Abstract
The dynamic changes and structural patterns of DNA methylation of genes without CpG islands are poorly characterized. The relevance of CpG to the non-CpG methylation equilibrium in transcriptional repression is unknown. In this work, we analyzed the DNA methylation pattern of the 5’-flanking of the myogenin gene, a positive regulator of muscle differentiation with no CpG island and low CpG density, in both C2C12 muscle satellite cells and embryonic muscle. Embryonic brain was studied as a non-expressing tissue. High levels of both CpG and non-CpG methylation were observed in non-expressing experimental conditions. Both CpG and non-CpG methylation rapidly dropped during muscle differentiation and myogenin transcriptional activation, with an active demethylation dynamics. Non-CpG demethylation occurred more rapidly than CpG demethylation. Demethylation spread from initially highly methylated short CpC-rich elements to a virtually unmethylated status. These short elements have a high CpC content and density, share some motifs and largely coincide with putative recognition sequences of some differentiation-related transcription factors. Our findings point to a dynamically controlled equilibrium between CpG and non-CpG active demethylation in the transcriptional control of tissue-specific genes. The short CpC-rich elements are new structural features of the methylation machinery, whose functions may include priming the complete demethylation of a transcriptionally crucial DNA region.
Introduction
Transcription is a crucial target of epigenetic modifications.Citation1 Although the specific order of molecular events remains unclear, the methylation of cytosines in the DNA is a key epigenetic modificationCitation2,Citation3 that is closely linked to the cascade of events that finally repress gene transcription.Citation4 A number of actors are involved in transcriptional repression, the shaping of DNA methylation patterns being dynamically performed by several concomitant mechanisms.Citation3,Citation5 DNA methyltransferases (Dnmts) are committed to the primary introduction of a methyl-group into DNA;Citation6 their action is counterbalanced by passive and/or active DNA demethylation mechanisms.Citation5,Citation7–Citation9 The effectors of this primary modification are methylation-influenced transcription factors, histone modification and chromatin remodelling enzymes, as well as multifunctional proteins involved in chromatin higher order organization.Citation10–Citation14 Both structuralCitation15–Citation18 and dynamicCitation8,Citation17,Citation19–Citation21 aspects are crucial for transcriptional modulation.
From the structural point of view, the CpG island is, so far, the only feature known to play a well-established role in epigenetic-mediated transcriptional repression, though the search for other CpG-based genomic sequences involved in methylation-controlled transcriptional modulation is in progress.Citation3,Citation18,Citation22,Citation23 The role of isolated CpG sites and the extension, or even the existence, of non-CpG methylation are topics that have only recently received more attention.Citation15 Non-CpG methylation has been demonstrated in the promoter of several genes in adult tissues or cell lines,Citation13,Citation24–Citation27 in fetal brain,Citation12,Citation28 in gametes and early embryo,Citation20,Citation29 in the myogenic myf-3 geneCitation30 and in mouse embryonic stem cells.Citation31 Finally, non-CpG methylation was also demonstrated in our recent experiments on the methylation of the Presenilin 1 gene in the brain of Alzheimer disease models.Citation32 Non-CpG methylation has invariably been associated with prevalent CpG methylation. When a functional assessment was performed, the silencing of the methylated gene was revealed, though it was generally impossible to distinguish between the functional role of CpG and that of non-CpG methylation. Although the regulation of methylating mechanisms has yet to be fully understood, several studies on the sequence specificity of Dnmts have revealed their ability to methylate non-CpG sites.Citation15,Citation16,Citation31,Citation33–Citation35
The issue of methylation dynamics is even more controversial than that of structural patterns since the dynamic aspect cannot be separated from as yet unclear issues regarding the methylation/demethylation equilibrium and active demethylation mechanisms. In the early phases of mouse spermatogenesis, evidence has emergedCitation21 of a sequence-specific interplay between de novo methylation and passive demethylation that changes the methylation/demethylation equilibrium. During early development, the mouse genome undergoes dynamic changes through an earlier active demethylation phase followed by a later passive demethylation phase.Citation36 A dynamic change in methylation patterns during the cell cycle of several genes has been described in HeLa cells.Citation19 Moreover, a cyclical change in methylation during transcription, with a surprisingly short timescale in minutes for both methylation and demethylation, has been described in both MCF-7Citation37 and MDA-MB231,Citation38 human breast cancer cells. Our previous data on the myogenin gene during muscle differentiation in C2C12 cells support the hypothesis of an equilibrium between active demethylation and re-methylation mechanisms.Citation8 Rapid (within hours) active dynamics and high structural specificity have been reported for mouse genes of three distinct gene categories and for an integrated transgene as an exogenous target.Citation39 Another controversial dynamics-related issue is the role and mechanisms of action of demethylases and active demethylation processes.Citation9 Several demethylases believed to be responsible for active dynamics have been proposed in mammals. The first was MBD2,Citation40 which was followed by 5-MCDG (MBD4),Citation41 GADD45aCitation42 and MBD3.Citation7 Recently, even Dnmt3a and Dnmt3b have been shown to play a dual role in methylation and active demethylation.Citation38
One almost unexplored field is that of the structural features of sequences undergoing active demethylation; indeed, since existing data are focused on CpG sites, the role of non-CpG moieties remains unknown. Few authors have attempted to associate the structural and dynamic aspects. One study demonstrated that the stability of CpG methylation and the expression patterns of an integrated proviral reporter gene, in murine erythroleukemia cells, depends on methylation density.Citation16 Another study demonstrated that specific CpG spacing is required for the stepwise and sequential methylation dynamics of the c-fos gene during the development and tissue differentiation of mouse liver.Citation43 Some models linking methylation/demethylation mechanisms, transcription and myogenic factors with muscle differentiation have previously been proposed.Citation8,Citation44,Citation45
The aim of this work was to study structural DNA methylation patterns and their dynamic behavior in the 5′-flanking region of the myogenin gene during mouse muscle differentiation. For this purpose, we used the C2C12 in vitro model of muscle satellite cells, which retains stem cell characteristics;Citation46 we also compared DNA methylation patterns in mouse embryonic brain (EmB) and embryonic muscle (EmM). We paid particular attention to non-CpG methylation and to the possible existence of specific sequence elements that are subjected to peculiar active demethylation dynamics.
Results
Differentiation and myogenin expression.
Both C2C12 and C2T18 differentiate after the shift to DM (). The differentiation ability of the C2T18 was greater than that of C2C12. Supplementation of 3-deaza-adenosine and homocysteine to C2C12 in DM (hypomethylating stimulus; DM+DH) induced enhanced differentiation (even more marked in C2T18, data not shown). The CK activity of the C2T18 clone was almost threefold higher in DM (in absence of DH) than that of C2C12 after the addition of DH. Furthermore, the CK activity of C2T18 in GM was similar to that of C2C12 in DM+DH.
Myogenin expression () parallels the differentiation pattern: it was induced in both C2C12 and C2T18 by DM and it is enhanced in C2C12 DM+DH. Myogenin expression was considerably higher in the highly differentiating C2T18 clone, which displayed high myogenin levels in GM at 48 h and even higher levels at 96 h (comparable to C2C12 levels in DM); C2T18 expression in DM at 96 h was higher than that of C2C12 DM+DH. Myogenin was well expressed in EmM, with a signal comparable to that of C2T18 in DM, while its expression was undetectable in EmB. The same expression pattern was detected by RT-PCR analysis (data not shown).
