Abstract
Programmed cell elimination is an important pathological mediator of disease. Multiple pathways to programmed cell death have been delineated, including apoptosis, autophagy, and programmed necrosis. Cross-talk between the signaling pathways mediating each process has made it difficult to define specific mechanisms of in vivo programmed cell death. For this reason, many “apoptotic” diseases may involve other death signaling pathways. Recent advances in genetic complementation using mouse knock-out models are helping to dissect apoptotic and necrotic cell death in different pathological states. The current state of research in this area is reviewed, focusing upon new findings describing the role of programmed necrosis induced by the mitochondrial permeability transition in mouse models of heart failure and diabetes.
Introduction
Like people, cells are born, they live for a time and (with a few notable exceptions) they die. Indeed, cell death is an essential part of tissue renewal. The linkage between cell death and birth is bi-directional. An unexpected loss of cells accelerates growth of similar cells in a healing process that is essential for functional homeostasis, whereas the need to replace senescent cells with new cells requires programmed elimination of older, damaged or less functional tissue. An appreciation of these two pathophysiological contexts led to the classical paradigm of cell death being either mandated or elective. Mandated cell death (often referred to as “necrosis”) is typically the consequence of trauma or tissue damage, and has therefore been anthropomorphically labeled cell murder; like murder, mandated cell death is not tidy. The cellular response to lethal injury involves cell swelling, loss of membrane integrity and release of chemical mediators that can cause collateral damage. From the victim's (cell/tissue/organ) perspective, this form of cell death is random and therefore unplanned. For the organism there is a crime scene to clean and cellular corpses to remove. Critical functions normally performed by the murdered cells are interrupted by their absence, and can be further disrupted by first responders (i.e., inflammation).
By contrast, the conventional view is that elective cell death is much cleaner for all concerned. This is certainly the case for apoptotic cell death, as originally conceived by Vogt in the 19th century and more strictly defined by Kerr, Wyllie and Curie in 1972.Citation1 The best anthropomorphism for apoptosis is probably “assisted cell suicide” as there is extensive molecular and cellular infrastructure that supports this form of elective cell termination. Apoptotic cell death is the deliberate result of processes initiated in response to developmental cues or external factors that are, themselves, non-lethal. In either case, the cell decides to remove itself, packs up its constituent proteins, organelles and DNA for easy removal and essentially “turns off and fades away”. As a consequence of this planning and effort, the untoward collateral effects associated with mandated cell death are largely avoided.
Elective cell death is essential for normal tissue development and homeostasis, and is therefore tightly regulated. Apoptotic cell death is an evolutionarily conserved process resulting from cascade activation of caspase cysteine proteases downstream of cytokine death receptors (the extrinsic apoptosis pathway) or transcriptionally and post-translationally regulated mitochondrial-localized Bcl2 family members (the intrinsic apoptosis pathway; reviewed in ref. Citation2). Dysregulation of programmed cell death perturbs normal tissue homeostasis and is implicated in cancer (too little apoptosis), Alzheimer disease (inappropriate apoptosis) and other chronic degenerative conditions.Citation3–Citation5
In recent years, this comfortable and straightforward paradigm of mandated versus elective cell death has been complicated by the recognition of multiple apoptotic and non-apoptotic pathways for programmed cell elimination. In particular, it is clear that cell death also occurs via a phenomenon of “programmed necrosis”, which is elective like apoptosis, but untidy like necrosis. To extend the previous anthropmorphisms, programmed necrosis is neither cell murder, nor assisted cell suicide, but is more like the cell figuratively throwing itself in front of an oncoming bus. The result is elective cell elimination in a manner that is functionally disruptive and activates inflammatory mechanisms. The molecular and biochemical pathways that regulate, initiate and mediate programmed cell necrosis are only now being defined, and it likely to be mediated by multiple non-apoptotic mechanisms leading to abrupt metabolic shut-down. It is ironic that John Foxton Ross Kerr, the father of modern apoptosis, originally used the term “programmed cell necrosis” to describe his concept of natural cell death. The various terms currently being used to describe non-apoptotic elective cell death (programmed necrosis, regulated necrosis, necroptosis) continue to be confusing as none are mechanistically based.Citation6,Citation7
Apoptosis and programmed necrosis are mediated by distinct signaling pathways, but the two processes can be difficult to differentiate in vivo. There is no specific histochemical marker for programmed necrosis. Dystrophic calcification and complement activation have been used to identify necrotic tissue, but these markers do not differentiate between standard and programmed tissue necrosis. On the other hand, conventional markers for apoptosis (cytochrome c release, activate caspases and TUNEL labeling) do not exclude programmed necrosis because mitochondrial swelling and outer membrane rupture that are the primary cause of programmed necrosis secondarily release cytochrome c into the cytosol. Although this activates apoptosis signaling, reversal of ATP production in cells undergoing necrosis prevents the orderly (and energy-requiring) dismantling of the cellular machinery, and so they don't die of apoptosis. Concomitant activation of apoptotic markers in cells undergoing programmed necrosis seems likely to have implicated apoptosis in diseases where programmed necrosis is a primary mechanism of elective cell elimination.
