertriglyceridemia with adipocyte abnormalities. J Biol Chem 2002; 277:8635 - 8647
- Pavlides S, Tsirigos A, Migneco G, Whitaker-Menezes D, Chiavarina B, Flomenberg N, et al. The autophagic tumor stroma model of cancer: Role of oxidative stress and ketone production in fueling tumor cell metabolism. Cell Cycle 2010; 9:3485 - 3505
- Shi Y, Pritchard KA Jr, Holman P, Rafiee P, Griffith OW, Kalyanaraman B, et al. Chronic myocardial hypoxia increases nitric oxide synthase and decreases caveolin-3. Free Radic Biol Med 2000; 29:695 - 703
- Toullec A, Gerald D, Despouy G, Bourachot B, Cardon M, Lefort S, et al. Oxidative stress promotes myofibroblast differentiation and tumour spreading. EMBO Mol Med 2010; 2:211 - 230
- Stathopoulos GT, Sherrill TP, Han W, Sadikot RT, Yull FE, Blackwell TS, et al. Host nuclear factor-kappaB activation potentiates lung cancer metastasis. Mol Cancer Res 2008; 6:364 - 371
- Joyce JA, Pollard JW. Microenvironmental regulation of metastasis. Nat Rev Cancer 2009; 9:239 - 252
- Mueller L, Goumas FA, Affeldt M, Sandtner S, Gehling UM, Brilloff S, et al. Stromal fibroblasts in colorectal liver metastases originate from resident fibroblasts and generate an inflammatory microenvironment. Am J Pathol 2007; 171:1608 - 1618
- Erez N, Truitt M, Olson P, Hanahan D. Cancer-associated fibroblasts are activated in incipient neoplasia to orchestrate tumor-promoting inflammation in an NFkappaB-dependent manner. Cancer Cell 2010; 17:135 - 147
- Gherghiceanu M, Hinescu ME, Popescu LM. Myocardial interstitial Cajal-like cells (ICLC) in caveolin-1 KO mice. J Cell Mol Med 2009; 13:202 - 206
- Le Lay S, Briand N, Blouin CM, Chateau D, Prado C, Lasnier F, et al. The lipoatrophic caveolin-1 deficient mouse model reveals autophagy in mature adipocytes. Autophagy 2010; 6:754 - 763
- Nomura H, Uzawa K, Yamano Y, Fushimi K, Ishigami T, Kouzu Y, et al. Overexpression and altered subcellular localization of autophagy-related 16-like 1 in human oral squamous-cell carcinoma: correlation with lymphovascular invasion and lymph-node metastasis. Hum Pathol 2009; 40:83 - 91
- Kleer CG, Bloushtain-Qimron N, Chen YH, Carrasco D, Hu M, Yao J, et al. Epithelial and stromal cathepsin K and CXCL14 expression in breast tumor progression. Clin Cancer Res 2008; 14:5357 - 5367
- Nadji M, Fresno M, Nassiri M, Conner G, Herrero A, Morales AR. Cathepsin D in host stromal cells, but not in tumor cells, is associated with aggressive behavior in node-negative breast cancer. Hum Pathol 1996; 27:890 - 895
- Tu C, Ortega-Cava CF, Chen G, Fernandes ND, Cavallo-Medved D, Sloane BF, et al. Lysosomal cathepsin B participates in the podosome-mediated extracellular matrix degradation and invasion via secreted lysosomes in v-Src fibroblasts. Cancer Res 2008; 68:9147 - 9156
- Moles A, Tarrats N, Fernandez-Checa JC, Mari M. Cathepsins B and D drive hepatic stellate cell proliferation and promote their fibrogenic potential. Hepatology 2009; 49:1297 - 1307
- Zhao H, Peehl DM. Tumor-promoting phenotype of CD90 hi prostate cancer-associated fibroblasts. Prostate 2009; 69:991 - 1000
- Singh H, Dang TD, Ayala GE, Rowley DR. Transforming growth factor-beta1 induced myofibroblasts regulate LNCaP cell death. J Urol 2004; 172:2421 - 2425
- Razani B, Zhang XL, Bitzer M, von Gersdorff G, Bottinger EP, Lisanti MP. Caveolin-1 regulates transforming growth factor (TGF)-beta/SMAD signaling through an interaction with the TGFbeta type I receptor. J Biol Chem 2001; 276:6727 - 6738
- Pavlides S, Whitaker-Menezes D, Castello-Cros R, Flomenberg N, Witkiewicz AK, Frank PG, et al. The reverse Warburg effect: aerobic glycolysis in cancer associated fibroblasts and the tumor stroma. Cell Cycle 2009; 8:3984 - 4001
- Sotgia F, Del Galdo F, Casimiro MC, Bonuccelli G, Mercier I, Whitaker-Menezes D, et al. Caveolin-1-/- null mammary stromal fibroblasts share characteristics with human breast cancer-associated fibroblasts. Am J Pathol 2009; 174:746 - 761
- Sato T. A modified method for lead staining of thin sections. J Electron Microsc (Tokyo) 1968; 17:158 - 159
- Schubert W, Frank PG, Woodman SE, Hyogo H, Cohen DE, Chow CW, et al. Microvascular hyperpermeability in caveolin-1 (−/−) knock-out mice. Treatment with a specific nitric-oxide synthase inhibitor, L-NAME, restores normal microvascular permeability in Cav-1 null mice. J Biol Chem 2002; 277:40091 - 40098
- Hanai J, Dhanabal M, Karumanchi SA, Albanese C, Waterman M, Chan B, et al. Endostatin causes G1 arrest of endothelial cells through inhibition of cyclin D1. J Biol Chem 2002; 277:16464 - 16469
- Sotgia F, Casimiro MC, Bonuccelli G, Liu M, Whitaker-Menezes D, Er O, et al. Loss of caveolin-3 induces a lactogenic microenvironment that is protective against mammary tumor formation. Am J Pathol 2009; 174:613 - 629