Taken together these results indicate that the C2T18 clone has greater myogenic potential than C2C12. The specific experimental conditions used in this work can be ranked, from both a differentiation and myogenin expression point of view, as follows: EmM ≥ C2T18 DM >> C2C12 DM+DH > C2T18 GM ≥ C2C12 DM >> C2C12 GM > EmB. The strong correlation between differentiation ability, myogenin expression and the terminal differentiation levels is in keeping with the role of myogenin as a positive regulator of muscle differentiation.Citation47 A significantly higher expression of myogenin in C2T18 (with high levels even in GM) than in C2C12 may underlie the enhanced differentiation ability of this clone.
Structural features of the 5′-flanking region.
No CpG island was detected within 10,000 bases upstream of the myogenin gene, according to both actually accepted definitions.Citation48,Citation49 Also, according to a more recent definition,Citation18 the myogenin promoter, consisting of the 1,092 upstream bases,Citation50 is a low-CpG promoter, with no 500 bp area with a CpG ratio above 0.48 (the maximum CpG ratio value found in the myogenin promoter was 0.31). The region analyzed for methylation, which is 258 bases long, contains 84 C moieties: 9 CpG, 42 CpC, 14 CpT, 19 CpA. This region was chosen because it surrounds the CCGG site in which we had previously shownCitation8 an actively modulated methylation pattern during differentiation. This region is also part of the 5′-flanking zone (from −1,092 to −134) that affects myogenin expression efficiency.Citation50 Myogenin can, therefore, be considered a non-CpG-island gene, with a low CpG density (in the analyzed zone 3.5% = 9 CpG out of 258 bp; in the overall promoter 1.4% = 15 CpG out of 1,092 bp) in its 5′-flanking region. The nine CpG dinucleotides include seven CCG (one of which is CCGG) and two ACG.
Overall methylation of CpG and non-CpG moieties.
Analyses performed by either methylation-sensitive restriction endonucleases (Sup. Table 2 and Sup. Fig. 1) or bisulphite () revealed the presence of highly modulated CpG and non-CpG methylation in the myogenin 5′-flanking region during differentiation. Dramatic changes in the methylation levels, and in both the temporal and spatial patterns, were detected in the different experimental conditions (). A temporal decrease in methylation was observed for each experimental condition associated with a highly heterogeneous spatial methylation pattern (i.e., methylation distribution along the analyzed region). The balance between temporal variations in homogeneous experimental conditions and spatial variation is analyzed statistically in Supplemental Table 4; this analysis indicates that variations in the methylation pattern are more closely related to temporal variations than to a demethylation gradient based on the position of C moieties in the 5′ flanking region sequence.
The highest level of overall methylation () was exhibited by EmB (75.4%) and the lowest level by C2C12 DM+DH 24 h (1.4%). High methylation levels were exhibited by the most undifferentiated condition of the least myogenic cell line, i.e., C2C12 in GM (52.7%); the corresponding undifferentiated condition of the most myogenic clone, C2T18 GM, displayed a lower level of overall methylation (32.5%). Overall methylation levels of C2C12 in DM at early times (2 h, with or without DH, respectively 34.4% and 32.4%) were comparable to those of the C2T18 GM: starting GM methylation levels of the most myogenic clone C2T18 were comparable to the methylation levels achieved by C2C12, which has a lower myogenic potential, only after the shift to DM. EmM displayed considerably lower levels of overall methylation (9.0%). Overall methylation levels of C2C12 in DM at late times (24 h, with or without DH), as well as those of C2T18 at early (20 min) and late (1 h) times, were lower than those of EmM (respectively 1.4, 2.9, 5.6 and 4.6%).
The overall methylation level was partitioned to distinguish CpG from non-CpG methylation (). A dramatically high proportion of non-CpG methylation was found in both EmB and in the most undifferentiated muscle condition (C2C12 GM). A high non-CpG methylation level was also found in early (2 h) differentiated C2C12 DM (with or without DH) as well as in C2T18 GM. In EmM and in both early and late C2T18 DM, the non-CpG/CpG methylation ratio decreased, though non-CpG methylation still prevailed. In each of these eight experimental conditions, non-CpG methylation was higher than CpG methylation. A prevalence of CpG over non-CpG methylation only occurred in the late C2C12 DM experimental conditions (with or without DH), where non-CpG methylation was absent. The decrease in overall methylation was largely due to the methyl loss of the CpC moieties, the contribution of CpG being negligible. A large variability was found in the overall methylation of each experimental condition, as evidenced by the large error bars. We speculated that this is probably due to a heterogeneous methylation pattern within the myogenin region analyzed (see below). The loss of statistical significance in some of the differences in methylation may due in part be to this variability.
Spatial and cluster analysis.
As non-linear spreading of demethylation was detected during differentiation and myogenin transcriptional activation ( and Sup. Table 4), the search for a more complex spatial pattern of demethylation was undertaken. For this purpose, the bisulphite experimental data were subjected to cluster analysis so as to group either experimental conditions (Sup. Fig. 2) or cytosines ().
The clustering of experimental conditions by overall methylation levels allowed the identification of clusters of homogeneously differentiating and myogenin expressing experimental conditions (Sup. Fig. 2). This finding not only lends support to experimental evidence of a strong correlation between methylation, differentiation and myogenin expression but also validates the clustering algorithm.
Cytosine clustering () allowed the identification of 14 single non-grouping C (sng-C) and six different clusters (numbered from the top to the bottom of the dendrogram from 1 to 6) of C dispersed along the 5′-flanking region (). Clusters 4 and 6 are composed, respectively, of seven and four adjacent C (adj-C, ). Some sub-clusters, composed of adjacent C, were also identified within clusters 1, 2, 3 and 5: 1a, composed of five C; 1b, composed of four C; 1c, composed of four C; 2a, composed of 20 C; 2b, composed of four C; 3a, composed of eight C; 5a, composed of four C. No CpG sites grouped in clusters, all being included within the sng-C. In cluster 1, grouped C localized at both the 5′ and the 3′ end of the region, while in cluster 2, grouped C localized in the middle and at the 3′ end (see C numbering in ). Moreover, groups of C in close proximity often grouped in different clusters (e.g., sub-cluster 1a and cluster 4, sub-clusters 1b, 3a and 5a; sub-cluster 2a, cluster 6, sub-clusters 1c and 2b; ). The demethylation dynamics of the clusters and sng-C is analyzed below.
Dynamics of sng-C and cluster methylation.
In C2C12 (A), different cluster and sng-C overall methylation levels were observed at the start (time 0 = GM); the highest values, 17 and 10%, were observed respectively in clusters 2 and 1, whereas those in the other clusters ranged from 8.8 to 2.7%. Following the shift to differentiation conditions and the addition of DH, general demethylation in these initial levels progressed, after 24 h, to a virtual absence of methylation. Only sng-C displayed a detectable methylation value after 24 h (1.5%). Between these extreme conditions there is an intermediate state, 2 h after stimulation, that is characterized by marked differences between clusters. During these first two hours, the overall methylation dramatically dropped from 17 to 0.3% in cluster 2 and from 10 to 2.6% in cluster 1. By contrast, the overall methylation levels of other clusters remained constant.