Here, new findings linking programmed necrosis mediated via the mitochondrial permeability transition with diabetes and heart failure are explored. The striking benefits that accrued from genetic interruption of programmed necrosis in mouse models of these two human conditions should prompt a re-evaluation of widely held notions that apoptotic cell death is the major effector of pathological cell loss.
Pancreatic β-cell Deficiency in Diabetes; Apoptosis or Programmed Necrosis?
A simple operational definition of diabetes is: any condition in which endogenous insulin production and secretion are insufficient to meet metabolic demands. Mechanistically, this includes conditions where there is an absolute paucity of insulin in individuals of normal body mass (type 1 diabetes),Citation8 or where there is an abundance of insulin in individuals whose need is still greater, due to increased body mass and/or insulin resistance (type 2 diabetes).Citation9,Citation10 Insulin is produced exclusively by β-cells located within the pancreatic Islets of Langerhans, and these cells have the ability to undergo hyperplasia in response to metabolic stress.Citation11 Both forms of diabetes are causally linked to (absolute or relative) β-cell deficiency,Citation12 and in many instances attributed to increased β-cell apoptosis.Citation13–Citation15 However, targeting β-cell apoptosis has either been incompletely successful in preventing diabetes,Citation16 or has unmasked latent oncogenic potential.Citation17 These results point to a parallel programmed death pathway in pancreatic β-cells.Citation18
We recently used genetic complementation in a mouse diabetes model to interrogate the role of programmed β-cell necrosis caused by opening of the mitochondrial permeability transition pore (MPTP). The MPTP is a non-selective pore in the mitochondrial inner membrane that opens in response to increased local calcium concentration.Citation19 Proton diffusion through the MPTP dissipates the normal mitochondrial pH gradient and inner membrane electrochemical potential (ΔΨm) that are required for ATP production through oxidative phosphorylation.Citation20 MPTP opening therefore reverses ATP synthesis, and the mitochondrion becomes a net ATP consumer. The loss of cellular ATP subsequent to MPTP opening ignites a cascade of metabolic dysfunction that can ultimately lead to cell necrosis.
Another consequence of MPTP opening is osmotic mitochondrial swelling that disrupts outer mitochondrial membranes, permitting cytochrome c to be released into the cytosol from the mitochondrial inter-membranous space. Although MPTP-dependent cytochrome c release can activate the apoptotic caspase cascade, cell death by apoptosis requires ATP that is critically deficient after the mitochondrial permeability transition. Thus, cell death subsequent to the mitochondrial permeability transition proceeds via metabolic shutdown and programmed necrosis, rather than apoptosis. Because cytochrome c release and its sequelae occur in both apoptosis and MPTP-dependent programmed cell necrosis, they do not reliably indicate one or the other cell death pathway.
The structural components of the MPTP are not completely clear.Citation21,Citation22 Core MPTP elements consist of the voltage-dependent anion channel (VDAC), the adenine nucleotide translocator (ANT) and cyclophilin D (CyP-D). The VDAC was long considered to be an essential component of the MPTP,Citation23–Citation25 but genetic models have convincingly refuted this notion; the VDAC is dispensable for both Bcl2 family member-mediated apoptosis and MPTP-dependent necrosis.Citation26 Likewise, gene ablation studies in mice showed that mitochondria lacking ANT can undergo permeability transition.Citation27 ANTs may function as phosphate- and calcium-sensitive MPTP regulators. In contrast, three independent groups have reported that genetic ablation of CyP-D is sufficient to eliminate the mitochondrial permeability transition without affecting conventional mitochondrial-mediated apoptosis.Citation6,Citation28,Citation29 For this reason, genetic Ppif ablation has been used to demonstrate the role of MPTP opening and programmed cell necrosis in mouse models of Alzheimer's disease, muscular dystrophy, diabetes mellitus and heart failure.Citation30–Citation33
Our studies of MPTP-mediated programmed β-cell death in diabetes used a genetic mouse model, the Pdx1 hemizygous mouse.Citation32 PDX1 (pancreatic and duodenal homeobox factor 1; also called insulin promoter factor 1, IPF1) is a necessary transcription factor for pancreas development. Loss of function human Pdx1 mutations are linked with pancreatic agenesis, heritable maturity onset diabetes of the young and type 2 diabetes.Citation34–Citation36 Likewise, germ-line homozygous Pdx1 gene ablation in mice produces lethal pancreatic agenesis,Citation37 whereas heterozygous germ-line or β-cell specific Pdx1 null mutations result in a diabetes phenotype.Citation38,Citation39 Several cellular mechanisms have been proposed for loss of insulin-producing pancreatic β-cells in the Pdx1 deficient model of diabetes, including decreased proliferation,Citation40 and increased programmed cell death through apoptosis and autophagy.Citation13,Citation41,Citation42 The prevailing opinion has been that increased β-cell death, and not decreased proliferation, is the more important mechanism, with apoptosis most often implicated.Citation14 As indicated above however, the standard markers of apoptosis do not discriminate between apoptosis and programmed necrosis. Therefore, it was possible that programmed β-cell necrosis in Pdx1 insufficiency had been mis-identified as apoptosis based on findings of cytochrome c release and β-cell TUNEL positivity. We examined this possibility in cultured mouse insulinoma-derived β-cells (MIN6 cells), using shRNA to suppress Pdx1 expression, TUNEL as a measure of apoptosis signaling, rhodamine 123 to measure integrity of the MPTP and propidium iodide to assess cell death in a pathway-independent manner.Citation32 As expected, Pdx1 suppression increased TUNEL positivity and β-cell death. Importantly, it also opened β-cell MPTPs, which is not a direct effect of apoptosis. To determine if MPTP opening was a primary or secondary effect of Pdx1 suppression, we treated the cells with cyclosporin A, a pharmacological inhibitor of CyP-D (the essential protein for MPTP opening). Cyclosporin A prevented MPTP opening and significantly reduced Pdx1-deficient β-cell TUNEL positivity and death. This result suggested that MPTP opening could be responsible for as much as half of cultured β-cell death induced by Pdx1 deficiency.
