Abstract
Okazaki fragment processing is an integral part of DNA replication. For a long time, we assumed that the maturation of these small RNA-primed DNA fragments did not necessarily have to occur during S phase, but could be postponed to late in S phase after the bulk of DNA synthesis had been completed. This view was primarily based on the arrest phenotype of temperature-sensitive DNA ligase I mutants in yeast, which accumulated with an almost fully duplicated set of chromosomes. However, many temperature-sensitive alleles can be leaky, and the re-evaluation of DNA ligase I-deficient cells has offered new and unexpected insights into how cells keep track of lagging strand synthesis. It turns out that if Okazaki fragment joining goes awry, cells have their own alarm system in the form of ubiquitin that is conjugated to the replication clamp PCNA. Although this modification results in mono- and poly-ubiquitination of PCNA, it is genetically distinct from the known post-replicative repair mark at lysine 164. In this Extra View, we discuss the possibility that eukaryotic cells utilize different enzymatic pathways and ubiquitin attachment sites on PCNA to alert the replication machinery to the accumulation of single-stranded gaps or nicks behind the fork.
Introduction
In humans, more than 30,000,000 Okazaki fragments (OFs) have to be initiated (assuming an average size of ∼200 nt/OF), processed and ligated during a single round of DNA replication.Citation1 It is easy to imagine that failure to properly join OFs would cause a large number of single-strand breaks (SSBs)—at a frequency that would likely overwhelm the SSB repair machinery and thus be lethal. It is therefore not surprising that to date only one individual with a severe defect in DNA ligase I, the enzyme that joins OFs during DNA replication, has been identified and described.Citation2,Citation3 The individual carried compound heterozygous mutations in the gene and retained approximately 5% ligase activity.Citation4,Citation5 Importantly, this reduction caused growth retardation, immunodeficiency, UV sensitivity and lymphoma.Citation2,Citation3 Most of these phenotypes are compatible with the known function of DNA ligase I in DNA replication and nucleotide excision repair.Citation6 The only exception is the immunodeficiency, which went hand-in-hand with a profound lack of immunoglobulin class switching.Citation2,Citation3 Although this latter defect was not recapitulated in a mouse model carrying a homozygous mutation of the viable missense allele, DNA ligase I-deficient animals displayed growth retardation, abnormal erythropoiesis and increased genome instability.Citation7,Citation8 Moreover, they developed an unexpectedly wide range of spontaneous tumors, such as lymphomas, adeno- and other carcinomas. The importance of proper OF maturation is further underscored by the fact that mutations in other enzymes that participate in OF processing, such as the flap endonuclease Fen1 (e.g., a E160D mutation), which retains partial endonuclease activity,Citation9 have been directly linked to cancer in humans.Citation10 Interestingly, recent evidence suggests that the E160D Fen1 mutation, similar to the DNA ligase I deficiency, causes early-onset lymphoma in mice.Citation11 This raises the question of whether cells harbor a system capable of monitoring perturbations during OF processing. In addition, it remains unclear if abnormalities in lagging strand maturation affect replication fork progression. This is exactly what we set out to explore in a recent study that was published earlier this year.Citation12
Post-Replicative Repair and DNA Damage Induction by Okazaki Fragment Processing Defects
Today we have a relatively good understanding of the post-replication repair (PRR) pathways involved in sensing DNA lesions, like ultraviolet (UV) irradiation-induced thymine dimers,Citation13 in the leading or lagging strand template.Citation14–Citation16 PRR seemed for many years somewhat of a misnomer, as most studies analyzed the pathway in the context of an active replication fork.Citation17–Citation21 However, recent studies have come full circle demonstrating that PRR can occur outside of S phase, after replication has been largely completed.Citation22,Citation23 The first description of PRR dates back to 1968 when Rupp and Howard-Flanders described discontinuities in the newly synthesized DNA of nucleotide excision repair (NER) defective E. coli.Citation24 Shortly thereafter, the concept was extended to mammalian cells.Citation25 During the past 42 years much progress has been made toward understanding error-prone and error-free PRR, respectively.Citation26 Nevertheless, most studies published to date utilize UV or other mutagens like methyl methanesulfonate (MMS) to create large adducts in the DNA that prompt collisions with the replicative polymerases, thereby causing transient replication fork arrest.Citation21,Citation27–Citation29 This is usually accompanied by formation of long stretches of replication protein A (RPA)-coated, single-stranded (ss) DNA.Citation30 These RPA-coated regions facilitate PRR activation by ubiquitination of PCNA at lysine 164,Citation18,Citation31 and also trigger the S phase checkpoint.Citation30 Both events are genetically separable and are thought to occur in parallel.Citation20,Citation32 Although the abovementioned experimental approach utilizing UV-irradiation or MMS has provided a powerful model to dissect the molecular steps of these pathways, it has obvious limitations, because forks that accumulate ssDNA are probably not the only structures that arise in replication-defective mutants. Aberrant Okazaki fragment processing, for example, can cause nicks—in a wide range of varieties: “clean” nicks due to ligation deficiency, “dirty” nicks that