Abstract
The CtBP transcriptional corepressors promote cancer cell survival and migration/invasion. CtBP senses cellular metabolism via a regulatory dehydrogenase domain, and is antagonized by p14/p19ARF tumor suppressors. The CtBP dehydrogenase substrate 4-methylthio-2-oxobutyric acid (MTOB) can act as a CtBP inhibitor at high concentrations, and is cytotoxic to cancer cells. MTOB induced apoptosis was p53-independent, correlated with the derepression of the pro-apoptotic CtBP repression target Bik, and was rescued by CtBP over-expression or Bik silencing. MTOB did not induce apoptosis in mouse embryonic fibroblasts (MEFs), but was increasingly cytotoxic to immortalized and transformed MEFs, suggesting that CtBP inhibition may provide a suitable therapeutic index for cancer therapy. In human colon cancer cell peritoneal xenografts, MTOB treatment decreased tumor burden and induced tumor cell apoptosis. To verify the potential utility of CtBP as a therapeutic target in human cancer, the expression of CtBP and its negative regulator ARF was studied in a series of resected human colon adenocarcinomas. CtBP and ARF levels were inversely-correlated, with elevated CtBP levels (compared with adjacent normal tissue) observed in greater than 60% of specimens, with ARF absent in nearly all specimens exhibiting elevated CtBP levels. Targeting CtBP may represent a useful therapeutic strategy in human malignancies.
Introduction
The transcriptional corepressors C-terminal binding protein (CtBP) 1 and 2 repress gene expression by interacting with DNA binding transcription factors and recruiting histone methyltransferases, histone deacetylases, polycomb group proteins and other chromatin remodeling proteins to targeted promoters.Citation1–Citation6 CtBP repression targets include epithelial and proapoptotic genes, consistent with its known roles in promoting cell survival and migration.Citation7,Citation8 Transcriptional repression requires CtBP's dehydrogenase/NADH binding domain and increases in intracellular NADH promote CtBP oligomerization and transcriptional repressor activity.Citation9–Citation14 Hypoxia and extracellular glucose levels, both stimuli affecting NADH levels, also modulate CtBP's effects on cell survival and migration.Citation7,Citation15 These findings have established CtBP as a redox sensor that regulates transcription based on the cell's metabolic environment.
A growing body of evidence suggests CtBP plays a role in human cancer.Citation16 CtBP promotes multiple pro-oncogenic activities, including EMT,Citation8 cell migration/invasion,Citation7,Citation17,Citation18 and cell survivalCitation8,Citation19 through repression of epithelial genes such as keratin-8 and E-cadherin and apoptosis genes such as Bik, Noxa, Puma and PERP.Citation8,Citation20 Additionally, CtBP targets multiple growth inhibitory tumor suppressors for repression, including PTEN, p16INK4a and p15INK4b.Citation8,Citation18,Citation21,Citation22
In addition to opposing the activities of multiple tumor suppressors, CtBP is also the target of inhibition by multiple tumor suppressors. In response to UV radiation, CtBP is phosophorylated by HIPK2 and targeted for ubiquitination and proteasomalmediated degradation,Citation23 resulting in apoptosis. The adenomatous polyposis coli tumor suppressor (APC) targets CtBP for degradation, and inactivation of this APC activity may be a necessary step in colonic adenoma formation.Citation24,Citation25 Finally, the tumor suppressor ARF can also downregulate CtBP by targeting it for proteasomal degradation, inducing Bik-dependent apoptosis.Citation19,Citation26 ARF is also capable of blocking CtBP-mediated repression of PTEN and by doing so, ARF can inhibit CtBP-dependent hypoxia-induced, cancer cell migration.Citation18
While the physiologic substrate for CtBP's dehydrogenase activity has not been definitively identified, the penultimate compound in the methionine salvage pathway, 2-keto-4-methylthiobutyrate (referred to hereafter as: 4-methylthio-2-oxobutyric acid; MTOB), is a specific and 80 to 5,000-fold better substrate for CtBP's dehydrogenase then other structurally similar α-ketoacids.Citation27 The catalysis of MTOB shows bi-phasic kinetics, with higher concentrations of MTOB inhibiting the reaction.Citation27
MTOB exhibits significant cytotoxicity against cancer cell lines and is also an inhibitor of ornithine decarboxylase (ODC), which links the methionine biosynthesis and polyamine pathways.Citation28 ODC inhibition is likely not responsible for MTOB's cytotoxicity in cancer cell lines, since supplementation with polyamines to bypass ODC inhibition did not reverse MTOB induced cell death.Citation28 MTOB is also a poor substrate for lactate dehydrogenase, suggesting that manipulation of MTOB levels should not perturb the cellular NAD+/NADH ratio in the way that manipulating pyruvate levels can, leaving the true cellular mechanism for MTOB toxicity unknown.Citation29
Based on its oncogenic properties and intrinsic enzymatic activity, CtBP represents a potential therapeutic target in neoplastic disease. The extent and mechanism of the CtBP substrate MTOB's cytotoxicity was therefore studied in vitro and in vivo. Treatment with MTOB induced p53-independent apoptosis in a variety of human cancer cell lines. MTOB displaced CtBP from the promoter of the pro-apoptotic gene Bik, increasing Bik expression in a dose-dependent manner. Overexpression of CtBP2 or RNAi mediated silencing of Bik, rescued MTOB toxicity. MTOB was demonstrated to be less toxic in primary and immortalized mouse embryonic fibroblasts (MEFs), than in those oncogenically transformed with c-Myc and mutant Ras. MTOB treatment also effectively induced apoptosis and reduced the tumor burden in human colon cancer cell peritoneal xenografts in nude mice. The expression of CtBP and its negative regulator ARF were examined in resected human colon adenocarcinomas and CtBP and ARF levels demonstrated a strong inverse correlation, with CtBP1/2 upregulated in greater than 60% of tumors compared with matched adjacent normal tissue. CtBP may therefore represent a physiologically relevant therapeutic target in human cancer.
Results
The CtBP substrate MTOB induces apoptosis in human colorectal cancer cell lines.
CtBP transcription repressors represent potentially attractive cancer drug targets since they encode a potentially “druggable” dehydrogenase domain and their silencing by RNAi leads to anti-cancer effects, including apoptosis and abrogation of cancer cell migration/invasion.Citation10,Citation17–Citation19 Based on the identification of MTOB as a CtBP substrate, as well as its reported cytotoxicity, the effect of MTOB on cell viability was determined in cell lines known to rely on CtBP for regulation of survival [HCT116 colon carcinoma wild type (+/+) or null (−/−) for p53 ()].Citation19,Citation29 After 72 hours of MTOB treatment, a dose-dependent loss in cell viability was observed, with a near-complete lethal effect at 10 mM (). There was no statistically significant difference in the effects of MTOB between HCT116+/+ and HCT116−/− cell lines, indicating that the loss of viability in these cells was p53-independent. Other cancer cell lines derived from pancreas, bone and colon were also tested, all showing varying degrees of sensitivity to MTOB's cytotoxic effects (Suppl. Fig. 1A).