Different starting cluster and sng-C group overall methylation levels (range 9.5 to 1.3%) were also observed in C2T18 () (time 0 = GM). All these levels were comparable to those observed in C2C12, with the exception of cluster 2, which exhibited a lower starting overall methylation level (3.2% in C2T18 compared with 17% in C2C12). Cluster 1 also displayed similar overall methylation levels in both C2T18 (9.5%) and in C2C12 (10%), the former being, however, the highest starting overall methylation level in C2T18. As observed in the C2C12 DM+DH condition, the initial overall methylation levels dropped in C2T18, with the lowest levels being reached as little as 20 min after stimulation and the overall methylation levels of all the clusters constantly below 2% until 60 min (the last time point). The C2T18 DM dynamics are thus markedly different from those of C2C12 DM+DH, which displayed overall methylation levels well above 2% at 2 h in all the clusters, except cluster 2, and reached its final demethylated status much later. In both C2T18 DM and C2C12 DM+DH, sng-C overall methylation levels were higher than those of the clusters, though it is noteworthy that values were well below 2% after only 20 min in the C2T18 DM, but were 9.4% after 2 h in the C2C12 DM+DH condition.
In C2C12 DM without hypomethylating drugs (), the initially high methylation levels (the same as those shown in at time 0 GM) dropped in the 24 h following the shift, leading to a virtual absence of methylation in all clusters with the exception of sng-C, which maintained a methylation level of 2.4%. Even in this case there was an intermediate state (2 h) in which each cluster displayed a slight decrease in its overall methylation as it proceeded towards its final level. In particular, the overall methylation in clusters 1 and 2, which were highly methylated at time 0, did not drop as dramatically in C2C12 DM in the first 2 h as in C2C12 DM+DH, although the difference in overall methylation between the starting level and 2 h was the highest even in this case.
Differences in the overall methylation levels between clusters were also marked in vivo in EmB (), the overall methylation order being: cluster 2 > cluster 1 > sng-C > cluster 3 > cluster 4 > cluster 5 > cluster 6. In terminally differentiated EmM, the least methylated cluster was cluster 2, which displayed, together with cluster 1, the greatest difference between non-expressing and expressing conditions, even in vivo. The overall methylation levels were also lower in EmM than in EmB in all the other clusters, the overall methylation order remaining substantially unchanged if compared with the starting levels. The overall methylation level in each cluster was higher in EmB than in cultured cells.
These data point to the existence of fast and slow demethylating clusters. Whether this dynamic difference is linked to the structure and/or composition of the clusters is analyzed below.
Cluster composition, peculiar sequence elements and recognition sites for transcription factors.
The base composition of each cluster and of the sng-C is significantly different (). The main feature of sng-C is their high CpG content (64.3%) (CpG are only represented in sng-C). The CpC content as well as the CpC/non-CpC ratio are highest in the fast demethylating clusters 1 and 2 (respectively 60.5% and 1.53 on pooled data), lowest in the sng-C (respectively 21.4% and 0.27) and intermediate in the slowly demethylating clusters 3, 4, 5 and 6 (respectively 48.1% and 0.93 on pooled data).
The sub-clusters with adjacent C identified () are composed mainly of CpC-rich elements, which have a peculiar and comparable structure (). The CpC dinucleotide and, consequently, the CpC-rich elements are more numerous and display a higher density in the fast demethylating clusters 1 and 2 than in other clusters ( and and ), constituting a remarkable feature of these fast demethylating clusters. If the myogenin promoter from the position −1,092 to the transcription start siteCitation50 was taken under consideration, the nine CpC-rich elements listed in were found to be represented only in the analyzed region of 258 bases, with the only exception being a single upstream CCT CC site (not analyzed by us). This marks a structural specificity of this zone.
As clusters 1 and 2 displayed peculiar demethylation dynamics (), the putative binding sites of DNA binding transcription factors overlapping sub-clusters 1a, 1b and 1c as well as 2a and 2b were analyzed (). A particularly high density of putative binding sites for Sp1 (trans-acting transcription factor 1), CTCF (CCCTC-binding factor) and Rreb1 (ras responsive element binding protein 1) was observed in correspondence of sub-cluster 2a. Two additional putative CTCF binding sites were found to overlap sub-cluster 1b and a sng-C. Several putative Sp1 binding sites were also observed in correspondence of sub-clusters 1b, 1c and 2b. An additional putative Rreb1 binding site was found to overlap cluster 4. Two Nfe2l2 (nuclear factor, erythroid derived 2, like 2) putative binding sites were found to overlap sub-clusters 1a and 1b. Five Snai1 (Snail homolog 1, Drosophila) putative binding sites were also observed: two overlapping sub-cluster 3a, one sub-cluster 2a and two sub-cluster 2b. A putative binding site for Cebpb (CCAAT/enhancer binding protein β) partially overlapping sub-cluster 2a, was also detected. Further information on these transcription factors is included in Supplemental Table 5.
Discussion
A peculiar aspect of our results is the higher level of non-CpG than CpG overall methylation observed in the vast majority of experimental conditions (8 out of 10); this finding is presumably linked to the origin of the C2C12 cells—muscle satellite cells, which retain stem cell properties,Citation46 as well as to the embryonic origin of both ex vivo muscle and brain. The highest non-CpG methylation level so far reported is 54.5%, detected in human spleen DNA.Citation51 Another studyCitation31 reported a non-CpG methylation of 15–20% of the total cytosine methylation in mouse ES cells, owing above all to Dnmt3a, which is highly expressed in ES cells. CpA and CpT methylation has been shown to be 250 times lower in sequencing libraries constructed from murine ES cells without Dnmt3a and 3b activities than in wild-type ES cells.Citation52 Our findings suggest that the role of non-CpG methylation has so far been greatly underestimated, although the questions of the sequence specificity of Dnmt and whether specific targets exist in the genome for different Dnmts remain unanswered. Recent findings point to the preferential methylation ability of the Dnmt3a/Dnmt3L complex on CpG pairs that are 8–10 base pairs apart,Citation53 which is a very common CpG pair periodicity in the human genome.Citation54 These studies highlight a potential role of DNA-sequence specificity for the Dnmt3a/Dnmt3L complex, as well as a functional role of this new structural element. Although fairly uniform hypomethylation was observed in all our muscle experimental conditions at the late times, there was a marked difference in the demethylation dynamics of the different CpN moieties. This variability in the methylation levels was particularly evident at the early experimental times, when initial demethylation occurred in particular clusters (). The final functional effect on transcription seems to be linked to the overall methylation level of groups of C, which is probably imposed by the bulk of methylating and demethylating activities.Citation8 Initial CpG and non-CpG methylation patterns appear to be more predictive of terminal myogenin expression and differentiation than the final methylation levels ( and ). Despite the low CpG density of the myogenin 5′-flanking, the functional impact of its methylation has already been evidenced as regards its developmental activation,Citation45 as well as by experiments imposing either hypomethylatedCitation8 or hypermethylatedCitation55 patterns, respectively inducing or repressing myogenin expression.