We used genetic complementation with ppif null mice (the ppif gene encodes CyP-D) to assess the role of MPTP-induced β-cell necrosis in murine diabetes caused by Pdx1 haplo-insufficiency. Pancreatic islets of Pdx1 haplo-insufficient mice are characteristically small, with strikingly reduced numbers of insulin-containing β-cells. Eliminating functional MPTPs through ppif ablation increased islet size and decreased TUNEL labeling. We also stained the pancreata for complement 9, a component of the complement membrane attack complex that is part of the inflammatory response to cell necrosis. Complement 9 staining was increased in Pdx1 haplo-insufficient islets, but was normalized by concomitant ppif ablation. This histological “rescue” was observed in both adult and neonatal pancreata, revealing a role for MPTP-mediated programmed β-cell necrosis during embryonic development and for β-cell homeostasis in the adult pancreas.
Finally, we determined the consequences of preventing MPTP-mediated programmed β-cell necrosis on the diabetic phenotype of adult Pdx1 deficient mice. Diabetes in this model is manifested by decreased circulating insulin and increased blood glucose levels measured either at baseline or in response to an acute glucose challenge. Ablation of ppif in Pdx1 haplo-insufficient mice almost completely normalized both of these parameters. Together, these studies implicate MPTP-mediated β-cell necrosis as an important component of pathological programmed β-cell death caused by Pdx1 deficiency, and support a co-equal role for conventional apoptosis through the mitochondrial pathway (). Prevention of diabetes with only partial normalization of β-cell mass in Pdx1-deficient mice lacking functional MPTP likely reflects physiological pancreatic insulin reserve.
Programmed Cardiac Myocyte Death in Heart Failure
Because adult cardiac myocytes are terminally differentiated and replication-deficient, normal tissue homeostatic mechanisms do not apply. Although these cells proliferate during cardiac development, shortly after birth the rate of cell division drops to levels that are difficult to quantify, but are certainly inadequate for meaningful tissue regeneration. Because there is no cardiac myocyte replacement, there is no normal level of programmed cardiac myocyte death. Even after tissue injury or under conditions of non-lethal stress, programmed cardiac myocyte elimination is pathological. Nevertheless, apoptosis is observed in cardiac myocytes of failing, ischemic and pressure overloaded hearts, and is implicated in progression to cardiomyopathy.Citation43 Indeed, a genetic program for programmed cell death is one of the hallmarks of the “compensatory” hypertrophic reaction to cardiac overload.Citation44,Citation45
Using microarray-based RNA profiling of Gq transgenic mice, our laboratory first described a genetic program for cardiac myocyte death in hypertrophied hearts. Gαq (the alpha sub-unit of the heterotrimeric Gq signaling protein) transduces signaling through cardiac myocyte hypertrophy pathways stimulated by angiotensin II, α1-adrenergic and endothelin receptors. The Gq mouse expresses four to five times the normal amount of myocardial Gαq, resulting in ligand-independent Gq signaling and autonomous cardiac hypertrophy with many of the pathological characteristics of pressure overload hypertrophy (but without pressure overload).Citation46 Shortly thereafter we further described progression to dilated cardiomyopathy mediated by Gαq-stimulated cardiomyocyte apoptosis,Citation47,Citation48 suggesting a link between cardiomyocyte hypertrophic growth and programmed death. Specifics of this linkage were established by transcriptional profiling, which identified increased levels of several death pathway genes in Gq hearts.Citation49,Citation50 One of these death genes encoded Nix (also called BNip3L), a member of the small BH3-like only subclass of mitochondrial-targeted Bcl2 apoptosis regulating proteins.
Because they are the sites of action for Nix and other Bcl2 family proteins, mitochondria have been called the “gatekeepers” of programmed cell death.Citation51 Bax and Bak are the essential pore-forming proteins that permeabilize mitochondrial outer membranes, leading to cytochrome c release and intrinsic apoptosis signaling. Pore-formation by Bax and Bak is facilitated by pro-apoptotic BH3 domain-only factors, including Nix/BNip3L that we identified in the heart. Nix and other BH3-only factors hetero-dimerize with anti-apoptotic Bcl2 and Bclxl, which prevents mitochondrial pore formation by Bax and Bak.Citation2 Dynamic regulation of Nix, Bax, Bak and other pro- or anti-apoptotic Bcl2 family proteins is characteristic of heart failure (reviewed in ref. Citation52). A protein kinase C/SP1-dependent mechanism for transcriptional upregulation appears to be responsible for increased Nix gene expression in cardiac hypertrophy.Citation53
We used cardiac transgenesis to determine the in vivo consequences of Nix upregulation in hearts, independent of Gq, cardiac hypertrophy or any hemodynamic stress or cardiac injury. Nix expression was directed specifically to cardiac myocytes using the MYH6 promoter, which is transcriptionally activated only after birth. The result of post-natal cardiac-specific Nix expression was mice that were born normal, but succumbed to progressive dilated cardiomyopathy on the seventh to tenth day of life. TUNEL staining was positive in 15–20% of cardiac myocytes, implicating massive programmed cardiac myocyte death in this form of fulminant heart failure.Citation50 A subsequent study in which Nix was conditionally expressed in the heart revealed synergism between Nix and pressure overload stress for inducing apoptotic heart failure in adult hearts,Citation54 uncovering functional coordination by Nix of transcriptional and physiological stimuli for programmed cardiomyocyte elimination.