Consistent with previous reports, cytotoxic MTOB concentrations were in the mM range in this 72 hr viability assay.Citation29 To determine if lower concentrations of MTOB might be effective over a longer-term assay period, a 7-day colony survival assay was performed using HCT116−/− cells. Cells were treated for 72 hours with MTOB and then replaced with fresh compound-free media for 96 hours prior to counting (). At seven days, the IC50 for MTOB was approximately 200 µM and 1 mM MTOB was 91% lethal, demonstrating significantly more potency for MTOB than seen at 72 hours and with a cellular IC50 closer to the IC50 for in vitro CtBP inhibition by MTOB.Citation27 Similar results were obtained using HCT116+/+ cells (data not shown).
In order to compare the efficacy of MTOB to other known cytotoxic agents, HCT116−/− or +/+ cells were treated with 1, 10 or 25 µM cisplatin in the presence or absence of 4 mM MTOB for 48 hours () and cell viability determined. Treatment with cisplatin resulted in a dose-dependent loss of viability in both cell lines, with the p53-null cells showing greater sensitivity at higher concentrations (). In the presence of 4 mM MTOB and 1 µM cisplatin, cell viability dropped from 76% to 41% in the HCT116−/− cells and from 79% to 52% in the HCT116+/+ cells, suggesting an additive effect (). At higher cisplatin doses, the difference between control and MTOB-treated cells disappeared in the p53-null cells, but was enhanced in the p53 wildtype setting, with the cytotoxicity of 25 µM cisplatin at 48 hours increasing from 46% viability to only 4% viability in the presence of MTOB (), suggesting synergy. MTOB was also compared to and combined with, 5-fluorouracil and doxorubicin, but no synergy was observed between MTOB and these agents in HCT116−/− and +/+ cells (Suppl. Fig. 1B).
MTOB displaces CtBP from the Bik promoter and induces Bik expression and apoptosis.
CtBP transcriptional repression is dependent on active regulation by the dehydrogenase domain.Citation11,Citation13,Citation14 MTOB at high concentrations may inhibit the dehydrogenase, impacting repression function.Citation27 Reduction of CtBP2 levels by RNAi has been previously shown to decrease its recruitment to BKLF binding sites in the promoter of the BH3 gene Bik, causing Bik upregulation and apoptosis in HCT116−/− cells.Citation26 In order to determine if MTOB had a similar effect on promoter recruitment of the CtBP co-repressor complex, the Bik promoter was analyzed by CtBP chromatin immunoprecipitation (ChIP) in the presence and absence of MTOB. HCT116−/− cells were treated with 4 mM MTOB or NaCl for 18 hours and the chromatin fraction harvested, immunoprecipitated with IgG, anti-CtBP1 or anti-CtBP2 antibodies and the ChIPs analyzed for bound Bik promoter fragment ().Citation26 Negative control PCR primers (NS) that amplify a fragment 10 kb upstream from the Bik promoter were used to establish specificity (). PCR amplification of input chromatin with Bik and NS primers showed equal amounts of amplification in both treated and untreated cells and there was little background binding of the Bik promoter in the IgG control ChIP (). Bik promoter binding was nearly completely suppressed in the anti-CtBP2 chip from MTOB-treated cells, while the anti-CtBP2 ChIP from NaCl-treated cells demonstrated easily detectable CtBP2 interaction with the Bik promoter (). The anti-CtBP1 ChIP showed slightly different results, with qualitatively less basal promoter binding in the NaCl-treated sample, but a still detectable loss of Bik promoter binding after MTOB treatment, as seen with CtBP2 (). Confirming these results with another CtBP repression target, MTOB treatment of MCF7 cells resulted in decreased CtBP binding to the E-cadherin promoter (Suppl. Fig. 2A), demonstrating that MTOB treatment can cause the dissociation of CtBP from multiple CtBP target promoters.
Having demonstrated that MTOB can displace CtBP1/2 from the Bik and E-cadherin promoters, MTOB's functional effect on CtBP repressor activity was investigated. HCT116−/− cells were treated with 10 mM MTOB or 10 mM NaCl and mRNA analyzed by qPCR for Bik expression (). The MTOB-treated cells showed a six-fold increase in relative Bik expression compared to vehicle treated cells (). Bik protein levels were checked in HCT116−/− cells 24 hours after treatment with vehicle, 1 mM or 4 mM MTOB (, left parts) and Bik levels increased in a dose-dependent manner. Likewise, treatment of MCF7 cells with MTOB led to the upregulation of E-cadherin protein levels, demonstrating that MTOB's ability to cause derepression of a CtBP target gene is neither gene- nor cell-type-specific (Suppl. Fig. 2B).
Given the known repression of pro-apoptotic genes, including Bik, by CtBP, the likely possibility that MTOB cytotoxicity was due to induction of apoptosis was investigated. Treatment of HCT116−/− cells with MTOB resulted in a dose-dependent increase in caspase activation, as measured by levels of cleaved caspase 3 (, right parts). Similar caspase activation was induced in MTOB-treated MCF7 cells (Suppl. Fig. 2C). Thus, MTOB impacts on CtBP2 occupancy on target promoters and resultant changes in gene expression were found to correlate with activation of apoptotic pathways.
MTOB modulates the CtBP/Bik pathway to induce apoptosis.
Having shown that MTOB treatment inhibits CtBP transcriptional repression of a target gene product, the hypothesis that CtBP is a primary target for MTOB was tested by attempting to rescue MTOB cytotoxicity with CtBP overexpression. HCT116−/− cells were stably transfected with human V5-tagged CtBP2 cDNA (CtBP2-V5) or empty vector (pCDNA), treated with 4 mM MTOB or vehicle for 72 hours and then stained with trypan blue (). Total cell viability was reduced by 66% in the control cells treated with 4 mM MTOB, while the CtBP2-V5 cells treated with 4 mM MTOB showed no significant loss in cell viability (, left part). These results were mirrored by the increase in the percentage of non-viable cells in the control transfectants from 12 to 46% in the vehicle and 4 mM MTOB-treated cells, respectively, while the CtBP2-V5 cells showed no difference between the untreated and treated cells, with 13 and 12% nonviable cells noted, respectively (, right part). CtBP2-V5 cells were thus completely resistant to MTOB-induced cell death. This resistance correlated well with Bik levels in the two cell lines. In the control transfectants treated with 4 mM MTOB, there was an induction of Bik compared to the untreated cells, whereas the CtBP2-V5 cells showed no Bik induction upon MTOB treatment (). Thus, rescue of MTOB-induced cell death by CtBP2 overexpression correlated with suppression of Bik induction.
To investigate whether MTOB-induced cell death was due in part or whole to reversal of CtBP repression of Bik, HCT116−/− cells stably expressing GFP shRNA or one of two different Bik shRNAsCitation26 were treated with vehicle or 4 mM MTOB for 72 hours and viability determined by trypan blue staining ( and D). ShGFP cells showed normal sensitivity to MTOB, with reduction in viability from 100 to 44% and an increase in dead cells from 7 to 39% (, left and right parts). Both shBik1 and shBik2 showed a significant ability to rescue MTOB-induced cell death, with viability post-MTOB increasing from 44% in shGFP cells to 73% and 94% in shBik1 (p = 0.001) and shBik2 (p = 0.002) cells, respectively (, left part). Cell death was also significantly reduced in shBik1 and 2 cells post-MTOB, with reductions in cell non-viability from 38% (shGFP cells) to 12% and 15% non-viability in shBik1 (p < 0.0001) and shBik2 (p = 0.0001) cells, respectively (, right part). Knockdown efficiencies for the two hairpins were unequal () with shBik1 having reduced knockdown (30%) compared to shBik2 (>90%), correlating with the more robust rescue of viability seen with shBik2 (). Thus, inhibition of CtBP2 repression of Bik is largely responsible for MTOB-induced cytotoxicity in HCT116−/− cells.