The effect on methylation, following stimulation, is not equivalent on all C. All nine CpG moieties, together with a further five non-CpG moieties, displayed marked differences in methylation modulation, thereby pointing to independent methylation behavior; all the other C exhibited more coordinated methylation modulation, grouping into six clusters (). The structural patterns of cluster demethylation were very similar in C2C12 and C2T18, the main differences and specificity being found in the starting methylation levels and in the actual timing of modulation (). Cluster 2 was the fastest demethylating cluster, followed by cluster 1; the behavior of clusters 3 to 6 was fairly homogeneous, with decreasing methylation levels reflecting starting levels. The cluster pattern in undifferentiated and differentiated tissues parallels that of cell lines; in particular, clusters 1 and 2 displayed the highest methylation levels in EmB and the lowest levels in EmM, the greatest difference in methylation between these two embryonic tissues being observed in cluster 2. The cluster demethylation dynamics are closely correlated with myogenin expression and, consequently, with terminal differentiation (). The most myogenic experimental condition, the C2T18 clone displayed, even in the basal conditions (GM), a cluster pattern (cluster 2 demethylation) that partially resembled that of C2C12 after 2 h of differentiation in the presence of a hypomethylating stimulus (an experimental condition with intermediate myogenin expression and differentiation). After 20 min, C2T18 displayed a cluster pattern (complete demethylation) that was achieved by C2C12 only at the very end (24 h with or without DH). C2C12 DM (without DH), the least myogenin expressing and differentiating condition, still displayed cluster 1 and 2 methylation 2 h after stimulation of differentiation, even though demethylation was already in progress and complete demethylation was reached later. The correlation observed between the cluster demethylation dynamics and the expression levels is particularly significant if we consider that the zone analyzed is part of the 5′-flanking region known to affect myogenin expression efficiency.Citation50
All the clusters (and sub-clusters), which were selected solely on the basis of their methylation pattern, were spatially separated from each other. In particular, in clusters 1 and 2, in which several sub-clusters of adj-C were defined, groups of C far along the analyzed region could be pooled together according to their methylation behavior (). This suggests that active demethylating activity is not linear and progressive, but instead starts from clustered, though dispersed, specific foci. Moreover, the base composition of each cluster and of the sng-C is not a random selection of bases from the overall 5′-flanking region. The highest CpC content seems to be a specific feature of the fast demethylating clusters 1 and 2 (). In these two clusters, several adj-C form characteristic CpC-rich elements () whose structure is similar and, in one case (sub-cluster 2a), duplicated. Starting from different methylation levels at early experimental times, some dispersed and specific CpC-rich elements, which appear to be more prone to active demethylation, demethylate faster than others. Although the final goal is complete demethylation within the region to promote chromatin decondensation, the CpC-rich elements appear to prime this effect by promoting simultaneous demethylation in multiple zones, possibly to increase its efficacy throughout the region. It has previoulsy been demonstrated,Citation16 by means of a proviral reporter premethylated at different densities, that a high methylation density propagates, whereas a low methylation density is unstable and is likely to evolve toward complete demethylation. The CpC-rich elements might prime a decrease in methylation density, thus constituting the structural basis for the transcriptional regulation of crucial genes that require both rapid and, owing to their low CpG content, additional modulation.
Several methylated elements based on the CpC dinucleotide have previously been described. There have been several reports of functional mCW and mCWG methylation, as well as of mCmCG and mCmCTT methylation.Citation12,Citation14,Citation15,Citation24 In a non-muscle system, methylated non-CpG residues have been foundCitation12 in a repeated element (mCmCTTmCTTmCmC) strictly resembling our CpC-rich elements in the human synaptotagmin X1 (syt11) gene, which is involved in neural function. When these residues were methylated, the binding activity of the Sp family protein to the region was significantly reduced. Transient transcription assays using artificially methylated promoter sequences have also shown that methylation reduces the expression of the reporter gene. Extensive non-CpG methylation involving CpC-rich regions has previously been observed in CC and CCC motifs of p53 in human non-small cell lung cancer,Citation26 in CCWGG motifs of p53 in bone marrow samples from patients with acute lymphoblastic leukemiaCitation56 as well as in the CMV promoter-enhancer following adenoviral gene delivery by intramuscular injection in rat.Citation57 Methylation at CmCWGG sites has also been observedCitation13 in the promoter of the B-cell-specific B29 (Igβ/CD79b) gene, in which it silences its in vivo transcriptional activity in primary effusion lymphoma and some myelomas; methylation of a central conserved CmCTGG B29 promoter site has also been shown to induce the switch from the binding of early B-cell transcription factor to a different methylation-dependent DNA-protein complex.
Peripheral blood leukocytes methylate the symmetric CCTGG motif within a region of the human myogenic myf-3 gene that is normally devoid of CpG methylation.Citation30 Both studiesCitation13,Citation30 reported an inverse correlation between CCTGG and CpG methylation, thereby highlighting CCWGG methylation as a new epigenetic mark in mammalian DNA, the existence of a methylating system for the CCTGG pentanucleotide that is independent of the CpG dinucleotide system, as well as a correlation between CCWGG methylation and transcriptional repression.Citation15 Methylation at non-CpG sites including CCG trinucleotides has also been observed.Citation24
We also identified some putative recognition sites for Sp1, CTCF, Rreb1, Nfe2l2, Snai1 and Cebpb transcription factors, which overlap some of the sub-clusters (Sup. Table 5). All these transcription factors are reported to be involved in differentiation, while five of them have been found to be involved in the patterning of DNA methylation; moreover, in some cases their binding has also been found to be influenced by DNA methylation. The subclusters and/or the CpC-rich elements may represent DNA regions that are specifically recognized by competing activators and repressors of transcription. Notably, it has been suggested that both CTCF and Sp1, whose DNA binding is influenced by methylation, act as either repressors or activators (depending on the target sites and, possibly, on the surrounding sequences); an intriguing model of c-myc transcriptional regulation by competitive binding of several distinct Sp1-like factors and different CTCF forms to the same promoter element has been proposed.Citation58
In conclusion, myogenin expression and, consequently, terminal muscle differentiation, appear to be linked to both the methylation starting levels and the fast demethylation of some specific clusters of C, that display homogeneous, dynamic behavior and are composed of peculiar sequence elements. The main structural features of these elements are the high content of the initially highly methylated CpC dinucleotide and the absence of the CpG dinucleotide. The main dynamic feature is the very fast demethylation of the CpC-rich elements coupled with the slower demethylation of the isolated CpG dinucleotides, both of which are highly suggestive of an active demethylation mechanism. The data reported in the present study support a model based on an interplay between demethylation and re-methylation activities that leads to a faster demethylation of CpC-rich elements and to a slower demethylation of CpG sites. The foci of rapidly demethylated DNA that are produced, possibly recognized by some methylation-dependent complex, prime the progressive, active demethylation within the region; this eventually leads to a fully demethylated status and, in turn, to an active chromatin conformation. This model may prove particularly suitable for the transcriptional regulation of non-CpG island genes, which have a low CpG density in their transcriptional control regions. The identification of the features of these DNA sequences may be relevant to functional genomics and should therefore be thoroughly pursued.
Materials and Methods
Chemicals.
Restriction enzymes were purchased from New England Biolabs, Inc., (Mississauga, Ontario, Canada) and from Promega (Madison, WI). Platinum Taq and buffer for PCR were obtained from Invitrogen (Milan, Italy). Oligonucleotides used as primers were synthesized by M-Medical Genenco (Firenze, Italy). All other chemicals were obtained from Sigma Chemical Co., (St. Louis, MO).