Based on the above results, we hypothesized that Nix (and the programmed cardiac myocyte death it produces) mediates progression to heart failure in pressure overload cardiac hypertrophy. To test this idea, we created cardiac-specific Nix gene knockout mice, subjected them to surgical pressure overload and compared cardiomyocyte TUNEL labeling, ventricular remodeling and cardiac function to identically pressure overloaded hearts of mice with intact Nix genes. Cardiac Nix knockout mice exhibited only half the typical rate of cardiomyocyte TUNEL positivity and myocardial fibrosis after pressure overloading and exhibited almost no ventricular dilation, wall thinning or decrease in contractile systolic function.Citation55 These data identified Nix as a critical inducible factor mediating programmed cardiac myocyte death in pressure overload hypertrophy and established a causal role for Nix in adverse ventricular remodeling and development of heart failure.
Delineation of Multiple Nix-Stimulated Cell Death Pathways
Like all pro-apoptotic Bcl2 family members, Nix stimulates caspase-dependent apoptosis resulting from mitochondrial outer membrane permeabilization. In cardiac myocytes and other cell types, Nix localizes to mitochondria and induces cytochrome c release that is followed by caspase activation and oligonucleosomal DNA degradation.Citation50 However, we recently observed that only ∼80% of transfected Nix localizes to mitochondria; the remainder is localized to endoplasmic reticulum (ER) or the analogous structure in cardiac myocytes, sarcoplasmic reticulum (SR).Citation56 We further observed a direct relationship between cardiac myocyte Nix levels and cardiomyocyte SR calcium stores: Nix overexpression increased SR calcium content by ∼20%, whereas Nix gene ablation decreased SR calcium content by ∼20%. Although SR calcium release is a critical regulator of myocyte contraction, we postulated that SR-localized Nix did not meaningfully regulate cardiac contractility. Rather, we felt that SR calcium might play a role in Nix-mediated cell death. Indeed, restoration of normal SR calcium levels in Nix knockout mice by genetic complementation with the phospholamban null mouse reversed the cardio-protective effects observed with Nix ablation, increasing programmed cardiomyocyte death, exaggerating the cardiomyopathy and increasing mortality. These results linked SR calcium to programmed cardiac myocyte death and heart failure produced by Nix.
We considered that reticular-mitochondrial calcium crosstalkCitation57 stimulated by SR-localized Nix could induce the “necrotic” pathway to programmed cardiomyocyte death. If this were the case, cardiomyocyte “apoptosis” we and others have reported based on TUNEL staining might instead be a secondary effect of mitochondrial rupture after MPTP opening, as we had observed in pancreatic β-cells (see above). However, little was known about the role of calcium and MPTP opening in programmed cardiac myocyte death. While calcium is used to stimulate MPTP opening in laboratory studies using isolated mitochondria, it has been suggested that the MPTP is relatively calcium insensitive in vivo due to stabilization by cytosolic factors.Citation58 Because dual Nix localization to mitochondria and reticulum confounds experimentation designed to identify calcium-stimulated non-apoptotic death pathways induced by reticular Nix, we created mitochondria-directed and ER/SR-directed Nix mutants and analyzed their effects after recombinant expression in Nix null embryonic fibroblasts.Citation33 Mitochondrial-directed Nix stimulated cell death marked by caspase activation, but normal ΔΨm, i.e., apoptosis. In contrast, reticular-directed Nix stimulated cell death preceded by MPTP opening and dissipation of ΔΨm, i.e., necrosis, but in which caspase activation attributed to cytochrome c release after MPTP opening and outer mitochondrial membrane rupture was also observed. Using the same experimental design as in our diabetes studies, we found that pharmacological (cyclosporin A) or genetic (CyP-D, Ppif ablation) inhibition of the MPTP prevented cell death induced by reticular Nix, but not by mitochondrial Nix.Citation33 Importantly, apoptosis induced by mitochondrial-directed Nix required Bax or Bak, whereas programmed cell necrosis induced by reticular-directed Nix showed no requirement for Bax and Bak. These results demonstrated that mitochondrial-directed Nix specifically activates conventional Bax/Bak-dependent mitochondrial pathway apoptosis, whereas reticular-directed Nix specifically induces MPTP-dependent programmed cell necrosis by increasing reticular calcium concentration and calcium delivery to mitochondria ().
