Abstract
Although many breast cancers respond to chemotherapy or hormonal therapy, lack of tumor eradication is a central clinical problem preceding the development of drug resistant tumors. Using the K14cre;Brca1F5-13/F5-13;p53F2-10/F2-10mouse model for hereditary breast cancer, we have previously studied responses of mammary tumors generated in to clinically relevant anti-cancer drugs, including cisplatin. The BRCA1- and p53-deficient tumors generated in this model are hypersensitive to cisplatin and never become resistant to this agent due to the large, irreversible deletion in Brca1. We show here that even dose-dense treatment with a maximum tolerated dose of cisplatin does not result in complete tumor eradication. To explain this result we have addressed the hypothesis that the lack of eradication of drug-sensitive tumors is due to increased in vivo chemotherapy resistance of tumor-initiating cells (TICs). Using the CD24 and CD49f cell surface markers which detect normal mouse mammary stem cells, we have identified tumor-initiating cells in BRCA1- and p53-deficient tumors. In addition to the "OLE_LINK14">Lin-/CD24+/CD49f+ subpopulation, we show that a larger population of Lin-/CD24+/CD49f- cells also has tumor-initiating capability in at least two serial orthotopic transplantations, suggesting that these are not more differentiated transit-amplifying cells. However, we did not find an enrichment of TICs in cisplatin-treated tumor remnants. We conclude that in this model the tolerance of the cisplatin-surviving cells cannot be attributed to special biochemical defense mechanisms of TICs.
Introduction
Heterozygous mutations in the breast cancer susceptibility genes BRCA1 or BRCA2 represent well-known genetic risk factors for the development of breast cancer, since the loss of BRCA1 or BRCA2 function compromises homology-directed DNA repair, ultimately resulting in genomic instability (reviewed in ref. Citation1). BRCA1-associated breast cancer appears to originate from aberrant luminal progenitor cells,Citation2 and nearly all tumors have a p53 mutation.Citation3 The molecular signature of BRCA1-associated breast cancer is basal-like.Citation4 Like patients with basal-like breast cancer, BRCA1 mutation carriers initially often respond well to chemotherapy,Citation5 but nevertheless face a poor prognosis in the absence of adjuvant chemotherapy.Citation6 A possible explanation for this paradox is the higher likelihood of relapse in patients in whom pathologic complete response was not achieved.Citation7
To study the chemotherapeutic response of BRCA1-associated breast cancer in a more clinically relevant in vivo model, we have previously used the K14cre;Brca1F5–13/F5–13;p53F2–10/F2–10 genetically engineered mouse model.Citation8 This model recapitulates several key features of human BRCA1-associated breast cancer, including a basal-like phenotype, lack of estrogen receptor, progesterone receptor and ERBB2 expression and a high degree of genomic instability.Citation9 Tumors can be transplanted orthotopically into syngeneic mice without loss of their genomic profile, morphology or sensitivity to drug,Citation8 and they are sensitive to the maximum tolerable dose (MTD) of cisplatin. However, like in their human counterparts, not all cells are eradicated, eventually resulting in the regrowth of tumors after drug pressure is taken off.
Cisplatin is a DNA adduct-forming chemotherapeutic that induces intra- and inter-strand DNA crosslinks (reviewed in ref. Citation10). Pt-DNA adducts are primarily removed by nucleotide excision repair (NER), which is functional in our tumor model. The major cytotoxic effect of Pt-DNA adducts appears to result from the formation of double-stranded DNA breaks (DSB). To repair this defect, error-prone non-homologous end-joining (NHEJ) can still be employed in BRCA1;p53-deficient tumors, whereas error-free repair by homologous recombination (HR) cannot, due to the lack of functional BRCA1 (reviewed in ref. Citation11 and Citation12). The requirement for HR to optimally repair platinuminduced DNA damage is demonstrated by the lack of acquired cisplatin resistance in the Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 model.Citation8,Citation13 Importantly, this requirement is also supported by clinical data: restoration of BRCA1 or BRCA2 function is a mechanism of acquired drug resistance in BRCA1 or BRCA2 mutation carriers with ovarian carcinomas.Citation14–Citation16 Such genetic reversion is impossible in our model due to the large intragenic Brca1 deletion. Moreover, occasional patients appear to have tumors with irreversible defects in HR that remain sensitive through multiple relapses.Citation17
Taking the preclinical and clinical data together, it is remarkable that such drug-sensitive tumors are usually not eradicated by chemotherapy. Several hypotheses have been proposed to explain this treatment failure and tumor recurrence. A popular hypothesis is that resistance is due to special defense mechanisms of a small subpopulation of cells within breast tumors, often designated “tumor-initiating cells (TICs)” or “cancer stem cells (CSCs)”.Citation18 In contrast to more differentiated tumor cells, TICs are hypothesized to survive therapy and hence reinitiate tumor growth after treatment. The complexity of the cancer stem cell paradigm has comprehensively been reviewed by Rosen and Jordan.Citation19 It is becoming clear that the definition of tumor-initiating cells often depends on the type of model and assay used, thus creating debate. An underinvestigated piece of this puzzle is the nature of residual therapy-surviving TICs in drug-sensitive breast cancer.
In the breast a range of cell surface markers has been tested to identify normal and cancer stem cells and to characterize a mammary epithelium hierarchy in both humans and mice (reviewed in ref. Citation20). Building on that information, we show here that the Lin−/CD24+/CD49f+ subpopulation of Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mouse mammary tumor cells has increased tumorigenicity in vivo upon orthotopic transplantation into syngeneic host animals. However, this TIC fraction is not increased in the tumor remnants that remain following cisplatin treatment, indicating that the highly tumorigenic Lin−/CD24+/CD49f+ cells are not preferentially resistant to cisplatin. Hence, the results of our model argue against the hypothesis that TICs have special biochemical defense mechanisms that would make them less vulnerable to drugs. We suggest that other mechanisms, such as cell cycle arrest of some of the cells with tumor-initiating capability, may prove to be of more importance.
Results
Treatment of mice bearing Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mammary tumors with a dose-dense cisplatin schedule does not result in a substantial increase in tumor eradication.