Cell cultures and tissues.
The experiments were performed on the C2C12 mouse muscle cell line and on its C2T18 clone, selected on account of its increased differentiation capacity, as previously described.Citation8 These cells are derived from muscle satellite cellsCitation59 and retain stem cell properties.Citation46 Briefly, cells were cultured in F14 medium supplemented with 50 µg/ml neomycin and 10% (growth medium, GM) or 1% (differentiation medium, DM) of fetal calf serum; C2C12 in DM were also treated with 3-deaza-adenosine and homocysteine (hypomethylating treatment: DM+DH). EmB and EmM (quadriceps femoris) tissues were isolated from CD1 mouse embryos (Ed7); three pools of two brains or muscles were used for each experimental condition. Cells were plated in GM and, after 24 h of growth, were stopped or shifted to DM or DM+DH (time 0); the subsequent collection times in DM and DM+DH are indicated in all the Figures. For the restriction enzyme analysis, the collection time in GM is different from time 0 (as shown in Sup. Fig. 1).
Differentiation assay.
Cells used for the enzymatic test, either in GM or DM, were rinsed twice with PBS and frozen at −80°C. After thawing, cells were scraped into 1 ml of 50 mM Tris/HCl (pH 7.2) and 1 mM dithiothreitol, sonified for 15 s in ice and centrifuged; supernatant was used for creatine kinase (CK, EC 2.7.3.2) dosage.Citation8 CK is a typical biochemical marker of terminal muscle differentiation contained in contractile muscle fibers; CK levels were measured by the CK-NAK kit (Abbott Diagnostics, Irving, TX). A parallel set of cell samples were fixed and stained by the May Grunwald-Giemsa method and the nuclei counted to calculate, in triplicate for each experimental condition, the mU CK/nucleus ratio ± sd.
RNA isolation and expression studies.
Total RNA extraction was performed by the RNAfast method (Molecular Systems; San Diego, CA). Northern blot for myogenin and 18S RNA and RT-PCR (data not shown) for myogenin and γ-actin assays were performed as previously described.Citation8
DNA digestion and PCR assays.
DNA digestion and PCR reactions were performed in triplicate as previously describedCitation8 (primer composition is reported in Sup. Table 1S). Briefly, 2 µg of genomic DNA were treated at 37°C overnight with 6 U, and for a further 6 h with another 4 U of the restriction endonucleases shown in Supplemental Table 2. The digested samples were amplified by multiplex PCR with pairs of primers (adjacent to the endonuclease recognition sites) specific for the 5′-flanking region, for the exon 3 and, in some samples (as indicated in Sup. Fig. 1), for exon 1. Exon 3, which possesses no recognition sites for these endonucleases, was used as an internal control. All the electrophoresis gels were acquired and analyzed using a computerized densitometer (Fluor-S, Bio-Rad). In each digestion experiment, parallel control samples (non-methylated PCR products obtained with the same primers as those used for the assay) were analyzed, to check for complete digestion.
DNA methylation studies by bisulphite modification and genomic sequencing.
The analyzed region is 258 bases long (primers excluded): from 1,106 to 1,401 (38 bases of primers) in the myogenin gene sequence (acc. N. M95800) 5′-flanking region. The analyzed strand is the plus (coding) strand. The primers used were those described previouslyCitation60 (see also Sup. Table 1). In particular, for the bisulphite analysis, the reverse primer is degenerated (with 3 G/A) and it works regardless of whether the 3 C on the target plus strand are transformed or not. The forward primer is G-rich (with only 1 degenerated C/T) and only targets the newly synthesized (by reverse primers) strand containing C moieties, as the original genomic DNA strand has bisulphite transformed C to U; this loss of recognition of the original genomic DNA strand is reinforced by the G moieties located at the 3′ end of the primer (particularly sensitive to mismatches). Bisulphite analysis of myogenin promoter methylation was performed, at least in duplicate, by standard proceduresCitation61 with modifications as previously described.Citation60 PCR products obtained after bisulphite treatment were cloned into pCR 2.1 plasmid with the “TA cloning” kit; 3 µl of the ligation products were used to transform INVβF competent cells (all from Invitrogen; Milan, Italy). Clones were sequenced by the cycle sequencing method using the ABI PRISM 3100 Avant genetic analyzer (Applied Biosystems). From 10 to 20 clones were analyzed for each replicate of the experimental condition; modified C residues were recognized by comparison with the original DNA sequence.
To ensure that cytosine conversion was complete, for both CpG and non-CpG methylation the following controls were performed. (1) Different bisulphite treatments were used on some duplicated samples with either low or high levels of CpG and non-CpG methylation: (i) EZ DNA methylation kit (Zymoresearch); (ii) EpiTect Bisulphite kit (Qiagen); (iii) bisulphite methods modified for high concentration and/or ammonium bisulphite (instead of sodium bisulphite).Citation62,Citation63 In all these cases, as well as after treatment with the afore-described method used for all the samples, the observed CpG and non-CpG methylation patterns were similar. (2) In each experimental series, both an unmethylated and SssI-methylated purified PCR product of the myogenin 5′-flanking analyzed region, obtained with the same primers as those used for restriction analysis, were used to check the correct transformation of C moieties. (3) Several samples had, within the analyzed region, both methylated and non-methylated cytosines (CpG and non-CpG), which were often adjacent. The non-methylated cytosines functioned as a transformation control for the methylated ones since there was no reason to assume that the transformed and non-transformed status of C moieties in close proximity was, in the case of the non-transformed C moieties, an experimental fault. (4) Different experimental conditions with presumed high or low methylation content (CpG and non-CpG) were always treated in the same experimental series; the low-methylation experimental conditions functioned as transformation controls for the high-methylation experimental conditions. (5) The methylation status was also assessed by methylation-sensitive restriction endonucleases (for the recognized sequences, which in some cases included non-CpG methylation), which identified the same methylation pattern as the bisulphite analysis (Sup. Fig. 1 and Sup. Table 2). (6) The bisulphite protocol is designed to analyze the plus strand using a G-rich forward primer that does not recognize the complementary C-rich genomic strand after bisulphite transformation, but does recognize the newly synthesized minus strand, which is a copy of the genomic transformed plus strand. There is the risk, if the C-rich region of the minus strand is not (or is only partially) transformed, that the G-rich forward primer will recognize the genomic minus strand; if so, however, all G included in the minus strand within the analyzed region will be copied as C in the plus strand and some C of the minus strand may be transformed, thereby originating a G →A change in the plus strand, a clone with no transformed C and, possibly, with just one transformed G that may be seen as an experimental fault; in our protocol fewer than 1% of clones displayed these implausible characteristics and were excluded from the analysis.
Definitions of the calculated methylation variables.
The methylation of a specific C moiety is the ratio between the number of clones where that moiety is methylated and the number of all the analyzed clones (for that moiety), expressed as percentage. The overall methylation is a measurement, expressed as a percentage, of the total methylation level of a specific group of C (a variable number of C grouped together according to a criterion); it is calculated as the number of methylated C of that group divided by the total number of analyzed C (including all other C not belonging to that group), including clone replicates. For more explanations and examples see Supplemental Materials and Methods, definitions.
Structural analysis of upstream sequence of myogenin.