To determine the relative contribution of Nix-mediated apoptosis versus programmed necrosis to in vivo heart failure we translated the above experimental design to the in vivo murine system and created cardiac-specific conditional transgenic mice expressing mitochondrial-directed or reticular-directed Nix mutants.Citation33 The in vivo dilated cardiomyopathy phenotypes and extent of cardiac myocyte TUNEL labeling (which does not differentiate between apoptosis and programmed necrosis) were similar in mitochondrial-directed and reticular-directed Nix mutant mice. However, cardiac myocyte necrosis visualized by anti-complement 9 staining was observed only in reticular-directed mutant Nix mice. Transmission electron microscopy likewise revealed mitochondrial swelling, matrix degeneration and outer membrane disruption only in cardiomyocytes from reticular-directed Nix mice. A causal link between MPTP opening in reticular-directed Nix-induced cardiac myocyte death was established by concomitant ablation of ppif (encoding CyP-D) in the Nix mutant cardiomyopathy models. ppif ablation prevented cardiomyocyte death (TUNEL labeling) and complement 9 staining in reticular-directed Nix expressing mice, normalizing mitochondrial ultrastructure and preventing the dilated cardiomyopathy. In contrast, ppif ablation did little to improve the apoptotic cardiomyopathy produced by mitochondrial-directed Nix. These findings show that an important aspect of Nix-mediated cell death is programmed necrosis mediated by SR-mitochondrial crosstalk. As other reports have implicated cardiac myocyte or sarcoplasmic reticular calcium levels in cardiac injury and heart failure progression,Citation59–Citation61 MTPT opening stimulated by reticular-mitochondrial calcium cross-talk may play a greater role than previously suspected in hypertrophy decompensation and the progression to overt heart failure.
Conclusions
It may be time to revise our perceptions of the role for apoptosis in chronic disease. In general, apoptosis seems to be a good thing. It is an essential feature of normal embryonic tissue development and adult tissue homeostasis. When normal apoptotic mechanisms are interrupted by natural or experimental mutation, the consequences of unopposed cell growth can be tumor or malignancy. Because the assays used to define apoptosis are not specific for that mechanism, “apoptosis” has been implicated and vilified in variety of chronic diseases associated with abnormal cell death. An increased appreciation that all programmed cell death is not apoptotic, and especially for the role of the mitochondrial permeability transition and programmed necrosis, may reveal that non-apoptotic programmed cell death plays a major role in pathological programmed cell elimination. Chronic diseases in which MPTP-dependent programmed cell death has been implicated include Alzheimer's disease,Citation30 muscular dystrophy,Citation31 amyotrophic lateral sclerosis,Citation62 heart failure,Citation33 and diabetes.Citation32 It is likely that this list will expand further.
The dichotomy between apoptotic and programmed necrotic cell death is mechanistically useful, but is almost certainly blurred in the in vivo context. As noted previously, a cell may initiate the process for apoptosis via permeabilization of mitochondrial outer membranes by Bax and Bak, but if there is insufficient ATP to complete the process, the ultimate consequence may be necrotic death from metabolic shutdown, or even a state of severe, but non-lethal dysfunction that some have describes as a “zombie” cell. It is also likely that some mitochondria within a single cell will undergo outer membrane permeabilization (apoptosis), others will undergo the permeability transition (necrosis), and some fraction will continue to function normally for at least a while. Under these circumstances, the determination of whether the cell will die, and the mechanism for programmed cell elimination, would be the aggregate of all relevant external factors and intracellular conditions.
As our understanding of programmed cell death continues to evolve, the following aspects deserve attention: First, researchers and reviewers should bear in mind that conventional markers of apoptosis are not specific for apoptosis. Mechanistic implications from TUNEL labeling may be no more valid than those from propidium iodide staining. Both label dead or dying cells without unambiguously defining a particular mechanistic pathway. Even observation of apoptotic bodies, oligonucleosomal DNA cleavage, and nuclear chromatin condensation (each of which is widely considered pathognomonic for apoptosis) does not exclude concomitant activation of alternate pathways to programmed cell elimination. Complicating this issue, there is no satisfactory marker for programmed cell necrosis, although complement activation is more abundant with necrosis than apoptosis. Second, we need to determine if there is a physiological role for MPTP opening and programmed cell necrosis, or if it is a purely pathological process. Finally, we need to identify the proximal causes of cell death after MPTP opening. Certainly ATP depletion starts the process, but is there a role for mitochondrial release of reactive oxygen species and calcium in addition to metabolic shutdown in programmed cell necrosis? Further examination of these issues may suggest novel approaches to interdict programmed cell death in disease.
Figures and Tables
Figure 1 Schematic depiction of programmed β-cell death pathways implicated in diabetes caused by Pdx1 haplo-insufficiency. Deficiency of transcription factor Pdx1 likely alters gene expression, activating pathways that induce apoptosis via mitochondrial outer membrane permeabilization (left) and programmed necrosis via the mitochondrial permeability transition (right). Pancreatic β-cell autophagy is also induced by Pdx1 insufficiency, but its role in programmed cell death is currently unclear.