We have previously shown that Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mouse mammary tumors are hypersensitive to the MTD of cisplatin (6 mg/kg i.v.) and never become fully resistant.Citation8 Nevertheless, these tumors are rarely eradicated by drug. We therefore tested whether dose-dense cisplatin treatment would lead to tumor eradication by applying an additional dose of 6 mg cisplatin per kg i.v at day 14. At this time animals had recovered from the initial treatment and tumors had shrunken markedly, in average by 92.5% (±12.2 SD, n = 18). The additional dose of cisplatin considerably increased the relapse-free survival (RFS) of mice (), resulting in a median RFS of 93 days, compared with 33 days in mice given only a single injection of cisplatin (p < 0.0004). However, even a dose-dense cisplatin treatment schedule did not completely eradicate the majority of Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumors (11/13 tumors), and the recurrent tumors remain sensitive to repeated rounds of cisplatin administration.Citation8
Identification of TICs in Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mammary tumors.
A plausible mechanism for survival of a small fraction of tumor cells after cisplatin treatment, is that a “remnant” fraction consisting of specialized cells, such as tumor-initiating cells (TICs) with stem cell-like properties, is insensitive to cisplatin and can in turn re-populate the tumor.
To identify the TICs in this tumor model, we examined a range of previously reported normal mammary and breast cancer stem cell markers, including CD24/Thy1, aldehyde dehydrogenase (ALDH), CD133 and CD24/CD49f.Citation21–Citation24 Of these, only the combination of CD24 and CD49f selected for cells with increased tumorigenic potential ( and ). Other markers did not, or were present in tumors at very low levels (data not shown). A representative FACS profile of a tumor stained with CD24 and CD49f antibodies, after removal of dead cells (propidium iodide positive) and fibroblastoid, endothelial and erythroid (Lin+) cells, is shown in . The Lin−/CD24+/CD49f+ subpopulation, representing 2–20% of total tumor cells, showed enhanced repopulation capacity in comparison with all other sorted fractions ( and ). Injection of as few as 100 Lin−/CD24+/CD49f+ tumor cells resulted in tumor outgrowth (3/8) with higher take when 250 cells were injected (12/16), whereas no tumors formed from corresponding dilutions of Lin−/CD24−/CD49f+, Lin−/CD24+/CD49f− or Lin−/CD24−/CD49f− populations. Purity of the sorted fractions was routinely checked and estimated to be >90% ().
Since tumor-initiating cells resemble normal stem cells in their ability to form spheres in culture under non-adherent conditions, we examined the ability of the Lin−/CD24+/CD49f+ fraction to form tumorspheres in vitro. Using culturing conditions described by Dontu et al.Citation25 we found that only Lin−/CD24+/CD49f+ cells formed tumorspheres after approximately 7 days in culture, whereas other fractions did not ( and B). Transplantation of cultured tumorspheres into the mammary fat pads of syngeneic animals resulted in tumor outgrowth after 4 weeks and these tumors retained the CD24, CD49f FACS profile of the donor tumor ().
Although our findings indicate that the Lin−/CD24+/CD49f+ cells drive tumorigenicity in this model, this was not the only population with tumorigenic capacity. Injection of higher cell numbers (n = 1,000), resulted in tumor outgrowth from the Lin−/CD24+/CD49f− fraction (9/21) and furthermore, injection of 5,000 Lin−/CD24−/CD49f+ cells, gave rise to tumors in 2 out of 7 mice ( and ). Tumor outgrowth from these fractions was significantly delayed in comparison with the highly tumorigenic Lin−/CD24+/CD49f+ cells (). The Lin−/CD24−/CD49f− cells did not generate any tumors, indicating that this fraction consisted largely of stromal cells.
Lin−/CD24+/CD49f− cells do not represent a transit-amplifying fraction.
The ability of Lin−/CD24−/CD49f+ and Lin−/CD24+/CD49f− fractions to generate tumors could be due to contamination with the tumorigenic Lin−/CD24+/CD49f+ subpopulation, or to the presence of transit-amplifying cells with limited proliferative ability. To examine these possibilities, we checked whether tumors that grew out after injection of sorted cells could reconstitute the cellular heterogeneity observed in the unsorted donor tumor. Secondary tumors from the sorted Lin−/CD24−/CD49f+ fraction had a FACS profile similar to the unsorted donor tumor and Lin−/CD24+/CD49f+ sorted tumor, with a variable increase in the number of CD49f positive cells (, C and D), suggesting that tumor formation from the Lin−/CD24−/CD49f+ population could be a result of minor contamination with Lin−/CD24+/CD49f+ cells. Tumors from the sorted Lin−/CD24+/CD49f− subpopulation remained largely negative for CD49f, in contrast to the donor tumor ( and B), suggesting that this fraction may contain more differentiated transit-amplifying tumor cells.
An important characteristic of transit-amplifying cells is their limited proliferative capacity. To determine the proliferative capacity of Lin−/CD24+/CD49f− tumor cells, tumors generated with this fraction were sorted and the Lin−/CD24+/CD49f− cells were injected into the mammary fat pad of a syngeneic animal. As indicated in and , Lin−/CD24+/CD49f− cells formed Lin−/CD24+/CD49f− tumors reproducibly upon secondary transplantation. These findings suggest the Lin−/CD24+/CD49f− cells are not likely to represent tumor cells with limited proliferative capacity, although the lack of CD49f expression found in these tumors may contribute to the lower tumorigenic potential of this fraction in vivo.
To further verify whether the Lin−/CD24+/CD49f− cells represent a more differentiated fraction, the histological profiles of all secondary tumors and their respective donor tumors were evaluated for a range of basal and luminal epithelial markers, including Keratin 5, 6, 8 and 14. As shown in , all secondary and donor tumors displayed similar levels of the markers examined. Moreover, all tumors remained negative for the stromal markers vimentin and smooth muscle actin (). CD44, an established human breast cancer stem cell marker, was equally highly expressed in tumors arising from all sorted subpopulations ().
Microarray analysis of donor and secondary tumors showed that they were indistinguishable in their gene expression profiles. The Lin−/CD24+/CD49f− subpopulation did not form a distinct group in the hierarchical clustering analysis (), in agreement with the histology findings. Collectively, these results suggest that Lin−/CD24+/CD49f− cells do not represent a fraction of more differentiated cells.
Cisplatin remnants are not enriched in the highly tumorigenic Lin−/CD 24+/CD 49f+ fraction.