The absence of a CpG island within 10,000 bases upstream of the mouse myogenin gene was assessed by the CpG island searcher software (cpgislands.usc.edu/). The observed/expected CpG ratio and the G + C content of the 1,092 bases of myogenin promoter was assessed by the CpG ratio and GC content plotter software (mwsross.bms.ed.ac.uk/public/cgi-bin/cpg.pl).
Transcription factor binding analysis.
The muscle specific Match software (www.gene-regulation.com/cgi-bin/pub/programs/match/bin/match.cgi) and the CTCFBSDB (insulatordb.utmem.edu/help.php) were used for the analysis of, respectively, the putative Sp1 and CTCF putative binding sites. The Consite software (asp.ii.uib.no:8090/cgi-bin/CONSITE/consite) was used for the analysis of the Rreb1, Sna1, Nfe2l2 and Cebpb transcription factor putative binding sites.
Statistical analysis.
Differentiation, expression and methylation were evaluated by ANOVA and Bonferroni's post-test by means of the STAT software. The mean square contingency Φ2 and Kendall's τ-c, used for the spatial analysis of methylation, were calculated by SPSS software.
Bisulphite experimental data were laid out in a matrix containing the methylation data of the C2C12, C2T18 and embryonic tissues; each datasheet cell represents the methylation percentage of a specific cytosine in a unique experimental condition (basal, time course from DM shifting as well as for C2C12 in DM, in the presence (+DH) or absence of hypomethylating agents). This lay out of the data permitted the cluster analysis, which was aimed at grouping the experimental conditions or cytosines. The cluster analysis was performed, using the centroid method, by means of SPSS software.
Animal Care Statement
All the experiments were performed in such a way as to sacrifice the minimum number of animals required and were approved by the author's institution (N. 1) according to the guidelines of Italian Ministry of Health (D.L. 92/116).
Abbreviations
| sng-C | = | single non-grouping C |
| Dnmt | = | DNA methyltransferase |
| sd | = | standard deviation |
| adj-C | = | sub-cluster of adjacent C |
| ES | = | embryonic stem |
| EmM | = | embryonic muscle |
| EmB | = | embryonic brain |
| DM | = | differentiation medium |
| DM+DH | = | differentiation medium + 3-deaza-adenosine and homocysteine |
| GM | = | growth medium |
| CK | = | creatine kinase |
| h | = | hours |
| min | = | minutes |
Figures and Tables
Figure 1 Differentiation and myogenin expression. (A) Differentiation ability in the C2C12 and C2T18 is shown as CK activity, relative to nuclei numbers in GM at day 4 after plating and in DM at day 7 after serum lowering; for C2C12, differentiation was also evaluated after addition of hypomethylating drugs (DM+DH). Standard deviations (sd) are reported. The overall significant difference between conditions was evaluated by ANOVA (p < 0.0001); the significance of differences between each experimental condition pair was evaluated by Bonferroni's post test (see Sup. Table 3A); the only non-significant differences are those between C2T18 GM and both C2C12 DM and C2C12 DM+DH. (B) Northern blot shows myogenin expression, at indicated time after plating in GM or after serum lowering in DM, in C2C12 (even with addition of hypomethylating drugs (DM+DH)) and C2T18, as well as in embryonic tissues. Ribosomal 18S RNA is also shown for normalization. Columns indicate the mean of densitometric O.D. ratio (and corresponding sd) between myogenin and 18S signals of three independent northern blots. The overall significant difference between conditions was evaluated by ANOVA (p < 0.0001); the significance of differences between each experimental condition pair was evaluated by Bonferroni's post test (see Sup. Table 3B); the only non-significant differences are those between the following pairs: embryonic muscle (EmM) and C2T18 DM, embryonic brain (EmB) and C2C12 GM 48 h, C2C12 DM and C2T18 GM 96 h.
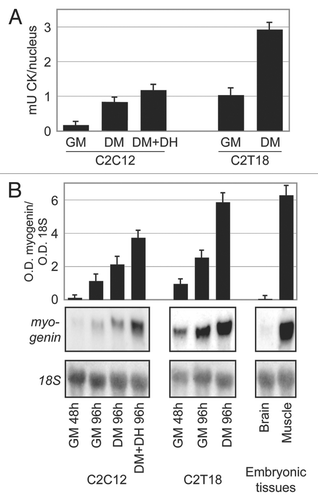
Figure 2 Summary of methylation percentages of all C moieties and all experimental conditions. The x-axis shows the 84 C moieties of the myogenin 5′-flanking region analyzed. The y-axis represents the percentage of methylation of each C. Only the experiments with controls of bisulphite modification (see Materials and Methods) with correct patterns were taken into account. The kind of CpN is indicated below the x-axis, according to the legend. The sub-cluster positions are indicated at the bottom of each graph.
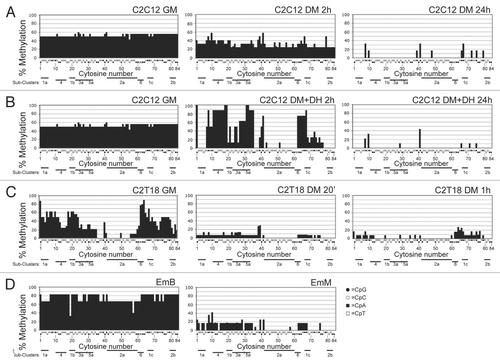
Figure 3 Overall methylation of CpG and non-CpG moieties. Cytosines were partitioned (from bottom to top of each bar) in CpG (black area), CpT (dark gray area), CpA (light gray area) and CpC (white area) moieties. For each experimental condition, the overall percentage of each CpN methylation (of the first C for CpC) is shown in the corresponding bar area; the sum of all CpN methylation is indicated by the total bar height and corresponds to the overall methylation percentage of all C; sd are also indicated. The overall differences are highly significant (ANOVA p < 0.0001). The heavy horizontal black lines group together the experimental conditions with overall methylation that was not statistically different (as evaluated by Bonferroni's t-test).
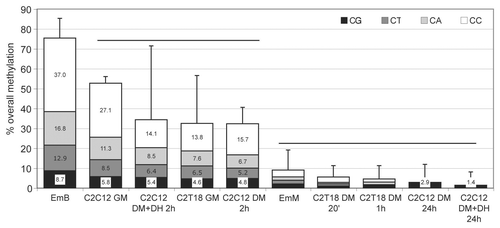
Figure 4 Cluster analysis of cytosines in myogenin 5′-flanking region. The cluster analysis shown is based on bisulphite data. The dashed line shows the chosen cut-off value.
Figure 5 Schematic representation of the 252 bp from the first to the last C moiety of the studied region of the myogenin 5′-flanking. Annotations regarding C numbering (both according to Genebank and progressive), sub-clusters (with gray background) and putative recognition sites of the CTCF, Sp1, Rreb1, Nfe2I2, Snai1 and Cebpb DNA binding transcription factors are shown.