exhibit adenylated 5′ ends if the ligation reaction is aborted prematurely or “flapped” nicks in the case of improper removal of RNA primers.Citation33,Citation34 This latter reaction requires the complicated interplay of multiple activities. Limited displacement synthesis by DNA polymerase (pol)-δ is required to initially create a short flap that is deleted one nucleotide at a time by Fen1.Citation35 Excessive strand displacement is counteracted by the intrinsic 3′-exonuclease activity of the catalytic subunit of pol-δ and its non-essential binding partner, Pol32.Citation36,Citation37 Cells defective in Fen1, appear to create larger flaps (in a Pol32-dependent manner) that can be processed by a second endonuclease, Dna2.Citation38–Citation40 Recent studies have also implicated other enzymes, such as exonuclease 1 and the helicase Pif1 in this backup system.Citation37,Citation41 Thus, the first two steps (strand displacement and flap removal) of OF processing (OFP) already involve an elaborate network of partially redundant pathways to assure proper production of a ligatable nick that can be sealed by DNA ligase I. Importantly, primer removal by Fen1 and OF ligation by DNA ligase I are coordinated by PCNA.Citation42 Both enzymes bind PCNA through a PCNA interacting peptide (PIP) box.Citation43–Citation45
Okazaki Fragment Processing Defects Trigger PCNA Ubiquitination
The fact that multiple OFP pathways exist suggests that cells have the ability to actively monitor maturation events during lagging strand synthesis. To address this question, we analyzed DNA ligase I-deficient cells.Citation46 In budding yeast, temperature-sensitive cdc9 mutants were among the first strains identified to have a cell cycle progression defect under non-permissive conditions as they arrested in late S/G2 phase.Citation47–Citation49 The arrest was dependent on the DNA damage responseCitation50 and cell viability required functional homologous recombination,Citation51 indicating that the nicks might be—at least partially—converted into double strand breaks (DSBs). These early studies suggested that OF ligation was uncoupled from replication fork progression and could occur very late in S phase after the bulk of the DNA had been replicated. This was also consistent with observations made much later that PCNA remained on chromatin after passage of the replication fork, providing anchor points for DNA ligase I on chromatin.Citation52 In fact, experiments with cdc9-1 cells that had two consecutive shifts, to the non-permissive temperature first and then back to the permissive temperature demonstrated that OF synthesis could be temporally separated from OF ligation.Citation46 All of these findings were consistent with the notion that DNA ligase I was not required until the end of S phase. However, our recent analysis of the cdc9-1 mutation in a different strain background suggested that the cells do not move through S phase with normal kinetics, but rather appeared to accumulate in mid-S phase.Citation12 Further exploration of a temperature-sensitive degron mutant (cdc9-td) verified that DNA replication was extensively delayed in the absence of DNA ligase I (). Although it still remains unclear how this delay was mediated (e.g., through reduced fork progression or inhibition of late-firing origins), it was dependent on the activation of the S phase checkpoint kinase Rad53.Citation53 Rad53 phosphorylation was not only Rad9- (and thus DNA damage) dependent,Citation54 but also required the mediator of the replication checkpoint, Mrc1,Citation55,Citation56 the yeast homolog of Claspin.Citation57 This finding implied that replication fork stalling was occuring.Citation58,Citation59 How this happened in a scenario in which none of the DNA polymerases were actively inhibited was puzzling. Even more surprising was the observation that the phosphorylation of Rad53 was amplified by a novel PRR-related pathway that triggered mono- and poly-ubiquitination of PCNA.Citation12 Mutation of lysine 164 of PCNA in cdc9 mutants had no effect on cell viability, arguing that neither error-prone translesion synthesisCitation19,Citation21,Citation60–Citation62 nor error-free replication by template switchCitation63–Citation66 played a role in this process. Instead, we observed synthetic lethality between cdc9-1 and a mutation in lysine 107 of PCNA, leading us to propose that ubiquitin is conjugated to lysine 107 in DNA ligase I-deficient cells. One aspect that we did not address in our recent study is whether PCNA is also modified by the ubiquitin-related SUMO peptide, which is attached to PCNA during S phase at lysine 164 and to a lesser extent at lysine 127.Citation18 In , we demonstrate that PCNA is indeed sumoylated in wild-type cells and cdc9 mutants. Because the mutants accumulated in S phase, they displayed stronger sumoylation signals. In addition, we observed higher molecular weight forms of sumoylated PCNA. This is consistent with the finding that PCNA carries SUMO chains in the face of profound replication stress.Citation67 Alternatively, it is also possible that a single PCNA monomer is ubiquitinated (at lysine 107) and sumoylated (at lysine 164) in cdc9 mutants. Sumoylation of PCNA is thought to repress homologous recombinationCitation68–Citation70 at stalled replication forks. This raises an interesting point because homologous recombination is required for the viability of cdc9 mutants and therefore its activity needs to be highly regulated. Future experiments are needed to clarify these issues. However, regardless of their outcome, it is important to mention that ubiquitination of PCNA is conserved in human cells that are depleted for DNA ligase I, even though lysine 107 of PCNA is not evolutionarily conserved.Citation12 This underscores the significance of our findings.