To evaluate the role of TICs in the cisplatin response, we first investigated whether Lin− cells in the remnants are more tumorigenic. About 10,000 Lin− cells were collected 10 days after cisplatin administration, when sufficient debulking of tumor mass was achieved and the size of the remnant was approximately 15 mm3. When these residual 10,000 Lin− cells were orthotopically transplanted, not a single tumor grew for T1–T3. We tested several protocols, including shorter tumor digestion time and collagenase treatment only. However, such milder tumor dissociations protocols failed to produce single cells. Since we know that tumors regrow from remnants, we conclude that tumor dissociation and FACS are too injurious for TICs that have survived recent cisplatin treatment. As a surrogate analysis, we compared the growth kinetics of untreated tumors and tumors that regrow from remnants. Based on our FACS analysis, we know that the percentage of Lin− cells in remnants is about 3-fold less than in untreated tumors [27.2% ± 9.8 SD (n = 5) versus 84.1% ± 4.7 SD (n = 5)] due to infiltrating stromal cells that are part of the wound healing reaction. Taking this into account, we do not see a significant difference in growth kinetics between remnants and untreated tumor fragments: remnants of about 10 to 15 mm3 (10–14 days after cisplatin treatment) take 24.8 days (±9.6 SD, n = 9) to grow into a 200 mm3 tumor, while 2 to 4 mm3 untreated tumor fragments take 23.3 days (±6.1 SD, n = 9) after transplantation to produce a 200 mm3 tumor.
An obvious indirect way to quantify TICs in remnants is to measure the Lin−/CD24+/CD49f+ fraction. The results of this analysis, presented in and , show that the highly tumorigenic Lin−/CD24+/CD49f+ fraction is not enriched in the remnants following drug treatment, suggesting mechanisms other than the resistance of “cancer stem cells” may determine the survival of Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumor cells after cisplatin treatment. Moreover, tumors formed from Lin−/CD24+/CD49f+ or Lin−/CD24+/CD49f− fractions remained sensitive to cisplatin treatment ().
To corroborate these unexpected results, we examined whether cisplatin remnants were enriched in tumor-initiating cells by using other previously reported tumor-initiating cell markers, including CD24/CD29 and ALDH.Citation25,Citation26 CD29 was present at moderate levels in these tumors () and furthermore, no enrichment in the Lin−/CD24med/CD29+ fraction was observed in the cisplatin remnants (). Similarly, cisplatin remnants did not contain elevated ALDH positivity as determined by the ALDEFLUOR® assay (Supp. Fig. 1).
Discussion
Using BRCA1- and p53-deficient mammary tumors we have investigated the frequency and properties of tumor-initiating Lin−/CD24+/CD49f+ cells in the tumor remnants found after cisplatin chemotherapy. The increased tumorinitiating capacity of this population was defined by the “gold standard” assay for “stemness,” which is the transplantation of sorted cell populations into (in our case syngeneic) host animals.Citation20 Moreover, only the Lin−/CD24+/CD49f+ cells formed tumorspheres in vitro using conditions developed to propagate mammary stem cells.Citation25 In our model the selection of dissociated Lin− mammary tumor cells with the CD24 and CD49f markers, which identify normal mouse mammary stem cells, worked best for enriching TICs. The TIC frequency of Lin−/CD24+/CD49f+ cells was calculated as 1/359. However, it is obvious that this is an underestimation of the true tumor-initiating capacity. On average, tumors grow from a hardly palpable cisplatin remnant that contains about 10,000 Lin− cells to a tumor with more than 200,000 Lin− cells (∼500 mm3) in about 30 days. The average doubling time in this tumor model is ∼3 days, which means that at least 200 of the 10,000 remnant Lin− tumor cells must be TICs; hence ∼1/50. Based on our limiting dilution transplantations it can be expected that the TIC frequency of Lin−/CD24+/CD49f+ is even higher, at least closer to ∼1/5. The difference with the measured TIC frequency can be explained by the damage that occurs during the dissociation and sorting of the tumor, required to generate and analyze single tumor cells.
We also tested other published markers including CD24/Thy1,Citation21 ALDHCitation22 and CD133,Citation23 and found that none of these outperformed Lin−/CD24+/CD49f+. Likewise, CD24 and CD49f were most useful to enrich for TICs in other mouse mammary tumor models as well.Citation27,Citation28 In the MMTV-Cre;Brca1F11/F11;p53+/−, WAP-Cre;Brca1F11/F11;p53F5–6/F5–6 and MMTV-Cre;Brca1F11/F11;p53F5–6/F5–6 mammary tumor models TICs were present in the CD29high;CD24medium fraction,Citation26,Citation27 whereas Zhang et al.Citation29 identified CD29high;CD24high as the TIC fraction in a p53−/− mouse breast tumor model.
Markers for TICs in mouse mammary tumors differ from those established for human breast cancer. Human breast cancer stem cells were first isolated and characterized by Al-Hajj et al.Citation30 who found that only CD44+/CD24−/low cells were able to grow tumors when injected into immunodeficient mice. This human breast cancer stem cell profile only partially matches that of enriched normal human mammary stem cells (MaSCs). Upon co-implantation of epithelial subsets with immortalized human breast fibroblasts in NOD/SCID/IL2Rγ−/− mice, only the CD49fhi/EpCAM− subpopulation had regenerative and limited self-renewal capacity.Citation2,Citation31 Whereas CD24 is mainly expressed in luminal cells of the human mammary gland, cells of both the basal and luminal lineages show CD44 expression.Citation32 Murine MaSC also have high levels of CD49f (and CD29), but CD24 is more a pan-epithelial marker in the mouse mammary gland.Citation24,Citation33,Citation34
Intriguingly, we found that tumors resulting from engrafted Lin−/CD24+/CD49f− cells reproducibly keep their Lin−/CD24+/CD49f− profile upon secondary transplantation. This suggests that Lin−/CD24+/CD49f− cells are not transit-amplifying but truly tumor-initiating, albeit less efficient than the Lin−/CD24+/CD49f+ ones since a higher number of cells is required. Unsupervised hierarchical cluster analysis of the gene expression profiles of Lin−/CD24+/CD49f− tumors did not distinguish them from tumors derived from unsorted or Lin−/CD24+/CD49f+ cells. This suggests there is no intrinsic difference between these tumors other than the expression of the CD49f cell surface marker. Since α6 integrin (CD49f) and β1 integrin (CD29) may play a role in connecting MaSC to the extracellular matrix (ECM),Citation20,Citation35 it is only logical to reason that á6 integrin (CD49f) helps to anchor tumor cells to the mammary fat pad stroma upon engraftment. This is probably advantageous for epithelial tumor cells to survive after the harsh procedure of enzymatic dissociation and sorting. This hypothesis is supported by the requirement of integrins, in particular β1 integrins, for the basal mammary epithelial cell-ECM interactions in mice,Citation36 and for the adhesion to matrigel and proliferation of human induced pluripotent stem cells.Citation37 Further optimization of the microenvironment to facilitate tumor-stromal cell interactions may identify a higher frequency of breast cancer cells with tumor-initiating ability, as shown by Quintana et al.Citation38 for melanoma cells.