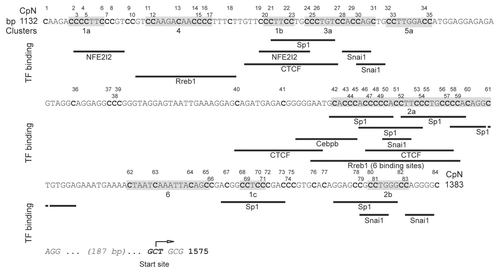
Figure 6 Modulation of overall cluster methylation. (A–C) The x-axis shows the timing after stimulation of differentiation and, where indicated, addition of DH, starting from undifferentiated conditions (indicated as time 0). The y-axis shows the percentage of overall methylation. Each line indicates the methylation level of each cluster (according to the legend), with the exception of the line in which all the sng-C (comprising all nine CpG and five non-CpG moieties) are pooled. (A) C2C12 with hypomethylating stimulus, (B) C2T18, (C) C2C12 without hypomethylating stimulus. (D) Columns indicate the percentage of overall methylation in EmB and EmM. Each column indicates the methylation level of each cluster (according to the legend), with the exception of the column in which all the sng-C (including all nine CpG and five non-CpG moieties) are pooled.
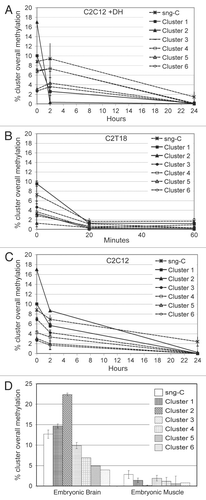
Table 1 Cluster composition features
Table 2 Peculiar short CpC-rich (at least 2 CpC) elements found and their general forms
Additional material
Download Zip (3.5 MB)Acknowledgements
This work was supported by a grant from the Sapienza Universita di Roma-Progetti di Università.
References
- Li B, Carey M, Workman JL. The role of chromatin during transcription. Cell 2007; 128:707 - 719
- Klose RJ, Bird AP. Genomic DNA methylation: The mark and its mediators. Trends Biochem Sci 2006; 31:89 - 97
- Suzuki MM, Bird A. DNA methylation landscapes: provocative insights from epigenomics. Nat Rev Genet 2008; 9:465 - 476
- Vaissiere T, Sawan C, Herceg Z. Epigenetic interplay between histone modifications and DNA methylation in gene silencing. Mutat Res 2008; 659:40 - 48
- D'Alessio AC, Szyf M. Epigenetic tete-a-tete: The bilateral relationship between chromatin modifications and DNA methylation. Biochem Cell Biol 2006; 84:463 - 476
- Turek-Plewa J, Jagodzinski PP. The role of mammalian DNA methyltransferases in the regulation of gene expression. Cell Mol Biol Lett 2005; 10:631 - 647
- Brown SE, Suderman MJ, Hallett M, Szyf M. DNA demethylation induced by the methyl-CpG-binding domain protein MBD3. Gene 2008; 420:99 - 106
- Lucarelli M, Fuso A, Strom R, Scarpa S. The dynamics of myogenin site-specific demethylation is strongly correlated with its expression and with muscle differentiation. J Biol Chem 2001; 276:7500 - 7506
- Ooi SK, Bestor TH. The colorful history of active DNA demethylation. Cell 2008; 133:1145 - 1148
- Filippova GN. Genetics and epigenetics of the multifunctional protein CTCF. Curr Top Dev Biol 2008; 80:337 - 360
- Douet V, Heller MB, Le SO. DNA methylation and Sp1 binding determine the tissue-specific transcriptional activity of the mouse Abcc6 promoter. Biochem Biophys Res Commun 2007; 354:66 - 71
- Inoue S, Oishi M. Effects of methylation of non-CpG sequence in the promoter region on the expression of human synaptotagmin XI (syt11). Gene 2005; 348:123 - 134
- Malone CS, Miner MD, Doerr JR, Jackson JP, Jacobsen SE, Wall R, et al. CmC(A/T)GG DNA methylation in mature B cell lymphoma gene silencing. Proc Natl Acad Sci USA 2001; 98:10404 - 10409
- Novik KL, Nimmrich I, Genc B, Maier S, Piepenbrock C, Olek A, et al. Epigenomics: Genome-wide study of methylation phenomena. Curr Issues Mol Biol 2002; 4:111 - 128
- Lorincz MC, Groudine M. C(m)C(a/t)GG methylation: a new epigenetic mark in mammalian DNA?. Proc Natl Acad Sci USA 2001; 98:10034 - 10036
- Lorincz MC, Schubeler D, Hutchinson SR, Dickerson DR, Groudine M. DNA methylation density influences the stability of an epigenetic imprint and Dnmt3a/b-independent de novo methylation. Mol Cell Biol 2002; 22:7572 - 7580
- Meissner A, Mikkelsen TS, Gu H, Wernig M, Hanna J, Sivachenko A, et al. Genome-scale DNA methylation maps of pluripotent and differentiated cells. Nature 2008; 454:776 - 780
- Weber M, Hellmann I, Stadler MB, Ramos L, Paabo S, Rebhan M, et al. Distribution, silencing potential and evolutionary impact of promoter DNA methylation in the human genome. Nat Genet 2007; 39:457 - 466
- Brown SE, Fraga MF, Weaver IC, Berdasco M, Szyf M. Variations in DNA methylation patterns during the cell cycle of HeLa cells. Epigenetics 2007; 2:54 - 65
- Imamura T, Kerjean A, Heams T, Kupiec JJ, Thenevin C, Paldi A. Dynamic CpG and non-CpG methylation of the Peg1/Mest gene in the mouse oocyte and preimplantation embryo. J Biol Chem 2005; 280:20171 - 20175
- Oakes CC, La SS, Smiraglia DJ, Robaire B, Trasler JM. Developmental acquisition of genome-wide DNA methylation occurs prior to meiosis in male germ cells. Dev Biol 2007; 307:368 - 379
- Fazzari MJ, Greally JM. Epigenomics: beyond CpG islands. Nat Rev Genet 2004; 5:446 - 455
- Saxonov S, Berg P, Brutlag DL. A genome-wide analysis of CpG dinucleotides in the human genome distinguishes two distinct classes of promoters. Proc Natl Acad Sci USA 2006; 103:1412 - 1417
- Chan QK, Khoo US, Chan KY, Ngan HY, Li SS, Chiu PM, et al. Promoter methylation and differential expression of pi-class glutathione S-transferase in endometrial carcinoma. J Mol Diagn 2005; 7:8 - 16
- Kitazawa S, Kitazawa R, Maeda S. Transcriptional regulation of rat cyclin D1 gene by CpG methylation status in promoter region. J Biol Chem 1999; 274:28787 - 28793
- Kouidou S, Agidou T, Kyrkou A, Andreou A, Katopodi T, Georgiou E, et al. Non-CpG cytosine methylation of p53 exon 5 in non-small cell lung carcinoma. Lung Cancer 2005; 50:299 - 307
- Teng C, Gladwell W, Raphiou I, Liu E. Methylation and expression of the lactoferrin gene in human tissues and cancer cells. Biometals 2004; 17:317 - 323
- Vu TH, Li T, Nguyen D, Nguyen BT, Yao XM, Hu JF, et al. Symmetric and asymmetric DNA methylation in the human IGF2-H19 imprinted region. Genomics 2000; 64:132 - 143
- Haines TR, Rodenhiser DI, Ainsworth PJ. Allele-specific non-CpG methylation of the Nf1 gene during early mouse development. Dev Biol 2001; 240:585 - 598
- Franchina M, Kay PH. Evidence that cytosine residues within 5′-CCT GG-3′ pentanucleotides can be methylated in human DNA independently of the methylating system that modifies 5′-CG-3′ dinucleotides. DNA Cell Biol 2000; 19:521 - 526