MTOB sensitivity correlates with the degree of cellular transformation.
To investigate MTOB's potential cytotoxic specificity for malignant cells, MTOB cytotoxicity was assessed in primary mouse embryonic fibroblasts (MEFs) with different genetic backgrounds. Treatment of primary MEFs with 4 or 10 mM MTOB for 48 hours revealed no differences in viability between treated and control cells (Suppl. Fig. 3A). In a 7-day colony assay, primary MEFs likewise showed no difference in survival between 4 mM MTOB-treated and untreated samples, confirming the lack of toxicity in the shorter experiment (, white bars). Immortalized MEFs from p19Arf-null mice, however, showed greater sensitivity to MTOB, with a 65% reduction in survival in the 7-day assay ( and black bars). Transformation of p19Arf-/- MEFs with c-Myc and activated Ras rendered cells substantially more vulnerable to MTOB treatment, with 4 mM MTOB reducing survival by 98% after 7 days (, lined bars). Thus, increasingly malignant cellular transformation renders cells sensitive to CtBP inhibition by MTOB.
MTOB is well tolerated and effective in vivo.
To begin to address MTOB's potential clinical utility in cancer therapy, MTOB was first assessed for any toxicity in the mouse. Nu/Nu mice (3 in each group) received either PBS or 750 mg/kg MTOB administered by intraperitoneal (IP) injection twice a week for four weeks and all mice were then sacrificed for necropsy. The mice showed no signs of illness or distress at the time of sacrifice. Tissues from the major organs from one of each group were read in a blinded fashion by an outside pathologist and all major organs from the MTOB-treated mouse looked no different histologically than those from the PBS treated mouse (data not shown). This data, combined with previous studies of MTOB toxicity in animal and human nutrition,Citation30–Citation33 supported the conclusion that MTOB has limited or no toxicity in normal cells and tissues in vitro and in vivo.
In order to determine MTOB's efficacy in vivo, a peritoneal xenograft model employing HCT116−/− cells was employed. Seven days after Nu/Nu mice received 3 × 106 HCT116−/− by IP injection, mice were randomized into control or treatment groups (10 each group) and IP injections with PBS or 750 mg/kg MTOB were begun three times a week for up to 8 weeks or until sacrifice due to tumor progression. Mice were assessed for tumor-free survival, tumor weight and production of ascites (). Mice treated with MTOB showed a median tumor-free survival of 38 days compared to 35 days for PBS-treated mice, with a log rank test p-value of 0.08 (). However, 48% of MTOB-treated mice were still tumor-free after all PBS-treated mice had visible tumor and 16% of the MTOB mice had no tumor at the end of the study, suggesting that from the standpoint of tumor-free survival, additional statistical power will be needed to establish whether a significant p-value can be achieved for this particular endpoint.
Tumor burden was determined by total tumor weight and total ascites volume at the time of sacrifice for tumor progression or study end and a representative portion of tumor from each mouse was fixed and stained with hematoxylin and eosin (H&E; and Suppl. Fig. 3B). H&E staining of tumor xenografts revealed poorly differentiated adenocarcinoma, with normal tissue (stomach, intestine, peritoneum) sometimes captured on the periphery (Suppl. Fig. 2B). All tumors had varying degrees of central necrosis and lymphocytic infiltrate. Tumor masses accumulated on the viscera and peritoneal surfaces with no apparent invasion of other organs. PBS-treated animals had tumors weighing an average of 4.7 grams, while MTOB-treated tumors were significantly smaller, weighing an average of 1.2 grams (p = 0.0007) (, left). Any ascites was aspirated prior to necropsy. Only one MTOB-treated animal developed ascites, while all but two of the PBS-treated mice did develop ascites. The average volume of ascites removed from PBS-treated mice was 5.7 mL, while an average of 1.25 mL was removed from the MTOB-treated mice and this was mainly attributed to just a single mouse (p = 0.04 for difference in ascites volume between PBS and MTOB treatments; , right). Two measures of disease burden therefore showed significant improvement with MTOB treatment, suggesting that MTOB can effectively reduce tumor burden in the HCT116−/− peritoneal xenograft model.
To assess MTOB-induced apoptosis in the tumors, TUNEL staining was performed and quantified on seven PBS- and seven MTOB-treated tumors, with a significant 11-fold increase in % TUNEL-positive cells in MTOB-treated tumors (p = 0.002 by Mann Whitney/Wilcoxon rank sum test) (). The range of apoptotic response to MTOB was variable, ranging from a 4 to 27-fold increase in % TUNEL-positive cells (, top) and this might possibly explain the range of tumor weights observed in MTOB-treated mice ().
A drawback to the peritoneal xenograft model is that tumor mass cannot be accurately measured over time prior to necropsy. In order to look at the effect of MTOB on tumor size over time, overall survival vs. tumor weight were plotted for PBS- and MTOB-treated mice (). The two groups were fit by linear regression (PBS slope = 0.26, MTOB slope = −0.008) and the slopes' variance from zero determined by an F-test of the model, with the PBS slope being significantly non-zero (p = 0.001), while the MTOB slope was not different from zero (p = 0.6). The two lines were then compared to each other using analysis of co-variance (ANCOVA), and they did vary from one another significantly, with p = 0.0009. Hence, PBS-treated mice grew tumors linearly proportional to their survival, while MTOB-treated mice showed no increase in tumor size over the time of the experiment. Taken together, these data suggest that MTOB is a potentially effective cancer therapeutic molecule and CtBP is a valid target for further therapeutic development.
CtBP and ARF levels vary coordinately in human colon tumors.
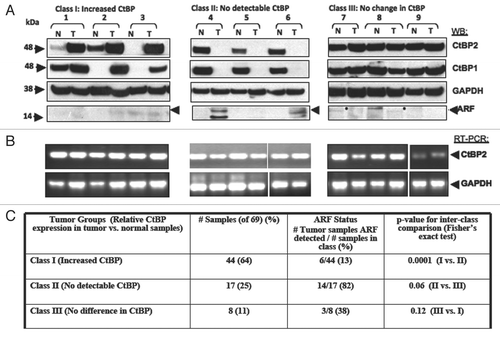
CtBP repressors represent potentially attractive cancer drug targets, but are only useful as such if expressed in human tumors. Recent studies have found increased levels of CtBP1 protein in adenomas from FAP patients.Citation24,Citation25 CtBP is regulated at the post-translational level by several tumors suppressors,Citation19,Citation23,Citation24 but there has been no systematic examination of CtBP1 and CtBP2 protein expression in human tumors. Since the cellular CtBP antagonist and tumor suppressor ARF is silenced due to methylation in ∼22–38% of colon cancers,Citation34 a series of 69 resected colon cancer specimens were analyzed for tumor-specific CtBP expression levels, to probe whether CtBP overexpression occurs in human tumors and especially in those tumors lacking ARF. Tumor specimens and corresponding adjacent normal tissue were thus analyzed for CtBP1/2, hARF and GAPDH protein levels by immunoblot () and CtBP2 and GAPDH mRNA levels by RT-PCR ().