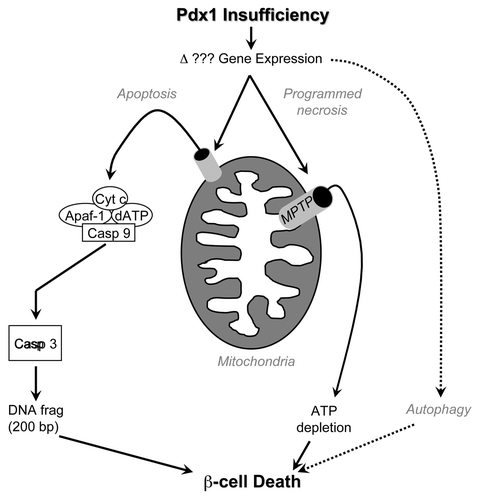
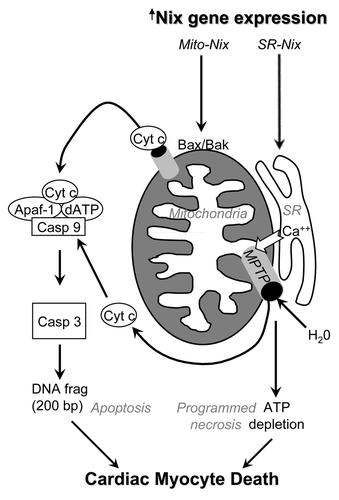
Figure 2 Schematic depiction of multiple Nix-induced pathways for cell death in heart failure. Transcriptionally upregulated Nix localizes either to mitochondria, where it interacts with Bax and Bak to induce apoptosis via outer membrane permeabilization, or to calcium-containing sarcoplasmic reticulum (SR), where it increases calcium stores and activates the mitochondrial permeability transition pore (MPTP) independent of Bax and Bak. Mitochondrial disruption subsequent to MPTP opening leads to metabolic shutdown and death via programmed necrosis.
References
- Kerr JF, Wyllie AH, Currie AR. Apoptosis: A basic biological phenomenon with wide-ranging implications in tissue kinetics. Br J Cancer 1972; 26:239 - 257
- Youle RJ, Strasser A. The BCL-2 protein family: Opposing activities that mediate cell death. Nat Rev Mol Cell Biol 2008; 9:47 - 59
- Vazquez A, Bond EE, Levine AJ, Bond GL. The genetics of the p53 pathway, apoptosis and cancer therapy. Nat Rev Drug Discov 2008; 7:979 - 987
- Shimohama S. Apoptosis in Alzheimer's disease—an update. Apoptosis 2000; 5:9 - 16
- Ohsako S, Elkon KB. Apoptosis in the effector phase of autoimmune diabetes, multiple sclerosis and thyroiditis. Cell Death Differ 1999; 6:13 - 21
- Baines CP, Kaiser RA, Purcell NH, Blair NS, Osinska H, Hambleton MA, et al. Loss of cyclophilin D reveals a critical role for mitochondrial permeability transition in cell death. Nature 2005; 434:658 - 662
- Hitomi J, Christofferson DE, Ng A, Yao J, Degterev A, Xavier RJ, et al. Identification of a molecular signaling network that regulates a cellular necrotic cell death pathway. Cell 2008; 135:1311 - 1323
- Notkins AL, Lernmark A. Autoimmune type 1 diabetes: resolved and unresolved issues. J Clin Invest 2001; 108:1247 - 1252
- Pimenta W, Korytkowski M, Mitrakou A, Jenssen T, Yki-Jarvinen H, Evron W, et al. Pancreatic beta-cell dysfunction as the primary genetic lesion in NIDDM. Evidence from studies in normal glucose-tolerant individuals with a first-degree NIDDM relative. JAMA 1995; 273:1855 - 1861
- Rhodes CJ. Type 2 diabetes-a matter of beta-cell life and death?. Science 2005; 307:380 - 384
- Kulkarni RN, Jhala US, Winnay JN, Krajewski S, Montminy M, Kahn CR. PDX-1 haploinsufficiency limits the compensatory islet hyperplasia that occurs in response to insulin resistance. J Clin Invest 2004; 114:828 - 836
- Mathis D, Vence L, Benoist C. b-cell death during progression to diabetes. Nature 2001; 414:792 - 798
- Johnson JD, Ahmed NT, Luciani DS, Han Z, Tran H, Fujita J, et al. Increased islet apoptosis in Pdx1+/− mice. J Clin Invest 2003; 111:1147 - 1160
- Thomas HE, Biden TJ. Bad news for beta-cell apoptosis. Diabetes 2009; 58:1725 - 1727
- Eizirik DL, Colli ML, Ortis F. The role of inflammation in insulitis and beta-cell loss in type 1 diabetes. Nat Rev Endocrinol 2009; 5:219 - 226
- Choi D, Radziszewska A, Schroer SA, Liadis N, Liu Y, Zhang Y, et al. Deletion of Fas in the pancreatic beta-cells leads to enhanced insulin secretion. Am J Physiol Endocrinol Metab 2009; 297:1304 - 1312
- Pelengaris S, Khan M, Evan GI. Suppression of Myc-induced apoptosis in beta cells exposes multiple oncogenic properties of Myc and triggers carcinogenic progression. Cell 2002; 109:321 - 334
- Cnop M, Welsh N, Jonas JC, Jorns A, Lenzen S, Eizirik DL. Mechanisms of pancreatic beta-cell death in type 1 and type 2 diabetes: many differences, few similarities. Diabetes 2005; 54:97 - 107
- Haworth RA, Hunter DR. The Ca2+-induced membrane transition in mitochondria. II. Nature of the Ca2+ trigger site. Arch Biochem Biophys 1979; 195:460 - 467
- Krasnikov BF, Zorov DB, Antonenko YN, Zaspa AA, Kulikov IV, Kristal BS, et al. Comparative kinetic analysis reveals that inducer-specific ion release precedes the mitochondrial permeability transition. Biochim Biophys Acta 2005; 1708:375 - 392
- Halestrap AP, Pasdois P. The role of the mitochondrial permeability transition pore in heart disease. Biochim Biophys Acta 2009; 1787:1402 - 1415