Consistent with the observation that a DNA ligase I deficiency triggers ubiquitin conjugation at a non-canonical lysine residue of PCNA in budding yeast, we have also uncovered genetic requirements for this process that clearly distinguish it from the classical DNA damage tolerance (DDT) pathways. For example, whereas DDT is dependent on Rad6 and Rad18, both proteins are dispensable for PCNA ubiquitination in cdc9 mutants. This makes sense in as far as Rad6 and Rad18 mediate predominantly translesion synthesis,Citation22 which would help little in facilitating the repair of nicks. Moreover, the poly-ubiquitin chains observed in cdc9 mutants are linked through lysine 29 and not lysine 63 as in PRR.Citation18 In the light of this result, it is not surprising that Ubc13, which together with Mms2 catalyzes lysine 63 linked ubiquitin chains,Citation71–Citation73 did not appear to play a role in this pathway.Citation12 What was puzzling, however, was the genetic requirement for MMS2 and UBC4. The human homolog of Ubc4, UbcH5A, synthesizes lysine 29 linked ubiquitin chains in vitro.Citation74 Although Ubc13 has been reported to function independently of Mms2,Citation75 no other studies have demonstrated a genetic function for Mms2 that is separable from Ubc13. How Mms2 fits into the PCNA ubiquitination pathway is not at all clear and will require additional genetic validation. Furthermore, biochemical studies are currently underway to dissect whether Mms2, Ubc4 and Rad5 cooperate in one physical complex—possibly together with other proteins—or not. At this point, we have no evidence to suggest a direct interaction between Mms2 and Ubc4. Moreover, we would like to emphasize that our data does not dispute nor contradict any of the established rules for the DDT and PRR pathways.Citation76 Rather, we interpret them to mean that both the repertoires of damaged DNA structures that cause PCNA ubiquitination as well as the pathways catalyzing these reactions are likely larger and more complex than anticipated. This is also underscored by recent observations that described the Rad6-independent ubiquitination of PCNA in response to chronic HU and MMS treatment at lysine 164.Citation77 Although we are still missing many pieces of the puzzle, it appears that different types of DNA damage may trigger different PCNA ubiquitination pathways that result in distinct attachment sites and/or differently linked ubiquitin chains (i.e., a “DNA damage code”; ).Citation12
Future Challenges
Taken together, our results suggest that lagging strand synthesis is actively monitored and that persistent nicks in newly replicated DNA (and maybe also those that arise during abortive repair or result from oxidative damage) can indeed be detected during S phase. Whether earlier steps in Okazaki fragment processing are surveyed in a similar manner remains obscure. Although we tested rad27 and dna2 mutants and neither displayed PCNA ubiquitination under non-permissive conditions,Citation12 it is highly likely that the respective enzymes substitute for one another in these particular mutants. Therefore, the lack of PCNA ubiquitination does not necessarily exclude the possibility that other OFP intermediates are recognized by the cell and trigger modification(s) of the PCNA clamp. Indeed, pol32 mutants cause lagging strand defects, which result in ss gaps.Citation23 Although it is not clear that these gaps are generated during OFP (e.g., they might occur during actual synthesis), they cause PCNA ubiquitination. The most burning question for us concerns the biological function of ubiquitinated PCNA in cdc9 mutants. What is the connection between the modified clamp, the slow replication dynamics and Rad53 activation? Replication fork slowdown would certainly aid in extending the time frame of an individual ligation reaction. In the same vein, modified PCNA may help to retain the enzyme on site until Okazaki fragments have been successfully joined and might counteract abortive ligation reactions due to the limited amounts of DNA ligase I present in the cell. The exciting new technical developments in producing ubiquitinated PCNA in vitroCitation78,Citation79 certainly provide invaluable tools toward finding answers to these questions.