Like most solid tumors in humans, BRCA1- and p53-deficient mouse mammary tumors are hardly ever eradicated by chemotherapy. Despite their high sensitivity to cisplatin, even a dose-dense treatment does not cure tumor-bearing animals. Our initial hypothesis to explain the chemotherapy failure was that mammary TICs were intrinsically resistant to therapeutic agents.Citation18,Citation39 If TICs comprise only a minority of the tumor cell population, then shrinkage of tumors after chemotherapy may reflect the effects of the chemotherapy on more differentiated cells in a tumor rather than on the relatively small TIC component. Special features of TICs that could contribute to resistance include increased DNA repair, a higher expression of drug efflux transporters or quiescence.Citation29,Citation40,Citation41 The hypothesis of increased chemotherapy resistance of human breast TICs is supported by the paclitaxel and 5-fluorouracil resistance of cultured CD44+/CD24−/low/EpCAM+ cells.Citation41 In addition, neoadjuvant chemotherapy has been reported to result in an increased fraction of CD44+/CD24−/low cells in breast cancer biopsies, accompanied by a higher tumorsphere-forming ability.Citation42 In contrast to these results, we did not find an enrichment of the Lin−/CD24+/CD49f+ or Lin−/CD24+/CD49f− fractions in Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mammary tumor remnants. The re-growth kinetics of the tumor from the remnants do not indicate TIC enrichment either. This strongly suggests that TICs in our mouse model do not have special biochemical features to protect them from being killed by cisplatin. An obvious explanation for the lack of TIC enrichment is the cisplatin hypersensitivity seen in our model. We have not observed the occurrence of acquired cisplatin resistance, which proves that no other resistance mechanism can compensate for the loss of BRCA1 function.Citation13 Hence, even if TICs possessed a general drug resistance mechanism (e.g., increased capability to repair DNA), this may not be sufficient to overcome the complete loss of BRCA1 function.
In contrast to our results, Shafee et al.Citation26 found that the CD29high;CD24med fraction was enriched in Brca1Δ11/Δ11;p53Δ5–6/Δ5–6 mouse mammary tumors that developed resistance to cisplatin by prolonged treatment with low doses of the drug. Why these cells were resistant is not known. The main difference with our tumor model is that Shafee et al. only delete exon 11 of Brca1 resulting in a hypomorphic allele encoding residual BRCA1 activity.Citation43 Resistant TICs could therefore have upregulated the expression of the hypomorphic Brca1 allele, but this possibility was not investigated.
Why residual TICs in our model survive the cisplatin onslaught is still unknown. We are currently investigating the possibility that surviving TICs arrest in their cell cycle and ‘hibernate’ until the drug is gone and DNA-platinum adducts are removed, e.g., by nucleotide excision repair. It is also possible that residual tumor cells have undergone reversible epigenetic alterations that confer drug tolerance, as observed in vitro by Sharma et al.Citation44 using various cell lines. Another hypothesis was suggested by Creighton et al.Citation45 who found that chemotherapy-surviving TICs have a mesenchymal molecular subtype and possibly undergo an epithelial-mesenchymal transition (EMT). Why such mesenchymal TICs would be more resistant to chemotherapy is still unclear. Since relapsing breast cancers also have an epithelial phenotype, it remains to be determined whether and how mesenchymal chemotherapy-surviving TICs switch back to an epithelial morphology. In our model EMT is an unlikely mechanism of chemotherapy survival, as neither Lin−/CD24+/CD49f+ cells nor relapsing Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumors show a mesenchymal phenotype characterized by spindle cell morphology or vimentin positivity.
In summary our study shows the utility of genetically engineered mouse models to test hypotheses for the failure of chemotherapy in eradicating some types of tumors. We hope that further characterization of the cells in this model that survive chemotherapy and then support tumor re-growth will provide information to identify such cells in human tumors and to develop methods to target them.
Materials and Methods
Generation of mammary tumors.
Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 mammary tumors were generated in K14cre;Brca1F5–13/F5–13;p53F2–10/F2–10 mice and genotyped as described.Citation9,Citation46 Orthotopic transplantations of tumor fragments or dissociated cells into syngeneic animals and caliper measurements of mammary tumors were reported previously.Citation8,Citation47 Tumor samples were snap-frozen in liquid nitrogen, fixed in both 4% formalin or embedded in Tissue-Tek Optimal Cutting Temperature and stored at −80°C. All animal experiments were approved by the Animal Ethics Committee of the Netherlands Cancer Institute.
Gene expression profiling.
Genome-wide expression analysis was carried out as described.Citation48 Unsupervised hierarchical clustering analysis was performed with the pvclust software (www.is.titech.ac.jp/∼shimo/prog/pvclust/). The hierachical clustering algorithm was based on Euclidean distance and complete linkage was applied to group tumor samples according to similarity in the pattern of gene expression. MIAME-compliant microarray data have been deposited in the ArrayExpress database (accession no. E-NCMF-33).
Cell preparation for sorting experiments.
Preparation of tumor cell suspensions was as previously performedCitation24 with the following modifications: tumors were mechanically dissociated with a razor blade and then sequentially digested with collagenase/hyaluronidase (300 units per mL and 100 units per mL, 2 hours at 37°C), trypsin-EDTA (0.05% trypsin/1 mM EDTA, 1 min at 37°C) and dispase (5 mg per mL, 2 min at 37°C) containing 0.1 mg per mL DNase I (Sigma). Finally, the cell mixture was resuspended in Hank's Buffer Salt Solution (HBSS) supplemented with 2% heat-inactivated fetal calf serum (termed HF buffer) and filtered through a 70 µm mesh. Reagents were from StemCell Technologies Inc., unless otherwise specified. For in vivo transplantation tumorigenicity assays of sorted populations, viable cell numbers were confirmed by microscope counts after staining with Trypan Blue. Cells were resuspended in HF buffer/matrigel mix (1:1) and injected into the left or right inguinal mammary fat pads.