- Ramsahoye BH, Biniszkiewicz D, Lyko F, Clark V, Bird AP, Jaenisch R. Non-CpG methylation is prevalent in embryonic stem cells and may be mediated by DNA methyltransferase 3a. Proc Natl Acad Sci USA 2000; 97:5237 - 5242
- Fuso A, Nicolia V, Pasqualato A, Fiorenza MT, Cavallaro RA, Scarpa S. Changes in Presenilin 1 gene methylation pattern in diet-induced B vitamin deficiency. Neurobiol Aging 2009; 32:187 - 199
- Aoki A, Suetake I, Miyagawa J, Fujio T, Chijiwa T, Sasaki H, et al. Enzymatic properties of de novo-type mouse DNA (cytosine-5) methyltransferases. Nucleic Acids Res 2001; 29:3506 - 3512
- Mund C, Musch T, Strodicke M, Assmann B, Li E, Lyko F. Comparative analysis of DNA methylation patterns in transgenic Drosophila overexpressing mouse DNA methyltransferases. Biochem J 2004; 378:763 - 768
- Gowher H, Jeltsch A. Enzymatic properties of recombinant Dnmt3a DNA methyltransferase from mouse: The enzyme modifies DNA in a non-processive manner and also methylates non-CpG [correction of non-CpA] sites. J Mol Biol 2001; 309:1201 - 1208
- Li E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat Rev Genet 2002; 3:662 - 673
- Kangaspeska S, Stride B, Metivier R, Polycarpou-Schwarz M, Ibberson D, Carmouche RP, et al. Transient cyclical methylation of promoter DNA. Nature 2008; 452:112 - 115
- Metivier R, Gallais R, Tiffoche C, Le PC, Jurkowska RZ, Carmouche RP, et al. Cyclical DNA methylation of a transcriptionally active promoter. Nature 2008; 452:45 - 50
- Oswald J, Engemann S, Lane N, Mayer W, Olek A, Fundele R, et al. Active demethylation of the paternal genome in the mouse zygote. Curr Biol 2000; 10:475 - 478
- Bhattacharya SK, Ramchandani S, Cervoni N, Szyf M. A mammalian protein with specific demethylase activity for mCpG DNA. Nature 1999; 397:579 - 583
- Zhu B, Zheng Y, Hess D, Angliker H, Schwarz S, Siegmann M, et al. 5-methylcytosine-DNA glycosylase activity is present in a cloned G/T mismatch DNA glycosylase associated with the chicken embryo DNA demethylation complex. Proc Natl Acad Sci USA 2000; 97:5135 - 5139
- Barreto G, Schafer A, Marhold J, Stach D, Swaminathan SK, Handa V, et al. Gadd45a promotes epigenetic gene activation by repair-mediated DNA demethylation. Nature 2007; 445:671 - 675
- Sachan M, Raman R. Developmental methylation of the coding region of c-fos occurs perinatally, stepwise and sequentially in the liver of laboratory mouse. Gene 2008; 416:22 - 29
- Figliola R, Busanello A, Vaccarello G, Maione R. Regulation of p57(KIP2) during muscle differentiation: role of Egr1, Sp1 and DNA hypomethylation. J Mol Biol 2008; 380:265 - 277
- Palacios D, Summerbell D, Rigby PW, Boyes J. Interplay between DNA methylation and transcription factor availability: implications for developmental activation of the mouse myogenin gene. Mol Cell Biol 2010; 30:3805 - 3815
- Asakura A, Komaki M, Rudnicki M. Muscle satellite cells are multipotential stem cells that exhibit myogenic, osteogenic and adipogenic differentiation. Differentiation 2001; 68:245 - 253
- Pownall ME, Gustafsson MK, Emerson CP Jr. Myogenic regulatory factors and the specification of muscle progenitors in vertebrate embryos. Annu Rev Cell Dev Biol 2002; 18:747 - 783
- Gardiner-Garden M, Frommer M. CpG islands in vertebrate genomes. J Mol Biol 1987; 196:261 - 282
- Takai D, Jones PA. Comprehensive analysis of CpG islands in human chromosomes 21 and 22. Proc Natl Acad Sci USA 2002; 99:3740 - 3745
- Yee SP, Rigby PW. The regulation of myogenin gene expression during the embryonic development of the mouse. Genes Dev 1993; 7:1277 - 1289
- Woodcock DM, Crowther PJ, Diver WP. The majority of methylated deoxycytidines in human DNA are not in the CpG dinucleotide. Biochem Biophys Res Commun 1987; 145:888 - 894
- Meissner A, Gnirke A, Bell GW, Ramsahoye B, Lander ES, Jaenisch R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res 2005; 33:5868 - 5877
- Jia D, Jurkowska RZ, Zhang X, Jeltsch A, Cheng X. Structure of Dnmt3a bound to Dnmt3L suggests a model for de novo DNA methylation. Nature 2007; 449:248 - 251
- Ferguson-Smith AC, Greally JM. Epigenetics: perceptive enzymes. Nature 2007; 449:148 - 149
- Fuso A, Cavallaro RA, Orrú L, Buttarelli FR, Scarpa S. Gene silencing by S-adenosylmethionine in muscle differentiation. FEBS Lett 2001; 508:337 - 340
- Agirre X, Vizmanos JL, Calasanz MJ, Garcia-Delgado M, Larrayoz MJ, Novo FJ. Methylation of CpG dinucleotides and/or CCWGG motifs at the promoter of TP53 correlates with decreased gene expression in a subset of acute lymphoblastic leukemia patients. Oncogene 2003; 22:1070 - 1072
- Brooks AR, Harkins RN, Wang P, Qian HS, Liu P, Rubanyi GM. Transcriptional silencing is associated with extensive methylation of the CMV promoter following adenoviral gene delivery to muscle. J Gene Med 2004; 6:395 - 404
- Klenova EM, Nicolas RH, Paterson HF, Carne AF, Heath CM, Goodwin GH, et al. CTCF, a conserved nuclear factor required for optimal transcriptional activity of the chicken c-myc gene, is an 11-Zn-finger protein differentially expressed in multiple forms. Mol Cell Biol 1993; 13:7612 - 7624
- Yaffe D, Saxel O. Serial passaging and differentiation of myogenic cells isolated from dystrophic mouse muscle. Nature 1977; 270:725 - 727
- Fuso A, Scarpa S, Grandoni F, Strom R, Lucarelli M. A reassessment of semiquantitative analytical procedures for DNA methylation: comparison of bisulfite- and HpaII polymerase-chain-reaction-based methods. Anal Biochem 2006; 350:24 - 31
- Clark SJ, Harrison J, Paul CL, Frommer M. High sensitivity mapping of methylated cytosines. Nucleic Acids Res 1994; 22:2990 - 2997
- Genereux DP, Johnson WC, Burden AF, Stoger R, Laird CD. Errors in the bisulfite conversion of DNA: modulating inappropriate- and failed-conversion frequencies. Nucleic Acids Res 2008; 36:e150
- Shiraishi M, Hayatsu H. High-speed conversion of cytosine to uracil in bisulfite genomic sequencing analysis of DNA methylation. DNA Res 2004; 11:409 - 415