Abbreviations
| PCNA | = | proliferating cell nuclear antigen |
| OF | = | Okazaki fragment |
| UV | = | ultraviolet |
| NER | = | nucleotide excision repair |
| MMS | = | methyl methane sulfonate |
| RPA | = | replication protein A |
| OFP | = | Okazaki fragment processing |
| PIP | = | PCNA interacting protein |
| DSBs | = | double strand breaks |
| DDT | = | DNA damage tolerance |
| PRR | = | post-replicative repair |
| SSBs | = | single strand breaks |
| pol | = | polymerase |
| ss | = | single strand |
| SUMO | = | small ubiquitin-like modifier |
Figures and Tables
Figure 1 DNA ligase I is required for S phase progression. Asynchronous cultures of ABy010 (GAL-UBR1 CDC9) and ABy008 (GAL-UBR1 cdc9-td) were grown at 28°C and arrested in G1 with α-factor. After 2 hours, Ubr1 expression was induced with 2% galactose for 30 min. Subsequently, cultures were shifted to 37°C in medium containing α-factor and 2% galactose. After 90 min, cells were released from G1 for 12 hours. DNA content at indicated time was monitored by flow cytometry and the vertical line indicates a 2C DNA content. The black arrows mark entry into S phase and completion of DNA replication in cdc9-td mutants, respectively.
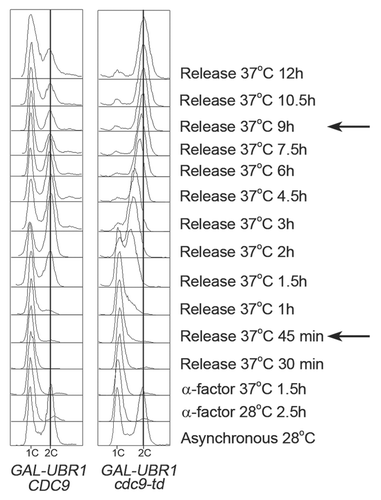
Figure 2 PCNA is sumoylated in cdc9 mutants. Wild-type (CDC9) and cdc9 mutants (cdc9-1 and cdc9-2) expressing PCNA-3HA were grown asynchronously at 25°C and shifted to 35°C for 3 hours. Whole-cell extracts were immunoprecipitated with anti-HA conjugated agarose beads (Sigma). Both unmodified and modified PCNA was detected with an anti-HA antibody (Covance, 16B12). Sumoylated PCNA was detected with an anti-SUMO antibody (gift from Xiaolan Zhao, Memorial Sloan-Kettering Cancer Center). The black dots mark prominent sumoylated PCNA bands. IgGH indicates the immunoglobulin heavy chain. Mono-ubiquitinated PCNA is visible in darker exposures around 42kDa in cdc9 mutants with HA-tagged PCNA (only visible as a faint band in this exposure). cdc9-1 is a more stringent allele than cdc9-2,Citation12 and thus sumoylation of PCNA is more prominent in this mutant.
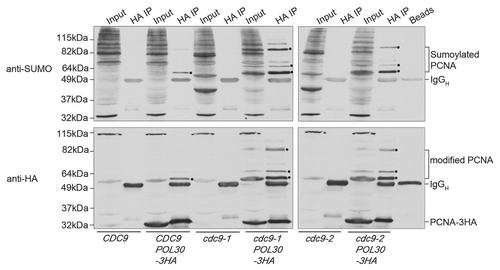
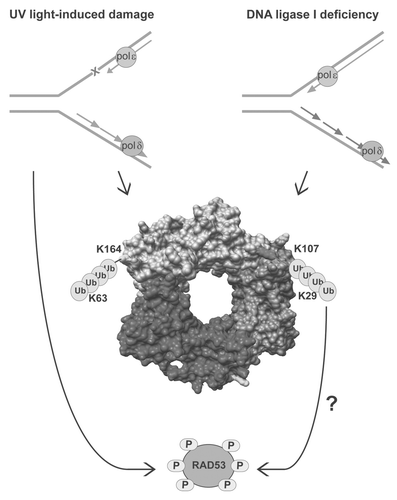
Figure 3 Damage-dependent PCNA ubiquitination in budding yeast. DNA synthesis occurs in a semi-discontinuous manner. Nascent DNA on the leading strand is synthesized by pol ε, whereas pol δ is required to synthesize Okazaki fragments (short arrows) on the lagging strand. In response to UV light-induced damage, which causes pyrimidine dimers on the template DNA (red X, left), PCNA is ubiquitinated at lysine (K) 164 and the poly-ubiquitin chain is linked through K63. Activation of the S phase checkpoint by UV damage is independent of PCNA ubiquitination at K164. In contrast, PCNA is ubiquitinated at K107 and the poly-ubiquitin chain is linked through K29 in response to DNA ligase I deficiency, which accumulates persistent nicks due to unligated Okazaki fragments (red arrows, right). PCNA ubiquitination at K107 appears to be a prerequisite for activation of the S phase checkpoint, illustrated by hyperphosphorylation of Rad53. The importance of the K29-linked poly-ubiquitin chain and how PCNA ubiquitination results in activation of the S phase checkpoint is still unknown. The S. cerevisiae homotrimeric PCNA structure (PDB ID 2OD8) was generated using Chimera software program.Citation80 Ub denotes ubiquitin. K164 and K107 on each of the subunits are indicated as yellow and red surfaces, respectively.