Cell staining for flow cytometry.
Antibodies used were CD24-fluorescein isothiocyanate (FITC, clone M1/69, Pharmingen, 1:50), CD49f-R-phycoerythrin (PE, clone GoH3, Pharmingen, 1:100), anti-Thy1-allophycocyanin (APC, clone HIS51, EBioscience), CD29 (PE, clone HMb1-1, EBioscience, 1:100) and CD133 (AlexaFluor®488, clone 13A4, EBioscience, 1:100). To remove cells of erythroid, endothelial and fibroblastoid origin from the mix, the cell suspension containing the above mentioned antibodies in HF was incubated for 15 minutes with biotinylated CD45 (1:100), TER119 (1:100), CD31 (1:300) and CD140a (1:300) antibodies (Ebioscience) on ice, followed by 20 min incubation with a Streptavidin-Cy5 conjugate (Invitrogen, 1:50) and finally, incubation with 1.34 µg propidium iodide (PI)/ml to check cell viability. Washing steps were performed with HF buffer. In all experiments, lineage+ (Lin+: CD45+, TER119+, CD31+, CD140a+) and dead cells (PI+) were discarded with a gating strategy (Supp. Fig. 2) during flow cytometry. The ALDEFLUOR® kit (StemCell Technologies, Durham, NC, USA) was used to label tumor cells with high aldehyde dehydrogenase (ALDH) activity according to manufacturer's instructions. All analyses and sorts were performed using a Cyan analyzer (Beckman Coulter; FITC = 530/30, PE = 575/25, PI = 613/20, Cy5 = 665/20; Summit 4.3.01 software) or FACSAria (Becton Dickinson; FITC = 530/30, PE = 585/42, PI = 695/40, Cy5 = 660/20; nozzle tip diameter 100 µm; 20 psi; 30 kHz; Diva 6.1.2 software), respectively. Side scatter and forward scatter profiles (height and width) were used to eliminate cell doublets. To ensure compatibility of the experiments single color controls (Lin-Cy5, CD24-FITC, CD49f-PE or PI) were carried out in each experiment and the compensation was calculated by the software.
In vitro tumorsphere assays.
For tumorsphere generation, single cells were plated on ultra-low attachment plates (Corning) in DMEM medium containing 20 ng basic fibroblast growth factor (Sigma), 20 ng epidermal growth factor (Sigma), 4 µg heparin (Sigma) per mL and B-27 supplement (1:50 dilution, Invitrogen). Cells were cultured at 37°C in 5% CO2.
Immunohistochemical analysis.
Immunohistochemical stainings of tumors were carried out by using Keratin 5 (Covance, #PRB-160P, 1:500), Keratin 6 (Covance, #PRB-169P, 1:500), Keratin 14 (Covance, #PRB-155P, 1:500), Keratin 8 (Sigma, #GW22752, 1:1,000), smooth muscle actin (Immunotech, #2111, 1:400), vimentin (Fitzgerald, #RDI-PROGP53, 1:100) and CD44 (BD Pharmingen, #550538, 1:100) antibodies diluted in phosphate saline buffer containing 1% bovine serum albumin (PBSA) and 0.05% Tween 20. Antigen accessibility was increased by boiling for 20 minutes in citrate buffer (pH 6.0, DAKO). After overnight probing at 4°C with primary antibodies, slides were incubated with biotinylated anti-mouse, anti-rat or anti-guinea pig secondary antibodies (DAKO) for 1 hour at room temperature. For detection, we used a standard Streptavidin-ABC amplified staining procedure with DAB (DAKO) and hematoxylin counterstaining. Positive and negative (no primary antibody) controls were included for each slide and staining procedure.
Financial Support
This work was supported by grants from the Dutch Cancer Society 2006-3566 (P.B., S.R., J.J.), 2009-4303 (S.R., J.J., P.B.), the European Union FP6 Integrated Project 037665-CHEMORES (P.B., S.R.) and CTMM Breast Care (J.J., S.R.).
Abbreviations
| ALDH | = | aldehyde dehydrogenase |
| APC | = | allophycocyanin |
| BRCA1 | = | breast cancer type 1 susceptibility protein |
| BRCA2 | = | breast cancer type 2 susceptibility protein |
| CSC | = | cancer stem cell |
| DAB | = | diaminobenzidine |
| DSB | = | DNA double-strand break |
| ECM | = | extracellular matrix |
| EDTA | = | ethylene-diamine-tetra-acetic acid |
| EpCAM | = | epithelial cell adhesion molecule |
| FITC | = | fluorescein isothiocyanate |
| HBSS | = | Hank's Buffer Salt Solution |
| HR | = | homologous recombination |
| K14 | = | keratin 14 |
| MaSC | = | mammary stem cell |
| MIAME | = | minimum information about a microarray experiment |
| MTD | = | maximum tolerable dose |
| NHEJ | = | non-homologous end-joining |
| PE | = | phycoerythrin |
| RFS | = | relapse-free survival |
| TIC | = | tumor-initiating cell |
Figures and Tables
Figure 1 Response of Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumors to dose-dense cisplatin treatment. (A) Example of 2 animals carrying an orthotopically transplanted Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumor (in this case T1). When the tumor reached a volume of 200 mm3 animals were treated with 6 mg cisplatin per kg i.v. on day 0 (blue) or on days 0 and 14 (green). The same treatments were resumed when tumors grew to 100% of their original size (indicated by blue rhombi and green squares). After 6 doses of cisplatin, animals had to be sacrificed due to kidney failure. The growth of the untreated control is shown in pink. (B) Relapse-free survival of 31 animals treated as outlined in (A). Animals are carrying 18 (blue, cisplatin day 0) or 13 (green, cisplatin days 0 and 14) individual orthotopically transplanted Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumors. p value was determined using the Wilcoxon test.