Three CtBP and ARF expression patterns were observed (; Class I–III; summarized in ). The majority of tumors (44/69, 64%) expressed substantially higher levels of CtBP1/2 than adjacent normal tissue and ARF was undetectable in 38/44 of these class I samples (86%; a faint background band is seen migrating just below the ARF position in many samples; representative cases in , #1–3). 17 of the 69 (25%) tumors demonstrated a striking absence of CtBP1/2 and most of these (14/17, 82%) exhibited expression of p14ARF (class II, representative cases in , #4–6). Notably, the matched normal samples for Class II tumors invariably contained detectable and comparatively high levels of CtBP1/2 protein, as also seen in matched normal samples from Class III tissues (where CtBP levels were the same in normal and tumor tissue; , #7–9), but unlike the matched normal class I specimens (, #1–3). ARF was detected in 3/8 class III samples, but at much lower levels of expression compared to class II tumors and variably in tumor or normal tissue (e.g., filled circles, #7 and 8, ). The inverse correlation between ARF and CtBP was statistically significant for comparison of the ARF/CtBP expression pattern between samples in Class I vs. II (p = 0.0001), but only trended to significance for comparisons of class I or class II samples vs. class III samples (Class II vs. III p = 0.06, and Class I vs. III p = 0.12) likely because of the smaller numbers of samples in classes II and III. Examining the inverse correlation of ARF and CtBP expression across all tumor samples revealed a highly significant chi square value of 26.7 (2 degrees of freedom, p < 0.00001).
RT-PCR analysis of normal and tumor samples for CtBP2 mRNA showed no consistent or significant difference between normal and tumor specimens across all classes (), suggesting that the loss of CtBP expression in Class II tumors and the increase in CtBP expression in Class I tumors was post-transcriptional. Taken together, the analysis of a series of colorectal cancer resection specimens demonstrated an inverse relationship between ARF and CtBP protein expression in ∼75% of tumors, which is consistent with previous results from cell line-based studies.Citation19 Additionally a majority (64%) of colon tumors exhibited levels of CtBP expression greater than that seen in normal tissue, suggesting that a CtBP inhibitor, such as MTOB, could be of utility in colon cancer therapy and of most utility and specificity in class I colon tumors.
Discussion
CtBP is one of only a very few transcription factors that harbor an intrinsic enzymatic, and thus druggable, functional domain. CtBP is a potential therapeutic target in cancer, therefore, due to its numerous pro-oncogenic properties.Citation7,Citation16,Citation17,Citation19 Here we have shown that the CtBP substrate MTOB regulates CtBP activities in vivo. Repression of a specific CtBP gene product, Bik, was alleviated with MTOB treatment, coinciding with decreased CtBP occupancy on the target promoter. CtBP2 rescue of MTOB-induced cell death was complete, as was rescue by RNAi knockdown of the BH3-only protein Bik, supporting the hypothesis that MTOB-induced cell death is caused by interfering with CtBP repression of Bik. Immortalized and transformed MEFs were sensitized to MTOB cytotoxic effects in comparison to normal MEFs, indicating that MTOB may exhibit its toxic effects specifically in cancer cells. In vivo testing of MTOB in colon cancer peritoneal xenografts demonstrated it to be a safe, well-tolerated therapy, with an ability to limit tumor growth and ascites production and possibly prolong survival.
In an effort to validate the usefulness of CtBP as a therapeutic target in human cancer, we have shown that CtBP1/2 protein is overexpressed relative to normal tissue in 64% of resected colorectal tumors, likely due to concomitant ARF loss and increased CtBP protein stability. Only a minority of tumors (25%) exhibited low CtBP levels, and in this group of tumors ARF and CtBP expression were inversely correlated in ∼80% of specimens. Thus, abnormally high levels of CtBP were associated with greater than 50% of colon adenocarcinoma specimens, suggesting that a substantial fraction of colon cancers may exhibit heightened sensitivity to anti-CtBP therapy.
Bik has been implicated as a human tumor suppressor, with epigenetic silencing, deletions or mutations detected in renal, colon, brain, and lymphatic malignancies.Citation35–Citation38 Bik upregulation is linked to apoptosis induced by a variety of anti-neoplastic agents, such as cisplatin, doxorubicin, 5′-aza-2′deoxycytidine (DNA methyltransferase 1 inhibitor), and the proteasome inhibitor bortezomib.Citation39 Bik overexpression has been reported to reduce chemotherapy resistance in vitro,Citation40 as well as dramatically reduce tumor formation in mouse models in vivo.Citation41,Citation42 Thus, molecules that can induce Bik expression, such as MTOB or another CtBP inhibitor, could be useful in the treatment of a wide spectrum of malignancies.
In addition to upregulating pro-apoptotic gene targets, CtBP inhibition could have additional therapeutic value. CtBP was recently identified as a transcriptional activator of the multi-drug resistance transporter (MDR1; Pg-p).Citation43 In a chemoresistant cell line, sensitivity was reestablished with RNAi depletion of CtBP, corresponding with reduced CtBP binding of the MDR1 promoter. MTOB induction of chemotherapy sensitivity to other therapeutics, outside of its intrinsic induction apoptosis, may be clinically useful as well.
The evidence to date suggests that MTOB or other compounds that target CtBP are viable candidates for therapeutic development. MTOB is toxic to a variety of cancer cell lines derived from many different tissues (reviewed in ref. Citation29). In our study, MTOB toxicity was relatively specific for transformed cells, with early passage mouse embryonic fibroblasts showing near complete resistance to the compound. MTOB has been evaluated as a methionine supplement in livestock feed,Citation32 as well as used in the treatment of uremic patients, without reported toxicity.Citation30,Citation31,Citation44 The oral doses used in both humans and animals were on the order of grams/day, with chicks showing no toxicity after receiving 15 g/kg daily,Citation32 suggesting the compound is well tolerated by healthy tissues in animals, as well as humans.
As to why MTOB is selectively more toxic to transformed cells is not yet clear. CtBP1/2 hetero- and homozygous knockout MEFs grow normally, but are hypersensitive to apoptotic stress.Citation8,Citation45 The constitutive apoptogenic stresses found in malignant cellsCitation46 might cause them to rely on enhanced or constitutive CtBP activity, whereas non-transformed cells are not under constant stress, and thus may be less dependent on CtBP activity for survival. It is therefore unclear whether improvement in MTOB's inhibition of CtBP with a more potent chemical inhibitor would improve or reduce the therapeutic window of CtBP inhibition.
The evidence for a role of CtBP in human cancer is growing. CtBP has been shown to regulate processes important for oncogenesis, tumor maintenance, and progression/metastasis.Citation16 We have demonstrated that the CtBP substrate and methionine salvage intermediate, MTOB, preferentially kills cancer cells. We have identified CtBP, and its repression of Bik, as the putative target responsible for MTOB-induced cell death. MTOB can be successfully, and safely, delivered to tumor cells in vivo. These findings suggest that the dehydrogenase activity of CtBP proteins may represent a viable drug target, and further detailed investigation of the mechanism of CtBP inhibition by MTOB and its clinical usefulness is warranted.