Acknowledgements
We thank Dr. Xiaolan Zhao for the anti-SUMO antibody and Dr. Eric A. Hendrickson for reading the manuscript. We acknowledge the assistance of the Flow Cytometry Core Facility at the University of Minnesota. Molecular graphics images were produced using the UCSF Chimera package from the Resource for Biocomputing, Visualization and Informatics at the University of California, San Francisco (supported by NIH P41 RR-01081). This work was supported by NIH grant GM074917 to A.K.B., who is a Scholar of the Leukemia and Lymphoma Society.
References
- Hubscher U, Seo YS. Replication of the lagging strand: a concert of at least 23 polypeptides. Mol Cells 2001; 12:149 - 157
- Barnes DE, Tomkinson AE, Lehmann AR, Webster AD, Lindahl T. Mutations in the DNA ligase I gene of an individual with immunodeficiencies and cellular hypersensitivity to DNA-damaging agents. Cell 1992; 69:495 - 503
- Webster AD, Barnes DE, Arlett CF, Lehmann AR, Lindahl T. Growth retardation and immunodeficiency in a patient with mutations in the DNA ligase I gene. Lancet 1992; 339:1508 - 1509
- Prigent C, Satoh MS, Daly G, Barnes DE, Lindahl T. Aberrant DNA repair and DNA replication due to an inherited enzymatic defect in human DNA ligase I. Mol Cell Biol 1994; 14:310 - 317
- Mackenney VJ, Barnes DE, Lindahl T. Specific function of DNA ligase I in simian virus 40 DNA replication by human cell-free extracts is mediated by the amino-terminal non-catalytic domain. J Biol Chem 1997; 272:11550 - 11556
- Ellenberger T, Tomkinson AE. Eukaryotic DNA ligases: Structural and functional insights. Annu Rev Biochem 2008; 77:313 - 338
- Harrison C, Ketchen AM, Redhead NJ, O'Sullivan MJ, Melton DW. Replication failure, genome instability and increased cancer susceptibility in mice with a point mutation in the DNA ligase I gene. Cancer Res 2002; 62:4065 - 4074
- Bentley DJ, Harrison C, Ketchen AM, Redhead NJ, Samuel K, Waterfall M, et al. DNA ligase I null mouse cells show normal DNA repair activity but altered DNA replication and reduced genome stability. J Cell Sci 2002; 115:1551 - 1561
- Frank G, Qiu J, Somsouk M, Weng Y, Somsouk L, Nolan JP, et al. Partial functional deficiency of E160D flap endonuclease-1 mutant in vitro and in vivo is due to defective cleavage of DNA substrates. J Biol Chem 1998; 273:33064 - 33072
- Zheng L, Dai H, Zhou M, Li M, Singh P, Qiu J, et al. Fen1 mutations result in autoimmunity, chronic inflammation and cancers. Nat Med 2007; 13:812 - 819
- Larsen E, Kleppa L, Meza TJ, Meza-Zepeda LA, Rada C, Castellanos CG, et al. Early-onset lymphoma and extensive embryonic apoptosis in two domain-specific Fen1 mice mutants. Cancer Res 2008; 68:4571 - 4579
- Das-Bradoo S, Nguyen HD, Wood JL, Ricke RM, Haworth JC, Bielinsky AK. Defects in DNA ligase I trigger PCNA ubiquitylation at Lys 107. Nat Cell Biol 2010; 12:74 - 79
- Setlow RB, Swenson PA, Carrier WL. Thymine dimers and inhibition of DNA synthesis by ultraviolet irradiation of cells. Science 1963; 142:1464 - 1466
- Lee KY, Myung K. PCNA modifications for regulation of post-replication repair pathways. Mol Cells 2008; 26:5 - 11
- Bergink S, Jentsch S. Principles of ubiquitin and SUMO modifications in DNA repair. Nature 2009; 458:461 - 467
- Ulrich HD. Regulating post-translational modifications of the eukaryotic replication clamp PCNA. DNA Repair (Amst) 2009; 8:461 - 469
- Moldovan GL, Pfander B, Jentsch S. PCNA, the maestro of the replication fork. Cell 2007; 129:665 - 679
- Hoege C, Pfander B, Moldovan GL, Pyrowolakis G, Jentsch S. RAD6-dependent DNA repair is linked to modification of PCNA by ubiquitin and SUMO. Nature 2002; 419:135 - 141