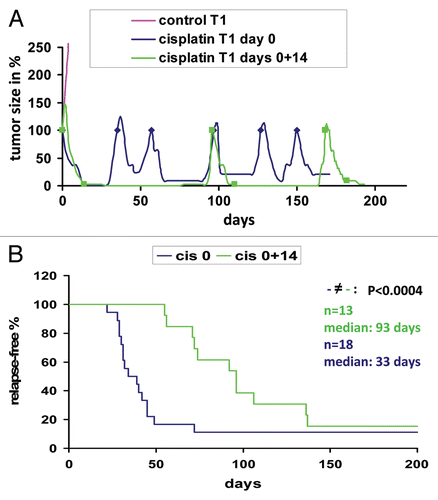
Figure 2 Fractionation of Brca1Δ5–13/Δ5–13;p53Δ2–10/Δ2–10 tumors using FACS. (A) Sorting of live (propidium iodide-negative), Lin- (non-endothelial, non-fibroblastic and non-hematopoietic) cells with CD24- and CD49f-specific antibodies. (B) Tumorigenicity of limiting dilutions of sorted fractions after orthotopic transplantation into syngeneic animals. (C) The FACS-sorted Lin−/CD24+/CD49f+ tumor fraction was collected (left) and re-analyzed by FACS (right) to verify the purity of the collected fraction.
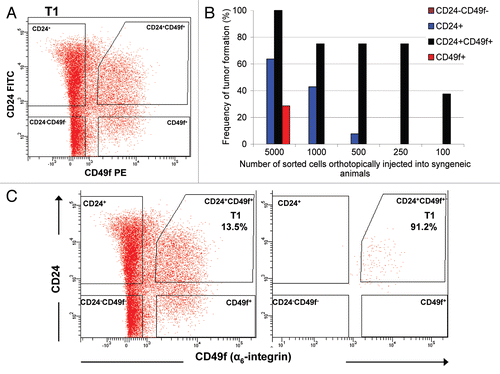
Figure 3 Enhanced tumorsphere-forming ability of Lin−/CD24+/CD49f+ cells in comparison with other sorted subpopulations. (A) Morphology of tumorspheres grown in ultra-low attachment plates from Lin−/CD24+/CD49f+ fraction and other sorted cells in culture. Lin−/CD24+/CD49f− cells occasionally formed small cell aggregates but did not generate tumorspheres. Images were taken on day 10 after plating of freshly sorted cells. Bars represent 50 µm. (B) Quantification of tumorsphere-initiating cells based on data generated from culturing sorted cells under non-adherent conditions from three independent sorted tumors. (C) FACS profile of a tumor generated upon orthotopic transplantation of 50 CD24+/CD49f+ spheres into a syngeneic female mouse.
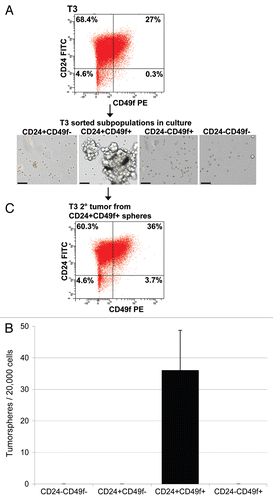
Figure 4 Characteristics of subpopulation-generated tumors compared to the donor tumor. (A) Time to tumor outgrowth upon transplantation of sorted subpopulations based on a combination of Lin−, CD24 and CD49f markers. Graph depicts time to tumor incidence for tumors that grew from 17 Lin−/CD24+/CD49f−, 32 Lin−/CD24+/CD49f+ and 2 Lin−/CD24−/CD49f+ subpopulations. Statistical analysis performed to determine any differences among the three groups was one-way analysis of variance (ANOVA). (B–M) Histological profiling by immunostaining for stromal markers vimentin (Vim) and smooth muscle actin (SMA) and the tumor-initiating cell marker, CD44. The parts show tumor sections from parental tumor T2 (B, F and J) and tumors resulting from the inoculation of 1,000 T2 Lin−/CD24+/CD49f− cells (C, G and K), 1,000 T2 Lin−/CD24+/CD49f+ cells (D, H and L) and 5,000 T2 Lin−/CD24−/CD49f+ cells (E, I and M). Bar represents 50 µm.
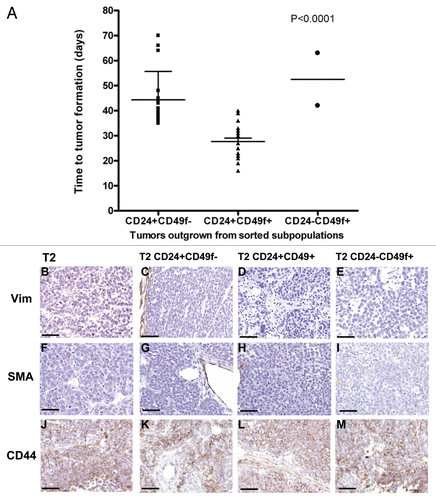
Figure 5 Histological and FACS profiles of subpopulation-derived tumors compared to the donor tumor. Donor tumor T2 (A, E, I, M and Q) and tumors resulting from orthotopic transplantation of 1,000 T2 Lin−/CD24+/CD49f− cells (B, F, J, N and R), 1,000 T2 Lin−/CD24+/CD49f+ cells (C, G, K, O and S) or 5,000 T2 Lin−/CD24−/CD49f+ cells (D, H, L, P and T) are shown. Single color controls (Lin-Cy5, CD24-FITC, CD49f-PE or PI) were included in each FACS experiment, with compensation done by the software. Bars represent 50 µm.
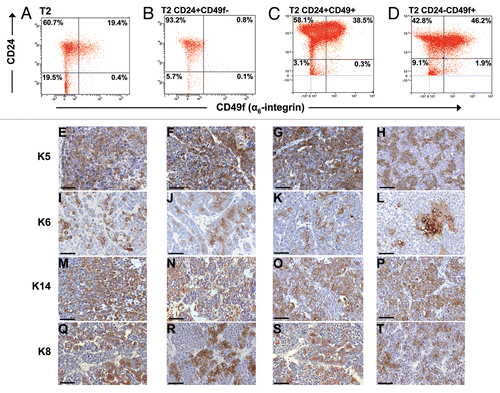
Figure 6 Lin−/CD24+/CD49f+ and Lin−/CD24+/CD49f− fractions repeatedly form tumors after multiple consecutive rounds of sorting and transplantation. (A) FACS plot of tumor T3 grown from spheres and subsequent outgrown tumors from orthotopically transplanted Lin−/CD24+/CD49f− and Lin−/CD24+/CD49f+ cell fractions. Tumors were analyzed on different days. In compliance with Alexander et al.Citation49 single color controls (Lin-Cy5, CD24-FITC, CD49f-PE or PI) were included in each experiment and the compensation was done by the software. (B) Unsupervised hierarchical cluster analysis of gene expression data from tumors outgrown after sorting. The populations used for orthotopic transplantation are indicated in parenthesis.