Materials and Methods
Plasmids and viral expression vectors.
V5-tagged CtBP2 expression plasmid pcDNA-V5-CtBP2 has been described.Citation19 c-Myc and RasV12 expression plasmids were generously provided by Dr. Ron DePinho.
Chemicals.
4-methylthio-2-oxobutyric Acid (Sigma) was dissolved in cell-specific media, 0.2 um filtered, and diluted to final concentration before addition to cultured cells.
Cell culture and transfection.
HCT116 human colon cancer cells (ARF silenced) with (−/−) and without (+/+) targeted deletion of p53,Citation47 were grown in McCoy's 5A medium with 10% fetal bovine serum and 100 U/ml penicillin, 100 µg/ml of streptomycin. MEFs were cultured in Dulbecco's modified Eagle's medium (DMEM) supplemented with 10% fetal bovine serum, 100 U/ml penicillin, 100 µg/ml of streptomycin, and incubated in humidified 5% CO2 at 37°C. MCF7 human breast carcinoma cells were grown in the same medium, with glucose levels reduced to 1 g/L. MCF10A human breast adenoma cells were grown in DMEM F-12 media as described.Citation48 Mammalian expression plasmids were transfected using Fugene (Roche). HCT116−/− cells stably expressing shRNAs targeting Bik were described previously.Citation26
Immunoblotting.
Cells were lysed in lysis buffer (20 mM HEPES pH 7.4, 0.5% Triton X-100, 2 mM MgCl2, 10 µM ZnCl2, 2 mM NEM, 1 mM PMSF, 240 mM NaCl) containing protease inhibitor tablets (Roche).Citation7 Antibodies used were as follows: CTBP1, CtBP2 (BD Biosciences), hARF (Novus), V-5 (Invitrogen), GAPDH (Advanced Immuno), E-cadherin, hBik and cleaved caspase 3 (Cell Signalling). Anti-rabbit IgG-horseradish peroxidase and anti-mouse IgG-horseradish peroxidase conjugates (Jackson Immunoresearch) were detected on immunoblots by ECL (GE Healthcare).
Cell viability, colony and relative survival assays.
For viability determination, cells were analyzed by trypan blue staining or MTT assay. For trypan blue studies, cells were trypsinized, washed and mixed 1:1 with trypan blue solution and counted with a hemocytometer. For MTT assays, cells were combined with MTT reagent (Calbiochem) according to manufacturers instructions, and developed for one hour before determination of A595.
Colony assays were performed in duplicate in six-well plates. Cells were plated at a limiting dilution and treated for 72 hours with no additive or MTOB-containing media. After 72 hours, media was replaced with fresh media without MTOB every two days, until fixation with methanol and acetic acid. After fixation, colonies were stained with Giemsa, and then either counted or resolubilized in methanol and SDS followed by determination of A595 as described.Citation49
Chromatin immunoprecipitation assay (ChIP).
108 cells per ChIP assay were washed once in PBS and treated with 1% formaldehyde in cold PBS for 10 min at 4°C with continuous shaking. Glycine (final concentration 125 mM) was added to quench the formaldehyde for 5 min at 4°C with continuous shaking. Cells were then harvested and washed twice with ice-cold PBS. Nuclei were isolated by incubating the cells in nucleus isolation buffer (5 mM PIPES pH 8.0, 85 mM KCl, and 0.5% NP-40) for 20 to 30 min on ice. The nuclei were harvested at 4°C by centrifuging the cell suspension at 7,000x g for 5 min, and resuspended in 2 ml of RIPA buffer (150 mM NaCl, 1% v/v NP-40, 0.5% sodium deoxycholate, 0.1% SDS, 50 mM Tris pH 8.0, 5 mM ethylenediamine tetra-acetic acid (EDTA)) containing protease inhibitors. Chromatin was fragmented to approximately 200–700 bp by sonication. Nuclear debris was removed by centrifuging the lysates at 4°C for 15 min at 14,000 rpm. The lysate was precleared by incubation with protein G Sepharose beads for 30 min at 4°C and immunoprecipitation was performed overnight at 4°C with the respective antibody. Protein G Sepharose beads were added and the immunocomplexes were allowed to bind to the beads for 2 hours at 4°C. The beads were then washed once each with RIPA buffer, RIPA buffer with 500 mM NaCl, immunoprecipitation wash buffer (10 mM Tris pH 8.0, 250 mM LiCl, 0.5% NP40, 0.5% sodium deoxycholate, 1 mM EDTA), and finally with TE (10 mM Tris pH 8.0, 1 mM EDTA). Beads were resuspended in 200 µl of elution buffer (50 mM Tris, pH 8.0, 10 mM EDTA, 1% SDS) with Proteinase K (20 mg/ml) and incubated overnight at 55°C. DNA was extracted using phenol-chloroform, precipitated in the presence of glycogen by ethanol, allowed to air-dry, and dissolved in TE. Immunoprecipitated DNA was diluted 10-fold prior to PCR. The following primer sets were used to amplify different regions of the genes indicated: Bik promoter primer set (fragment endpoints −551 bp to −693 bp relative to Bik transcription start site) in which tandem BKLF-binding sites are present; sense 5′-TAT ACC AGG GCT GGA GTT AGG TCC-3′/antisense 5′-CTC ACG TGC AGA CCT GGT GAG A-3′; non-specific primers (NS) (fragment endpoints −9.5 to −9.3 kb relative to the Bik transcription start site) sense 5′-CCT AAG AAG CTG GCC ACA GCT C-3′/antisense 5′-CCA TCA TGT TGG CCA GAA TGG TCT C-3′.
Tumor sample analysis.
Whole cell lysates (for protein analysis) and total RNA from tumor and associated normal samples (colon adenocarcinoma resection specimens, UMass Memorial Cancer Center Tumor Bank, anonymous deidentified samples with stages I–IV represented) were prepared using a PARIS® kit (Ambion). RT-PCR was performed using the Stratascript® RT kit (Stratagene) using primers for CtBP2 and GAPDH as described.Citation26
MTOB treatment of peritoneal xenograft tumors.
Mouse toxicity and xenograft studies were approved by the UMass Medical School Institutional Animal Care and Use Committee. For the toxicity study, male Nu/Nu mice (Charles River Laboratories; 3 reach group) were injected IP with PBS or 750 mg/kg MTOB prepared in PBS twice weekly for 4 weeks, and then sacrificed for necropsy and histopathological analysis of all major organs. Slides were read in a blinded fashion by D. Garlick, UMass Memorial Cancer Center Veterinary Pathology Core. Male Nu/Nu mice were then inoculated with 3 × 106 HCT116−/− cells in PBS by intraperitoneal injection. One week after inoculation, mice were randomized into 2 groups (n = 10 mice/treatment group) and injected three times a week (M,W,F) with either PBS or 750 mg/kg MTOB prepared in PBS. Mice were necropsied at time of death or sacrificed when determined by a third party to be moribund or too burdened by ascites. Tumor-free survival was plotted as a Kaplan-Meier curve using Prism Graphpad software (GraphPad Software). Any ascites was aspirated before necropsy and volume quantitated in ml. Total peritoneal tumor mass was measured by weight in grams. Tumor weight and ascites in MTOB- vs. PBS-treated mice were analyzed by unpaired student t-test. Tumor weight as a function of survival time was analyzed by an F-test of both linear regression models and an analysis of covariance (ANCOVA), to determine the difference between the PBS and MTOB-treated groups.