- Stelter P, Ulrich HD. Control of spontaneous and damage-induced mutagenesis by SUMO and ubiquitin conjugation. Nature 2003; 425:188 - 191
- Frampton J, Irmisch A, Green CM, Neiss A, Trickey M, Ulrich HD, et al. Postreplication repair and PCNA modification in Schizosaccharomyces pombe. Mol Biol Cell 2006; 17:2976 - 2985
- Kannouche PL, Wing J, Lehmann AR. Interaction of human DNA polymerase eta with monoubiquitinated PCNA: a possible mechanism for the polymerase switch in response to DNA damage. Mol Cell 2004; 14:491 - 500
- Daigaku Y, Davies AA, Ulrich HD. Ubiquitin-dependent DNA damage bypass is separable from genome replication. Nature 2010; 465:951 - 955
- Karras GI, Jentsch S. The RAD6 DNA damage tolerance pathway operates uncoupled from the replication fork and is functional beyond S phase. Cell 2010; 141:255 - 267
- Rupp WD, Howard-Flanders P. Discontinuities in the DNA synthesized in an excision-defective strain of Escherichia coli following ultraviolet irradiation. J Mol Biol 1968; 31:291 - 304
- Lehmann AR. Postreplication repair of DNA in ultraviolet-irradiated mammalian cells. J Mol Biol 1972; 66:319 - 337
- Bridges B. Error-prone DNA repair and translesion synthesis: focus on the replication fork. DNA Repair (Amst) 2005; 4:618 - 619
- Tercero JA, Diffley JF. Regulation of DNA replication fork progression through damaged DNA by the Mec1/Rad53 checkpoint. Nature 2001; 412:553 - 557
- Stokes MP, Michael WM. DNA damage-induced replication arrest in Xenopus egg extracts. J Cell Biol 2003; 163:245 - 255
- Niimi A, Brown S, Sabbioneda S, Kannouche PL, Scott A, Yasui A, et al. Regulation of proliferating cell nuclear antigen ubiquitination in mammalian cells. Proc Natl Acad Sci USA 2008; 105:16125 - 16130
- Zou L, Elledge SJ. Sensing DNA damage through ATRIP recognition of RPA-ssDNA complexes. Science 2003; 300:1542 - 1548
- Davies AA, Huttner D, Daigaku Y, Chen S, Ulrich HD. Activation of ubiquitin-dependent DNA damage bypass is mediated by replication protein A. Mol Cell 2008; 29:625 - 636
- Yang XH, Zou L. Dual functions of DNA replication forks in checkpoint signaling and PCNA ubiquitination. Cell Cycle 2009; 8:191 - 194
- Tomkinson AE, Vijayakumar S, Pascal JM, Ellenberger T. DNA ligases: structure, reaction mechanism and function. Chem Rev 2006; 106:687 - 699
- Rossi ML, Bambara RA. Reconstituted Okazaki fragment processing indicates two pathways of primer removal. J Biol Chem 2006; 281:26051 - 26061
- Garg P, Stith CM, Sabouri N, Johansson E, Burgers PM. Idling by DNA polymerase delta maintains a ligatable nick during lagging-strand DNA replication. Genes Dev 2004; 18:2764 - 2773
- Jin YH, Garg P, Stith CM, Al-Refai H, Sterling JF, Murray LJ, et al. The multiple biological roles of the 3′—>5′ exonuclease of Saccharomyces cerevisiae DNA polymerase delta require switching between the polymerase and exonuclease domains. Mol Cell Biol 2005; 25:461 - 471
- Stith CM, Sterling J, Resnick MA, Gordenin DA, Burgers PM. Flexibility of eukaryotic Okazaki fragment maturation through regulated strand displacement synthesis. J Biol Chem 2008; 283:34129 - 34140
- Lee KH, Kim DW, Bae SH, Kim JA, Ryu GH, Kwon YN, et al. The endonuclease activity of the yeast Dna2 enzyme is essential in vivo. Nucleic Acids Res 2000; 28:2873 - 2881
- Budd ME, Choe W, Campbell JL. The nuclease activity of the yeast DNA2 protein, which is related to the RecB-like nucleases, is essential in vivo. J Biol Chem 2000; 275:16518 - 16529