- Javadov S, Karmazyn M, Escobales N. Mitochondrial permeability transition pore opening as a promising therapeutic target in cardiac diseases. J Pharm Exp Ther 2009; 330:670 - 678
- Szabo I, De Pinto V, Zoratti M. The mitochondrial permeability transition pore may comprise VDAC molecules. II. The electrophysiological properties of VDAC are compatible with those of the mitochondrial megachannel. FEBS Lett 1993; 330:206 - 210
- Beutner G, Ruck A, Riede B, Welte W, Brdiczka D. Complexes between kinases, mitochondrial porin and adenylate translocator in rat brain resemble the permeability transition pore. FEBS Lett 1996; 396:189 - 195
- Marzo I, Brenner C, Zamzami N, Susin SA, Beutner G, Brdiczka D, et al. The permeability transition pore complex: a target for apoptosis regulation by caspases and bcl-2-related proteins. J Exp Med 1998; 187:1261 - 1271
- Baines CP, Kaiser RA, Sheiko T, Craigen WJ, Molkentin JD. Voltage-dependent anion channels are dispensable for mitochondrial-dependent cell death. Nat Cell Biol 2007; 9:550 - 555
- Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, Jones DP, et al. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 2004; 427:461 - 465
- Nakagawa T, Shimizu S, Watanabe T, Yamaguchi O, Otsu K, Yamagata H, et al. Cyclophilin D-dependent mitochondrial permeability transition regulates some necrotic but not apoptotic cell death. Nature 2005; 434:652 - 658
- Schinzel AC, Takeuchi O, Huang Z, Fisher JK, Zhou Z, Rubens J, et al. Cyclophilin D is a component of mitochondrial permeability transition and mediates neuronal cell death after focal cerebral ischemia. Proc Natl Acad Sci USA 2005; 102:12005 - 12010
- Du H, Guo L, Fang F, Chen D, Sosunov AA, McKhann GM, et al. Cyclophilin D deficiency attenuates mitochondrial and neuronal perturbation and ameliorates learning and memory in Alzheimer's disease. Nat Med 2008; 14:1097 - 1105
- Millay DP, Sargent MA, Osinska H, Baines CP, Barton ER, Vuagniaux G, et al. Genetic and pharmacologic inhibition of mitochondrial-dependent necrosis attenuates muscular dystrophy. Nat Med 2008; 14:442 - 447
- Fujimoto K, Chen Y, Polonsky KS, Dorn GW 2nd. Targeting cyclophilin D and the mitochondrial permeability transition enhances β-cell survival and prevents diabetes in Pdx1 deficiency. Proc Natl Acad Sci USA 2010; 107:22 10214 - 10219
- Chen Y, Lewis W, Diwan A, Cheng EH, Matkovich SJ, Dorn GW 2nd. Dual autonomous mitochondrial cell death pathways are activated by Nix/BNip3L and induce cardiomyopathy. Proc Natl Acad Sci USA 2010; 107:9035 - 9042
- Stoffers DA, Zinkin NT, Stanojevic V, Clarke WL, Habener JF. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat Genet 1997; 15:106 - 110
- Stoffers DA, Stanojevic V, Habener JF. Insulin promoter factor-1 gene mutation linked to early-onset type 2 diabetes mellitus directs expression of a dominant negative isoprotein. J Clin Invest 1998; 102:232 - 241
- Macfarlane WM, Frayling TM, Ellard S, Evans JC, Allen LI, Bulman MP, et al. Missense mutations in the insulin promoter factor-1 gene predispose to type 2 diabetes. J Clin Invest 1999; 104:33 - 39
- Jonsson J, Carlsson L, Edlund T, Edlund H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994; 371:606 - 609
- Brissova M, Shiota M, Nicholson WE, Gannon M, Knobel SM, Piston DW, et al. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J Biol Chem 2002; 277:11225 - 11232
- Gannon M, Ables ET, Crawford L, Lowe D, Offield MF, Magnuson MA, et al. pdx-1 function is specifically required in embryonic beta cells to generate appropriate numbers of endocrine cell types and maintain glucose homeostasis. Dev Biol 2008; 314:406 - 417
- Sharma A, Zangen DH, Reitz P, Taneja M, Lissauer ME, Miller CP, et al. The homeodomain protein IDX-1 increases after an early burst of proliferation during pancreatic regeneration. Diabetes 1999; 48:507 - 513
- Johnson JD, Bernal-Mizrachi E, Alejandro EU, Han Z, Kalynyak TB, Li H, et al. Insulin protects islets from apoptosis via Pdx1 and specific changes in the human islet proteome. Proc Natl Acad Sci USA 2006; 103:19575 - 19580