TUNEL staining.
After weighing, peritoneal xenograft tumors were fixed in formalin. Paraffin-embedded tumor sections were analyzed for apoptosis using a peroxidase based In Situ Cell Death Detection Kit (Roche). The staining was automated using the Discovery XT (Ventana Medical Systems). TUNEL-positive cells from 5 fields from each slide, excluding necrotic fields, were counted. The Mann-Whitney/Wilcoxon rank-sum test was used to determine statistical significance in fold change of % TUNEL-positive cells using Prism Graphpad software. Images were taken using an Olympus BX41 microscope (Olympus) with 200x or 400x magnification. The images were acquired with an Evolution MP 5.0 camera (Media Cybernetics) and Q Capture Pro 5.1 (QImaging) image acquisition software.
Real-time quantitative PCR.
mRNA transcripts for human Bik and GAPDH were analyzed by RQ-PCR using SYBR green (Applied Biosystems) on an ABI 7300 (Applied Biosystems). Relative amounts of the mRNA transcripts were calculated using the ΔΔCT method with GAPDH mRNA as the internal reference. The primer sets used were Bik (sense: 5′-TCC TAT GGC TCT GCA ATT GTC A-3′/antisense: 5′-GGC AGG AGT GAA TGG CTC TTC-3′); GAPDH (sense 5′-ATC ACC ATC TTC CAG GAG CGA-3′/antisense 5′-GCC AGT GAG CTT CCC GTT CA-3′).
Abbreviations
| CtBP | = | C-terminal binding protein |
| MTOB | = | 4-methylthio-2-oxo butyrate |
| ARF | = | alternate reading frame |
| Bik | = | Bcl-2 interacting killer |
| MEF | = | mouse embryonic fibroblast |
| NADH | = | nicotinamide adenine dinucleotide |
Figures and Tables
Figure 1 The CtBP substrate MTOB is cytotoxic to colon cancer cell lines. (A) The colon adenocarcinoma cell lines HCT116 wild type (+/+) or p53-null (−/−), were treated for 72 hours with MTOB or normal media (Control). Cell viability was determined by MTT assay and normalized to the untreated samples. The average of three separate experiments is shown, and error bars equal the standard error of the mean (SEM). (B) HCT116−/− cells were plated at low density and treated with normal media or media containing the indicated amount of MTOB for 72 hours, and then replaced with compoundfree media for four days. Colony formation was assessed by GIEMSA staining, and all wells were normalized to the untreated control. Experiments were done three times in duplicate, with a representative plate shown. Error bars represent the SEM. (C) HCT116−/− and +/+ cells were treated with the indicated concentrations of cisplatin in the presence or absence of 4 mM MTOB for 48 hours, and cell viability assessed by MTT assay. Error bars represent the SEM.
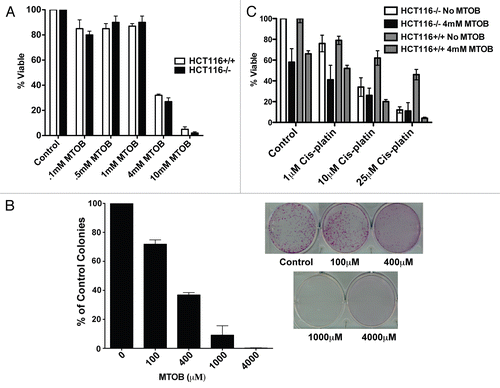
Figure 2 MTOB modulates CtBP transcriptional functions. (A) MTOB displaces CtBP2 from the Bik promoter. HCT116−/− cells were treated for 18 hours with 4 mM MTOB or NaCl (control), and CtBP2 and CtBP1 association with the Bik promoter assessed by ChIP using control IgG, anti-CtBP 1 or 2 antibodies and formaldehyde crosslinked chromatin. DNA retained in the ChIPs was analyzed by PCR using either Bik promoter primers that amplify a fragment containing BKLF sites (Bik) or a fragment of DNA located 10 kb upstream of the Bik transcription start site (NS). (B) MTOB increases expression of Bik mRNA. Quantitative real-time PCR using RNA prepared from HCT116 p53−/−cells treated with NaCl (control) or 10 mM MTOB. Error bars are equal to the SEM. (C) MTOB regulates Bik expression. HCT116 p53−/− cells were treated with control media (−) or media containing 1 mM or 4 mM MTOB. Bik, cleaved caspase 3, and GAPDH levels were determined by immunoblotting of cell lysates prepared 24 hr after treatment.
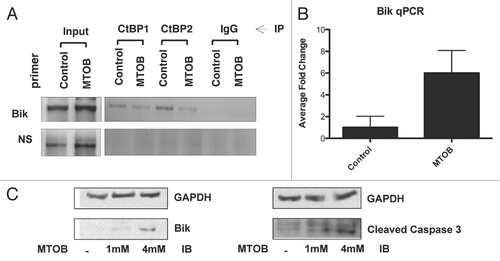
Figure 3 MTOB-induced cell death is dependent on inhibition of CtBP-mediated repression of Bik. (A) HCT116−/− cells were transfected with CtBP2-V5 or empty pCDNA3 vector, and transfected cells were enriched by G418 selection for one week to form pooled cell lines. Once selected, cells were plated and treated for 72 hours with either 4 mM MTOB or normal media (control). Cell viability was then determined by trypan blue assay. Experiments were performed in triplicate and error bars indicate the SEM. (B) Cell lysates were immunoblotted to detect expression of CtBP2, CtBP2-V5 and Bik in the presence (+) and absence (−) of 4 mM MTOB. (C) HCT116−/− cells stably expressing one of two shRNAs targeting Bik or GFP under the control of the Tet-repressor were plated in the presence of doxycycline for 24 hours before being treated with normal media or 4 mM MTOB. After 72 hours, cells were harvested and cell viability determined by trypan blue assay. Error bars represent the SEM. (D) Cell lysates were immunoblotted to detect expression of Bik in the presence of shGFP or one of two hairpins targeting Bik, shBik1 and shBik2. Bik levels were quantified by densitometry, and normalized for GAPDH.