- Ayyagari R, Gomes XV, Gordenin DA, Burgers PM. Okazaki fragment maturation in yeast. I. Distribution of functions between FEN1 and DNA2. J Biol Chem 2003; 278:1618 - 1625
- Rossi ML, Pike JE, Wang W, Burgers PM, Campbell JL, Bambara RA. Pif1 helicase directs eukaryotic Okazaki fragments toward the two-nuclease cleavage pathway for primer removal. J Biol Chem 2008; 283:27483 - 27493
- Karanja KK, Livingston DM. C-terminal flap endonuclease (rad27) mutations: lethal interactions with a DNA ligase I mutation (cdc9-p) and suppression by proliferating cell nuclear antigen (POL30) in Saccharomyces cerevisiae. Genetics 2009; 183:63 - 78
- Maga G, Hubscher U. Proliferating cell nuclear antigen (PCNA): a dancer with many partners. J Cell Sci 2003; 116:3051 - 3060
- Sakurai S, Kitano K, Yamaguchi H, Hamada K, Okada K, Fukuda K, et al. Structural basis for recruitment of human flap endonuclease 1 to PCNA. EMBO J 2005; 24:683 - 693
- Vijayakumar S, Chapados BR, Schmidt KH, Kolodner RD, Tainer JA, Tomkinson AE. The C-terminal domain of yeast PCNA is required for physical and functional interactions with cdc9 DNA ligase. Nucleic Acids Res 2007; 35:1624 - 1637
- Bielinsky AK, Gerbi SA. Chromosomal ARS1 has a single leading strand start site. Mol Cell 1999; 3:477 - 486
- Johnston LH, Nasmyth KA. Saccharomyces cerevisiae cell cycle mutant cdc9 is defective in DNA ligase. Nature 1978; 274:891 - 893
- Johnston LH. The cdc9 ligase joins completed replicons in baker's yeast. Mol Gen Genet 1983; 190:315 - 317
- Culotti J, Hartwell LH. Genetic control of the cell division cycle in yeast. 3. Seven genes controlling nuclear division. Exp Cell Res 1971; 67:389 - 401
- Weinert TA, Kiser GL, Hartwell LH. Mitotic checkpoint genes in budding yeast and the dependence of mitosis on DNA replication and repair. Genes Dev 1994; 8:652 - 665
- Montelone BA, Prakash S, Prakash L. Spontaneous mitotic recombination in mms8-1, an allele of the CDC9 gene of Saccharomyces cerevisiae. J Bacteriol 1981; 147:517 - 525
- Shibahara K, Stillman B. Replication-dependent marking of DNA by PCNA facilitates CAF-1-coupled inheritance of chromatin. Cell 1999; 96:575 - 585
- Branzei D, Foiani M. Interplay of replication checkpoints and repair proteins at stalled replication forks. DNA Repair (Amst) 2007; 6:994 - 1003
- Sweeney FD, Yang F, Chi A, Shabanowitz J, Hunt DF, Durocher D. Saccharomyces cerevisiae Rad9 acts as a Mec1 adaptor to allow Rad53 activation. Curr Biol 2005; 15:1364 - 1375
- Osborn AJ, Elledge SJ. Mrc1 is a replication fork component whose phosphorylation in response to DNA replication stress activates Rad53. Genes Dev 2003; 17:1755 - 1767
- Naylor ML, Li JM, Osborn AJ, Elledge SJ. Mrc1 phosphorylation in response to DNA replication stress is required for Mec1 accumulation at the stalled fork. Proc Natl Acad Sci USA 2009; 106:12765 - 12770
- Kumagai A, Dunphy WG. Claspin, a novel protein required for the activation of Chk1 during a DNA replication checkpoint response in Xenopus egg extracts. Mol Cell 2000; 6:839 - 849
- Alcasabas AA, Osborn AJ, Bachant J, Hu F, Werler PJ, Bousset K, et al. Mrc1 transduces signals of DNA replication stress to activate Rad53. Nat Cell Biol 2001; 3:958 - 965
- Tanaka K, Russell P. Mrc1 channels the DNA replication arrest signal to checkpoint kinase Cds1. Nat Cell Biol 2001; 3:966 - 972
- Watanabe K, Tateishi S, Kawasuji M, Tsurimoto T, Inoue H, Yamaizumi M. Rad18 guides pol eta to replication stalling sites through physical interaction and PCNA monoubiquitination. EMBO J 2004; 23:3886 - 3896