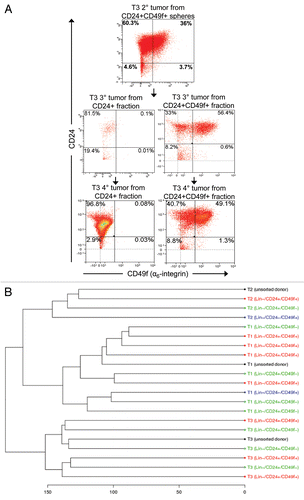
Figure 7 Tumor-initiating cells are not enriched in the tumor remnants that survive cisplatin treatment. FACS analyses of cells dissociated from untreated tumors and tumor remnants 10 days after i.v. injection of 6 mg cisplatin (CDDP) per kg, represented graphically in (A) and summarized in . (B) Repeated cisplatin sensitivity of tumors generated from sorted cells in comparison with the donor tumor. Arrows indicate cisplatin administration (6 mg/kg i.v.) after each tumor reached a size that was 100% of the initial tumor. (C) The same analysis as in (A), using a CD29 antibody instead of CD49f.
Table 1 Tumor incidence summary of the graph shown in
Table 2 Lin−/CD24+/CD49f+ and Lin−/CD24+/CD49f− fractions repeatedly form tumors after multiple consecutive rounds of sorting and transplantation
Table 3 Summary of FACS analyses of cells dissociated from untreated tumors and tumor remnants 10 days after i.v. injection of 6mg cisplatin (CDDP) per kg
Additional material
Download Zip (132.4 KB)Acknowledgements
We would like to thank Janneke Jaspers, Guotai Xu and Serge Zander for critical reading of the manuscript. Arno Velds was very helpful in processing the gene expression profiling data.
References
- Turner N, Tutt A, Ashworth A. Hallmarks of ‘BRCAness’ in sporadic cancers. Nat Rev Cancer 2004; 4:814 - 819
- Lim E, Vaillant F, Wu D, Forrest NC, Pal B, Hart AH, et al. Aberrant luminal progenitors as the candidate target population for basal tumor development in BRCA1 mutation carriers. Nat Med 2009; 15:907 - 913
- Holstege H, Joosse SA, van Oostrom CT, Nederlof PM, de Vries A, Jonkers J. High incidence of protein-truncating TP53 mutations in BRCA1-related breast cancer. Cancer Res 2009; 69:3625 - 3633
- Sorlie T, Tibshirani R, Parker J, Hastie T, Marron JS, Nobel A, et al. Repeated observation of breast tumor subtypes in independent gene expression data sets. Proc Natl Acad Sci USA 2003; 100:8418 - 8423
- Kriege M, Seynaeve C, Meijers-Heijboer H, Collee JM, Menke-Pluymers MB, Bartels CC, et al. Sensitivity to first-line chemotherapy for metastatic breast cancer in BRCA1 and BRCA2 mutation carriers. J Clin Oncol 2009; 27:3764 - 3771
- Goffin JR, Chappuis PO, Bégin LR, Wong N, Brunet JS, Hamel N, et al. Impact of germline BRCA1 mutations and overexpression of p53 on prognosis and response to treatment following breast carcinoma: 10-year follow up data. Cancer 2003; 97:527 - 536
- Carey LA, Dees EC, Sawyer L, Gatti L, Moore DT, Collichio F, et al. The triple negative paradox: primary tumor chemosensitivity of breast cancer subtypes. Clin Cancer Res 2007; 13:2329 - 2334
- Rottenberg S, Nygren AO, Pajic M, van Leeuwen FW, van der Heijden I, van de Wetering K, et al. Selective induction of chemotherapy resistance of mammary tumors in a conditional mouse model for hereditary breast cancer. Proc Natl Acad Sci USA 2007; 104:12117 - 12122
- Liu X, Holstege H, van der Gulden H, Treur-Mulder M, Zevenhoven J, Velds A, et al. Somatic loss of BRCA1 and p53 in mice induces mammary tumors with features of human BRCA1-mutated basal-like breast cancer. Proc Natl Acad Sci USA 2007; 104:12111 - 12116
- Kelland L. The resurgence of platinum-based cancer chemotherapy. Nat Rev Cancer 2007; 7:573 - 584
- Jackson SP, Bartek J. The DNA-damage response in human biology and disease. Nature 2009; 461:1071 - 1078
- Branzei D, Foiani M. Regulation of DNA repair throughout the cell cycle. Nat Rev Mol Cell Biol 2008; 9:297 - 308
- Borst P, Rottenberg S, Jonkers J. How do real tumors become resistant to cisplatin?. Cell Cycle 2008; 7:1353 - 1359
- Edwards SL, Brough R, Lord CJ, Natrajan R, Vatcheva R, Levine DA, et al. Resistance to therapy caused by intragenic deletion in BRCA2. Nature 2008; 451:1111 - 1115
- Swisher EM, Sakai W, Karlan BY, Wurz K, Urban N, Taniguchi T. Secondary BRCA1 mutations in BRCA1-mutated ovarian carcinomas with platinum resistance. Cancer Res 2008; 68:2581 - 2586
- Sakai W, Swisher EM, Karlan BY, Agarwal MK, Higgins J, Friedman C, et al. Secondary mutations as a mechanism of cisplatin resistance in BRCA2-mutated cancers. Nature 2008; 451:1116 - 1120
- Melichar B, Fridrichova P, Tomsova M, Malirova E. Repeat chemosensitivity of epithelial ovarian carcinoma in a BRCA1 mutation carrier to paclitaxel/platinum combination chemotherapy. Eur J Gynaecol Oncol 2009; 30:323 - 325
- Clarke MF, Dick JE, Dirks PB, Eaves CJ, Jamieson CH, Jones DL, et al. Cancer stem cells—perspectives on current status and future directions: AACR Workshop on cancer stem cells. Cancer Res 2006; 66:9339 - 9344
- Rosen JM, Jordan CT. The increasing complexity of the cancer stem cell paradigm. Science 2009; 324:1670 - 1673
- Visvader JE. Keeping abreast of the mammary epithelial hierarchy and breast tumorigenesis. Genes Dev 2009; 23:2563 - 2577
- Cho RW, Wang X, Diehn M, Shedden K, Chen GY, Sherlock G, et al. Isolation and molecular characterization of cancer stem cells in MMTV-Wnt-1 murine breast tumors. Stem Cells 2008; 26:364 - 371
- Ginestier C, Hur MH, Charafe-Jauffret E, Monville F, Dutcher J, Brown M, et al. ALDH1 is a marker of normal and malignant human mammary stem cells and a predictor of poor clinical outcome. Cell Stem Cell 2007; 1:555 - 567