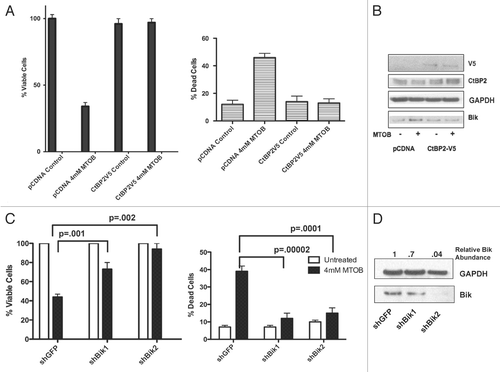
Figure 4 MTOB effectively and safely targets cancer cells in vivo. (A) Primary wild-type mouse embryonic fibroblasts (MEFs), immortalized ARF−/− MEFs, and ARF−/− MEFs stably transfected with mutant Ras and c-Myc vectors were treated for 3 days with 4 mM MTOB or normal media (control), followed by four days in normal media. Cell survival of MEF colonies was determined by A595 of solubilized Giemsa-stained cells expressed relative to the untreated cells. Experiments were performed 3 times in duplicate, and error bars represent the SEM. (B) Tumor-free survival (TFS) of Nu/Nu mice inoculated with HCT116−/− cells and treated one week later with PBS or 750 mg/kg MTOB three times a week for 7 weeks, unless mice were euthanized sooner due to progressive tumor growth. p = 0.08 for comparison of median TFS between PBS and MTOB treatment. (C) Tumor burden was assessed by measuring total peritoneal tumor weight at time of death or sacrifice (left), and by measuring the volume of ascites before necropsy (right). Treatment groups were compared by unpaired t-test with tumor weight and ascites both significantly less in the MTOB group (p = 0.007 and 0.04, respectively). Error bars indicate SEM. (D) Sections of paraffin-embedded tumor were analyzed for apoptosis by TUNEL staining. 5 high power fields were counted for 7 tumors each from PBS- or MTOB-treated animals, and the averages plotted (top). Differences between the two groups were analyzed for statistical significance by Mann-Whitney test, with p = 0.0001. A representative section of PBS and MTOB treated tumors are shown at 400x magnification. (E) Tumor weights for MTOB- and PBS-treated tumors were plotted against days of survival, analyzed by linear regression, and the slopes compared by ANCOVA, with a p value of 0.0009.
Figure 5 CtBP and p14ARF levels are inversely-correlated in human colorectal adenocarcinomas. (A) Samples from colorectal adenocarcinoma specimens were analyzed by immunoblot for levels of p14ARF, CtBP1/2 and GAPDH. Tumors were separated into three groups based on the CtBP levels in tumor vs. normal tissue. Representative blots from the three groups are shown. (B) RNA was isolated from the same tumors and RT-PCR performed to determine CtBP2 and GAPDH mRNA levels in tumor vs. normal tissue. (C) Summary of the three classes of colorectal tumors based on CtBP and ARF expression levels. The inverse correlation of ARF and CtBP1/2 expression was significant by exact Fischer's test with p = 0.0001 for Class I vs. Class II, p = 0.06 for Class II vs. Class III, but not siginificant for the comparison of Class I vs. Class III (p = 0.12).
Additional material
Download Zip (583.3 KB)Acknowledgements
The authors wish to thank C.C. Hsieh for assistance with statistical analysis, R. DePinho for Myc and Ras expression plasmids, M. Kelliher for helpful discussions, D. Garlick for pathology assistance, and K.M. Draheim for advice and assistance with quantitative RT-P.C.R. S.P. was supported by an ACS/UMASS Individual Research Grant, C.S. was supported by GM65347 from NIGMS, and S.R.G. was supported by an ACS Research Scholar Grant and CA143763 from NCI. The content of this paper is solely the responsibility of the authors and does not necessarily represent the official views of ACS, NIGMS, NCI or NIH.
References
- Subramanian T, Chinnadurai G. Association of class I histone deacetylases with transcriptional corepressor CtBP. FEBS Lett 2003; 540:255 - 258
- Chinnadurai G. CtBP family proteins: more than transcriptional corepressors. Bioessays 2003; 25:9 - 12
- Boyd JM, Subramanian T, Schaeper U, La Regina M, Bayley S, Chinnadurai G. A region in the C-terminus of adenovirus 2/5 E1a protein is required for association with a cellular phosphoprotein and important for the negative modulation of T24-ras mediated transformation, tumorigenesis and metastasis. EMBO J 1993; 12:469 - 478
- Schaeper U, Boyd JM, Verma S, Uhlmann E, Subramanian T, Chinnadurai G. Molecular cloning and characterization of a cellular phosphoprotein that interacts with a conserved C-terminal domain of adenovirus E1A involved in negative modulation of oncogenic transformation. Proc Natl Acad Sci USA 1995; 92:10467 - 10471
- Bergman LM, Blaydes JP. C-terminal binding proteins: emerging roles in cell survival and tumorigenesis. Apoptosis 2006; 11:879 - 888
- Kuppuswamy M, Vijayalingam S, Zhao LJ, Zhou Y, Subramanian T, Ryerse J, et al. Role of the PLDLS-binding cleft region of CtBP1 in recruitment of core and auxiliary components of the corepressor complex. Mol Cell Biol 2008; 28:269 - 281
- Zhang Q, Wang SY, Nottke AC, Rocheleau JV, Piston DW, Goodman RH. Redox sensor CtBP mediates hypoxia-induced tumor cell migration. Proc Natl Acad Sci USA 2006; 103:9029 - 9033
- Grooteclaes M, Deveraux Q, Hildebrand J, Zhang Q, Goodman RH, Frisch SM. C-terminal-binding protein corepresses epithelial and proapoptotic gene expression programs. Proc Natl Acad Sci USA 2003; 100:4568 - 4573
- Mani-Telang P, Sutrias-Grau M, Williams G, Arnosti DN. Role of NAD binding and catalytic residues in the C-terminal binding protein corepressor. FEBS Lett 2007; 581:5241 - 5246
- Fjeld CC, Birdsong WT, Goodman RH. Differential binding of NAD+ and NADH allows the transcriptional corepressor carboxyl-terminal binding protein to serve as a metabolic sensor. Proc Natl Acad Sci USA 2003; 100:9202 - 9207
- Zhang Q, Piston DW, Goodman RH. Regulation of corepressor function by nuclear NADH. Science 2002; 295:1895 - 1897
- Thio SS, Bonventre JV, Hsu SI. The CtBP2 co-repressor is regulated by NADH-dependent dimerization and possesses a novel N-terminal repression domain. Nucleic Acids Res 2004; 32:1836 - 1847
- Kumar V, Carlson JE, Ohgi KA, Edwards TA, Rose DW, Escalante CR, et al. Transcription corepressor CtBP is an NAD(+)-regulated dehydrogenase. Mol Cell 2002; 10:857 - 869
- Balasubramanian P, Zhao LJ, Chinnadurai G. Nicotinamide adenine dinucleotide stimulates oligomerization, interaction with adenovirus E1A and an intrinsic dehydrogenase activity of CtBP. FEBS Lett 2003; 537:157 - 160
- Kim JH, Youn HD. C-terminal binding protein maintains mitochondrial activities. Cell Death Differ 2009; 16:584 - 592
- Chinnadurai G. The transcriptional corepressor CtBP: a foe of multiple tumor suppressors. Cancer Res 2009; 69:731 - 734