- Zhuang Z, Johnson RE, Haracska L, Prakash L, Prakash S, Benkovic SJ. Regulation of polymerase exchange between Poleta and Poldelta by monoubiquitination of PCNA and the movement of DNA polymerase holoenzyme. Proc Natl Acad Sci USA 2008; 105:5361 - 5366
- Terai K, Abbas T, Jazaeri AA, Dutta A. CRL4(Cdt2) E3 ubiquitin ligase monoubiquitinates PCNA to promote translesion DNA synthesis. Mol Cell 2010; 37:143 - 149
- Torres-Ramos CA, Prakash S, Prakash L. Requirement of RAD5 and MMS2 for postreplication repair of UV-damaged DNA in Saccharomyces cerevisiae. Mol Cell Biol 2002; 22:2419 - 2426
- Blastyak A, Pinter L, Unk I, Prakash L, Prakash S, Haracska L. Yeast Rad5 protein required for postreplication repair has a DNA helicase activity specific for replication fork regression. Mol Cell 2007; 28:167 - 175
- Branzei D, Vanoli F, Foiani M. SUMOylation regulates Rad18-mediated template switch. Nature 2008; 456:915 - 920
- Minca EC, Kowalski D. Multiple Rad5 activities mediate sister chromatid recombination to bypass DNA damage at stalled replication forks. Mol Cell 2010; 38:649 - 661
- Windecker H, Ulrich HD. Architecture and assembly of poly-SUMO chains on PCNA in Saccharomyces cerevisiae. J Mol Biol 2008; 376:221 - 231
- Haracska L, Torres-Ramos CA, Johnson RE, Prakash S, Prakash L. Opposing effects of ubiquitin conjugation and SUMO modification of PCNA on replicational bypass of DNA lesions in Saccharomyces cerevisiae. Mol Cell Biol 2004; 24:4267 - 4274
- Papouli E, Chen S, Davies AA, Huttner D, Krejci L, Sung P, et al. Crosstalk between SUMO and ubiquitin on PCNA is mediated by recruitment of the helicase Srs2p. Mol Cell 2005; 19:123 - 133
- Branzei D, Sollier J, Liberi G, Zhao X, Maeda D, Seki M, et al. Ubc9- and mms21-mediated sumoylation counteracts recombinogenic events at damaged replication forks. Cell 2006; 127:509 - 522
- Eddins MJ, Carlile CM, Gomez KM, Pickart CM, Wolberger C. Mms2-Ubc13 covalently bound to ubiquitin reveals the structural basis of linkage-specific polyubiquitin chain formation. Nat Struct Mol Biol 2006; 13:915 - 920
- VanDemark AP, Hofmann RM, Tsui C, Pickart CM, Wolberger C. Molecular insights into polyubiquitin chain assembly: crystal structure of the Mms2/Ubc13 heterodimer. Cell 2001; 105:711 - 720
- Hofmann RM, Pickart CM. Noncanonical MMS2-encoded ubiquitin-conjugating enzyme functions in assembly of novel polyubiquitin chains for DNA repair. Cell 1999; 96:645 - 653
- Mastrandrea LD, You J, Niles EG, Pickart CM. E2/E3-mediated assembly of lysine 29-linked polyubiquitin chains. J Biol Chem 1999; 274:27299 - 27306
- Stewart GS, Panier S, Townsend K, Al-Hakim AK, Kolas NK, Miller ES, et al. The RIDDLE syndrome protein mediates a ubiquitin-dependent signaling cascade at sites of DNA damage. Cell 2009; 136:420 - 434
- Ulrich HD, Walden H. Ubiquitin signalling in DNA replication and repair. Nat Rev Mol Cell Biol 2010; 11:479 - 489
- Kats ES, Enserink JM, Martinez S, Kolodner RD. The Saccharomyces cerevisiae Rad6 postreplication repair and Siz1/Srs2 homologous recombination-inhibiting pathways process DNA damage that arises in asf1 mutants. Mol Cell Biol 2009; 29:5226 - 5237
- Freudenthal BD, Gakhar L, Ramaswamy S, Washington MT. Structure of monoubiquitinated PCNA and implications for translesion synthesis and DNA polymerase exchange. Nat Struct Mol Biol 2010; 17:479 - 484
- Chen J, Ai Y, Wang J, Haracska L, Zhuang Z. Chemically ubiquitylated PCNA as a probe for eukaryotic translesion DNA synthesis. Nat Chem Biol 2010; 6:270 - 272
- Pettersen EF, Goddard TD, Huang CC, Couch GS, Greenblatt DM, Meng EC, et al. UCSF Chimera—a visualization system for exploratory research and analysis. J Comput Chem 2004; 25:1605 - 1612