- Fujimoto K, Hanson PT, Tran H, Ford EL, Han Z, Johnson JD, et al. Autophagy regulates pancreatic beta cell death in response to Pdx1 deficiency and nutrient deprivation. J Biol Chem 2009; 284:27664 - 27673
- Whelan RS, Kaplinskiy V, Kitsis RN. Cell death in the pathogenesis of heart disease: mechanisms and significance. Annu Rev Physiol 2010; 72:19 - 44
- Dorn GW 2nd. Physiologic growth and pathologic genes in cardiac development and cardiomyopathy. Trends Cardiovasc Med 2005; 15:185 - 189
- Dorn GW 2nd. Apoptotic and non-apoptotic programmed cardiomyocyte death in ventricular remodelling. Cardiovasc Res 2009; 81:465 - 473
- D'Angelo DD, Sakata Y, Lorenz JN, Boivin GP, Walsh RA, Liggett SB, et al. Transgenic G-alphaq overexpression induces cardiac contractile failure in mice. Proc Natl Acad Sci USA 1997; 94:8121 - 8126
- Adams JW, Sakata Y, Davis MG, Sah VP, Wang Y, Liggett SB, et al. Enhanced Galphaq signaling: a common pathway mediates cardiac hypertrophy and apoptotic heart failure. Proc Natl Acad Sci USA 1998; 95:10140 - 10145
- Sakata Y, Hoit BD, Liggett SB, Walsh RA, Dorn GW 2nd. Decompensation of pressure-overload hypertrophy in G alpha q-overexpressing mice. Circulation 1998; 97:1488 - 1495
- Aronow BJ, Toyokawa T, Canning A, Haghighi K, Delling U, Kranias E, et al. Divergent transcriptional responses to independent genetic causes of cardiac hypertrophy. Physiol Genomics 2001; 6:19 - 28
- Yussman MG, Toyokawa T, Odley A, Lynch RA, Wu G, Colbert MC, et al. Mitochondrial death protein Nix is induced in cardiac hypertrophy and triggers apoptotic cardiomyopathy. Nat Med 2002; 8:725 - 730
- Wei MC, Zong WX, Cheng EH, Lindsten T, Panoutsakopoulou V, Ross AJ, et al. Proapoptotic BAX and BAK: a requisite gateway to mitochondrial dysfunction and death. Science 2001; 292:727 - 730
- Dorn GW 2nd, Kirshenbaum LA. Cardiac reanimation: targeting cardiomyocyte death by BNIP3 and NIX/BNIP3L. Oncogene 2008; 27:158 - 167
- Galvez AS, Brunskill EW, Marreez Y, Benner BJ, Regula KM, Kirshenbaum LA, et al. Distinct pathways regulate proapoptotic Nix and BNip3 in cardiac stress. J Biol Chem 2006; 281:1442 - 1448
- Syed F, Odley A, Hahn HS, Brunskill EW, Lynch RA, Marreez Y, et al. Physiological growth synergizes with pathological genes in experimental cardiomyopathy. Circ Res 2004; 95:1200 - 1206
- Diwan A, Wansapura J, Syed FM, Matkovich SJ, Lorenz JN, Dorn GW 2nd. Nix-mediated apoptosis links myocardial fibrosis, cardiac remodeling and hypertrophy decompensation. Circulation 2008; 117:396 - 404
- Diwan A, Matkovich SJ, Yuan Q, Zhao W, Yatani A, Brown JH, et al. Endoplasmic reticulum-mitochondria crosstalk in NIX-mediated murine cell death. J Clin Invest 2009; 119:203 - 212
- Rizzuto R, Pozzan T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol Rev 2006; 86:369 - 408
- Juhaszova M, Zorov DB, Kim SH, Pepe S, Fu Q, Fishbein KW, et al. Glycogen synthase kinase-3beta mediates convergence of protection signaling to inhibit the mitochondrial permeability transition pore. J Clin Invest 2004; 113:1535 - 1549
- de Jesus Garcia-Rivas G, Guerrero-Hernandez A, Guerrero-Serna G, Rodriguez-Zavala JS, Zazueta C. Inhibition of the mitochondrial calcium uniporter by the oxo-bridged dinuclear ruthenium amine complex (Ru360) prevents from irreversible injury in postischemic rat heart. FEBS J 2005; 272:3477 - 3488
- Ruiz-Meana M, Abellan A, Miro-Casas E, Agullo E, Garcia-Dorado D. Role of sarcoplasmic reticulum in mitochondrial permeability transition and cardiomyocyte death during reperfusion. Am J Physiol Heart Circ Physiol 2009; 297:1281 - 1289
- Odagiri K, Katoh H, Kawashima H, Tanaka T, Ohtani H, Saotome M, et al. Local control of mitochondrial membrane potential, permeability transition pore and reactive oxygen species by calcium and calmodulin in rat ventricular myocytes. J Mol Cell Cardiol 2009; 46:989 - 997
- Martin LJ, Gertz B, Pan Y, Price AC, Molkentin JD, Chang Q. The mitochondrial permeability transition pore in motor neurons: involvement in the pathobiology of ALS mice. Exp Neurol 2009; 218:333 - 346