- Chen YW, Paliwal S, Draheim K, Grossman SR, Lewis BC. p19Arf inhibits the invasion of hepatocellular carcinoma cells by binding to C-terminal binding protein. Cancer Res 2008; 68:476 - 482
- Paliwal S, Kovi RC, Nath B, Chen YW, Lewis BC, Grossman SR. The alternative reading frame tumor suppressor antagonizes hypoxia-induced cancer cell migration via interaction with the COOH-terminal binding protein corepressor. Cancer Res 2007; 67:9322 - 9329
- Paliwal S, Pande S, Kovi RC, Sharpless NE, Bardeesy N, Grossman SR. Targeting of C-terminal binding protein (CtBP) by ARF results in p53-independent apoptosis. Mol Cell Biol 2006; 26:2360 - 2372
- Mirnezami AH, Campbell SJ, Darley M, Primrose JN, Johnson PW, Blaydes JP. Hdm2 recruits a hypoxia-sensitive corepressor to negatively regulate p53-dependent transcription. Curr Biol 2003; 13:1234 - 1239
- Mroz EA, Baird AH, Michaud WA, Rocco JW. COOH-terminal binding protein regulates expression of the p16INK4A tumor suppressor and senescence in primary human cells. Cancer Res 2008; 68:6049 - 6053
- Liu Y, El-Naggar S, Darling DS, Higashi Y, Dean DC. Zeb1 links epithelial-mesenchymal transition and cellular senescence. Development 2008; 135:579 - 588
- Zhang Q, Yoshimatsu Y, Hildebrand J, Frisch SM, Goodman RH. Homeodomain interacting protein kinase 2 promotes apoptosis by downregulating the transcriptional corepressor CtBP. Cell 2003; 115:177 - 186
- Nadauld LD, Phelps R, Moore BC, Eisinger A, Sandoval IT, Chidester S, et al. Adenomatous polyposis coli control of C-terminal binding protein-1 stability regulates expression of intestinal retinol dehydrogenases. J Biol Chem 2006; 281:37828 - 37835
- Phelps RA, Chidester S, Dehghanizadeh S, Phelps J, Sandoval IT, Rai K, et al. A two-step model for colon adenoma initiation and progression caused by APC loss. Cell 2009; 137:623 - 634
- Kovi RC, Paliwal S, Pande S, Grossman SR. An ARF/CtBP2 complex regulates BH3-only gene expression and p53-independent apoptosis. Cell Death Differ 17:513 - 521
- Achouri Y, Noel G, Van Schaftingen E. 2-Keto-4-methylthiobutyrate, an intermediate in the methionine salvage pathway, is a good substrate for CtBP1. Biochem Biophys Res Commun 2007; 352:903 - 906
- Subhi AL, Diegelman P, Porter CW, Tang B, Lu ZJ, Markham GD, et al. Methylthioadenosine phosphorylase regulates ornithine decarboxylase by production of downstream metabolites. J Biol Chem 2003; 278:49868 - 49873
- Tang B, Kadariya Y, Murphy ME, Kruger WD. The methionine salvage pathway compound 4-methylthio-2-oxobutanate causes apoptosis independent of downregulation of ornithine decarboxylase. Biochem Pharmacol 2006; 72:806 - 815
- Walser M. Ketoacids in the treatment of uremia. Clin Nephrol 1975; 3:180 - 186
- Mitch WE, Abras E, Walser M. Long-term effects of a new ketoacid-amino acid supplement in patients with chronic renal failure. Kidney Int 1982; 22:48 - 53
- Dilger RN, Kobler C, Weckbecker C, Hoehler D, Baker DH. 2-keto-4-(methylthio)butyric acid (keto analog of methionine) is a safe and efficacious precursor of L-methionine in chicks. J Nutr 2007; 137:1868 - 1873
- Stipanuk MH. The keto acid of methionine is a safe and efficacious substitute for dietary L-methionine: the answer from chick bioassays. J Nutr 2007; 137:1844 - 1845
- Burri N, Shaw P, Bouzourene H, Sordat I, Sordat B, Gillet M, et al. Methylation silencing and mutations of the p14ARF and p16INK4a genes in colon cancer. Lab Invest 2001; 81:217 - 229
- Chinnadurai G, Vijayalingam S, Rashmi R. BIK, the founding member of the BH3-only family proteins: mechanisms of cell death and role in cancer and pathogenic processes. Oncogene 2008; 27:20 - 29
- Daniel PT, Pun KT, Ritschel S, Sturm I, Holler J, Dorken B, et al. Expression of the death gene Bik/Nbk promotes sensitivity to drug-induced apoptosis in corticosteroid-resistant T-cell lymphoma and prevents tumor growth in severe combined immunodeficient mice. Blood 1999; 94:1100 - 1107
- Sturm I, Stephan C, Gillissen B, Siebert R, Janz M, Radetzki S, et al. Loss of the tissue-specific proapoptotic BH3-only protein Nbk/Bik is a unifying feature of renal cell carcinoma. Cell Death Differ 2006; 13:619 - 627
- Castells A, Ino Y, Louis DN, Ramesh V, Gusella JF, Rustgi AK. Mapping of a target region of allelic loss to a 0.5 cM interval on chromosome 22q13 in human colorectal cancer. Gastroenterology 1999; 117:831 - 837
- Zhu H, Zhang L, Dong F, Guo W, Wu S, Teraishi F, et al. Bik/NBK accumulation correlates with apoptosis-induction by bortezomib (PS-341, Velcade) and other proteasome inhibitors. Oncogene 2005; 24:4993 - 4999
- Radetzki S, Kohne CH, von Haefen C, Gillissen B, Sturm I, Dorken B, et al. The apoptosis promoting Bcl-2 homologues Bak and Nbk/Bik overcome drug resistance in Mdr-1-negative and Mdr-1-overexpressing breast cancer cell lines. Oncogene 2002; 21:227 - 238
- Xie X, Xia W, Li Z, Kuo HP, Liu Y, Ding Q, et al. Targeted expression of BikDD eradicates pancreatic tumors in noninvasive imaging models. Cancer Cell 2007; 12:52 - 65
- Zou Y, Peng H, Zhou B, Wen Y, Wang SC, Tsai EM, et al. Systemic tumor suppression by the proapoptotic gene bik. Cancer Res 2002; 62:8 - 12
- Jin W, Scotto KW, Hait WN, Yang JM. Involvement of CtBP1 in the transcriptional activation of the MDR1 gene in human multidrug resistant cancer cells. Biochem Pharmacol 2007; 74:851 - 859
- Walser M, Coulter AW, Dighe S, Crantz FR. The effect of keto-analogues of essential amino acids in severe chronic uremia. J Clin Invest 1973; 52:678 - 690
- Hildebrand JD, Soriano P. Overlapping and unique roles for C-terminal binding protein 1 (CtBP1) and CtBP2 during mouse development. Mol Cell Biol 2002; 22:5296 - 5307
- Lowe SW, Cepero E, Evan G. Intrinsic tumour suppression. Nature 2004; 432:307 - 315
- Bunz F, Dutriaux A, Lengauer C, Waldman T, Zhou S, Brown JP, et al. Requirement for p53 and p21 to sustain G2 arrest after DNA damage. Science 1998; 282:1497 - 1501
- Soule HD, Maloney TM, Wolman SR, Peterson WD Jr, Brenz R, McGrath CM, et al. Isolation and characterization of a spontaneously immortalized human breast epithelial cell line, MCF-10. Cancer Res 1990; 50:6075 - 6086
- Taniguchi T, Garcia-Higuera I, Xu B, Andreassen PR, Gregory RC, Kim ST, et al. Convergence of the fanconi anemia and ataxia telangiectasia signaling pathways. Cell 2002; 109:459 - 472