- Wright MH, Calcagno AM, Salcido CD, Carlson MD, Ambudkar SV, Varticovski L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res 2008; 10:10
- Stingl J, Eirew P, Ricketson I, Shackleton M, Vaillant F, Choi D, et al. Purification and unique properties of mammary epithelial stem cells. Nature 2006; 439:993 - 997
- Dontu G, Abdallah WM, Foley JM, Jackson KW, Clarke MF, Kawamura MJ, et al. In vitro propagation and transcriptional profiling of human mammary stem/progenitor cells. Genes Dev 2003; 17:1253 - 1270
- Shafee N, Smith CR, Wei S, Kim Y, Mills GB, Hortobagyi GN, et al. Cancer stem cells contribute to cisplatin resistance in Brca1/p53-mediated mouse mammary tumors. Cancer Res 2008; 68:3243 - 3250
- Vassilopoulos A, Wang RH, Petrovas C, Ambrozak D, Koup R, Deng CX. Identification and characterization of cancer initiating cells from BRCA1 related mammary tumors using markers for normal mammary stem cells. Int J Biol Sci 2008; 4:133 - 142
- Teissedre B, Pinderhughes A, Incassati A, Hatsell SJ, Hiremath M, Cowin P. MMTV-Wnt1 and -DeltaN89beta-catenin induce canonical signaling in distinct progenitors and differentially activate Hedgehog signaling within mammary tumors. PLoS One 2009; 4:e4537
- Zhang M, Behbod F, Atkinson RL, Landis MD, Kittrell F, Edwards D, et al. Identification of tumor-initiating cells in a p53-null mouse model of breast cancer. Cancer Res 2008; 68:4674 - 4682
- Al-Hajj M, Wicha MS, Benito-Hernandez A, Morrison SJ, Clarke MF. Prospective identification of tumorigenic breast cancer cells. Proc Natl Acad Sci USA 2003; 100:3983 - 3988
- Eirew P, Stingl J, Raouf A, Turashvili G, Aparicio S, Emerman JT, et al. A method for quantifying normal human mammary epithelial stem cells with in vivo regenerative ability. Nat Med 2008; 14:1384 - 1389
- Raouf A, Zhao Y, To K, Stingl J, Delaney A, Barbara M, et al. Transcriptome analysis of the normal human mammary cell commitment and differentiation process. Cell Stem Cell 2008; 3:109 - 118
- Shackleton M, Vaillant F, Simpson KJ, Stingl J, Smyth GK, Asselin-Labat ML, et al. Generation of a functional mammary gland from a single stem cell. Nature 2006; 439:84 - 88
- Sleeman KE, Kendrick H, Ashworth A, Isacke CM, Smalley MJ. CD24 staining of mouse mammary gland cells defines luminal epithelial, myoepithelial/basal and non-epithelial cells. Breast Cancer Res 2006; 8:7
- Raymond K, Deugnier MA, Faraldo MM, Glukhova MA. Adhesion within the stem cell niches. Curr Opin Cell Biol 2009; 21:623 - 629
- Taddei I, Deugnier MA, Faraldo MM, Petit V, Bouvard D, Medina D, et al. Beta1 integrin deletion from the basal compartment of the mammary epithelium affects stem cells. Nat Cell Biol 2008; 10:716 - 722
- Rowland TJ, Miller LM, Blaschke AJ, Doss EL, Bonham AJ, Hikita ST, et al. Roles of integrins in human induced pluripotent stem cell growth on matrigel and vitronectin. Stem Cells Dev 2009; 19:1231 - 1240
- Quintana E, Shackleton M, Sabel MS, Fullen DR, Johnson TM, Morrison SJ. Efficient tumour formation by single human melanoma cells. Nature 2008; 456:593 - 598
- Reya T, Morrison SJ, Clarke MF, Weissman IL. Stem cells, cancer and cancer stem cells. Nature 2001; 414:105 - 111
- Dean M, Fojo T, Bates S. Tumour stem cells and drug resistance. Nat Rev Cancer 2005; 5:275 - 284
- Fillmore CM, Kuperwasser C. Human breast cancer cell lines contain stem-like cells that self-renew, give rise to phenotypically diverse progeny and survive chemotherapy. Breast Cancer Res 2008; 10:25
- Li X, Lewis MT, Huang J, Gutierrez C, Osborne CK, Wu MF, et al. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J Natl Cancer Inst 2008; 100:672 - 679
- Huber LJ, Yang TW, Sarkisian CJ, Master SR, Deng CX, Chodosh LA. Impaired DNA damage response in cells expressing an exon 11-deleted murine Brca1 variant that localizes to nuclear foci. Mol Cell Biol 2001; 21:4005 - 4015
- Sharma SV, Lee DY, Li B, Quinlan MP, Takahashi F, Maheswaran S, et al. A chromatin-mediated reversible drug-tolerant state in cancer cell subpopulations. Cell 2010; 141:69 - 80
- Creighton CJ, Li X, Landis M, Dixon JM, Neumeister VM, Sjolund A, et al. Residual breast cancers after conventional therapy display mesenchymal as well as tumor-initiating features. Proc Natl Acad Sci USA 2009; 106:13820 - 13825
- Jonkers J, Meuwissen R, van der Gulden H, Peterse H, van der Valk M, Berns A. Synergistic tumor suppressor activity of BRCA2 and p53 in a conditional mouse model for breast cancer. Nat Genet 2001; 29:418 - 425
- Rottenberg S, Pajic M, Jonkers J. Studying drug resistance using genetically engineered mouse models for breast cancer. Methods Mol Biol 2010; 596:33 - 45
- Zander SA, Kersbergen A, van der Burg E, de Water N, van Tellingen O, Gunnarsdottir S, et al. Sensitivity and acquired resistance of BRCA1;p53-deficient mouse mammary tumors to the topoisomerase I inhibitor topotecan. Cancer Res 2010; 70:1700 - 1710
- Alexander CM, Puchalski J, Klos KS, Badders N, Ailles L, Kim CF, et al. Separating stem cells by flow cytometry: reducing variability for solid tissues. Cell Stem Cell 2009; 5:579 - 583