Abstract
Development of the cerebellum, a brain region regulating posture and coordination, occurs post-natally and is marked by rapid proliferation of granule neuron precursors (CGNPs), stimulated by mitogenic Sonic hedgehog (Shh) signaling. β-Arrestin (βArr) proteins play important roles downstream of Smoothened, the Shh signal transducer. However, whether Shh regulates βArrs and what role they play in Shh-driven CGNP proliferation remains to be determined. Here, we report that Shh induces βArr1 accumulation and localization to the nucleus, where it participates in enhancing expression of the cyclin dependent kinase (cdk) inhibitor p27, whose accumulation eventually drives CGNP cell cycle exit. βArr1 knock-down enhances CGNP proliferation and reduces p27 expression. Thus, Shh-mediated βArr1 induction represents a novel negative feedback loop within the Shh mitogenic pathway, such that ongoing Shh signaling, while required for CGNPs to proliferate, also sets up a cell-intrinsic clock programming their ultimate exit from the cell cycle.
Introduction
The cerebellum is a brain structure that regulates posture, coordination and fine movements. This tissue is marked by a stereotypical foliated architecture and is comprised of several specific neural subtypes, including interneurons referred to as granule neurons. CGNPs are understood to be a potential cell of origin for certain subclasses of the childhood brain tumor, medulloblastoma, particularly those marked by aberrant activation of the Shh signaling pathway.Citation1 Understanding mechanisms regulating CGNP cell cycle progression and cell cycle exit will ultimately shed light on how aberrant Shh pathway activity may contribute to transformation of these cells.
In mammals, much of cerebellar development takes place peri-natally. Cerebellar granule neuron precursors (CGNPs) undergo a rapid expansion phase in a proliferative region referred to as the external granule layer (EGL), a cell layer lining the dorsal surface of the cerebellum. After several rounds of cell division, CGNP cell bodies migrate through the underlying layer of Purkinje neurons to form the internal granule layer (IGL), where they will complete differentiation into interneurons.Citation2 Post-natal CGNP proliferation requires Shh signaling. Impairment of Shh signaling results in reduced proliferation in the EGL of neonatal mice.Citation3–Citation5 Neonatal mouse CGNPs in culture will also proliferate in response to exogenous Shh; however, in vitro and in vivo, even in the continued presence of Shh, CGNPs leave the cell cycle and differentiate, suggesting that CGNPs possess cell-intrinsic mechanisms that modulate the proliferative response to Shh. However, very little is understood about how CGNPs lose their ability to proliferate in response to Shh and commence differentiation. Failure to progress from a proliferative state to a differentiating state may contribute to medulloblastoma formation.
Shh signals by binding to the transmembrane protein Patched (Ptch) thereby relieving its repression of Smoothened (Smo), a G protein-coupled receptor-like protein that transduces Shh signaling by activating Gli transcription factors.Citation6–Citation8 In cell lines, arrestin proteins have been shown to play a role in Shh pathway activation. Arrestins (β-arrestin-1 and β-arrestin-2) were originally identified as negative regulators of G-protein coupled receptors; however, additional roles as scaffolding proteins in both the cytosol and in the nucleus have been described.Citation9–Citation11 Phosphorylation of receptors by G protein-coupled receptor (GPCR) kinases (GRK) mediates arrestin binding and subsequent disruption of G protein:GPCR interactions.Citation12 GRK2 has been shown to phosphorylate Smo in response to hedgehog and recruit βArr2.Citation13,Citation14 Furthermore, in response to hedgehog, both βArr1 and βArr2 have been shown to localize to the primary cilium, a critical cellular structure required for hedgehog signaling.Citation15
An unexpected function of βArr1 involves its sub-cellular localization. Unlike other members of the arrestin family that appear to be primarily cytoplasmic, βArr1 is found in both the cytoplasm and the nucleus.Citation16 Several years ago it was discovered that βArr1 can form a nuclear complex with CREB and p300 and that this interaction increases specific gene transcription. One of the genes whose transcription was enhanced by this mechanism was p27Kip1, a cyclin dependent kinase (CDK) inhibitor and differentiation marker for CGNPs.Citation10,Citation17 Here we show that in CGNPs, mitogenic signaling by Shh includes an intrinsic negative feedback loop, ultimately driving cell cycle exit through Shh-mediated bArr upregulation and nuclear translocation and subsequent βArr1-mediated increased p27 transcription.
Results
Shh mediates βArr1 accumulation and nuclear localization.
Using an NIH3T3 cell system, Kovakis et al. showed that knockdown of βArr1 or βArr2 diminishes the ability of Smo to translocate to the primary cilium and the ability of hedgehog to activate Gli transcription factors.Citation15 However, whether βArrs play roles in Shh signaling during cerebellum development is unknown. In order to determine whether Shh treatment affects βArr levels, we cultured CGNPs from post-natal (PN), day 5 mice in the presence or absence of Shh. In the absence of Shh CGNPs leave the cell cycle and differentiate; however, if Shh is present in the culture media, CGNPs will continue to proliferate for several days as evidenced by the presence of the proliferation marker Cyclin D2 (). Shh treatment had no effect on βArr2 levels, but increases both βArr1 total protein and PS412-βArr1 ( and B). Although βArr1 Ser412 phosphorylation has been shown to play an important role in mediating βArr1 function in GPCR signaling, its role in Shh signaling has not been studied.Citation18 Interestingly, increased protein levels of βArr1 do not correlate to increases in βArr1 mRNA ().
Although we saw moderate changes in βArr1 protein levels after treatment with Shh, the most dramatic effect was seen when we determined the sub-cellular localization of βArr1. Cultured CGNPs were treated with vehicle, Shh or Shh plus the Smo inhibitor cyclopamine (). Immuno-staining was performed using a validated βArr1 antibody (red); nuclei were visualized with DAPI (blue) (Sup. Fig. 1). Treatment with Shh caused a shift of βArr1 from the cytoplasm to the nucleus (, middle part). Treatment of CGNPs with Shh in conjunction with the Shh pathway inhibitor cyclopamine blocked nuclear translocation of βArr1 (, right). Cell counts of CGNPs with nuclear βArr1 show that approximately 50% of Shh-treated cells have nuclear βArr1 compared to only 15% of vehicle-treated cells (). Results of the immuno-staining were confirmed by sub-cellular fractionation of CGNPs followed by western blot analysis (); cyclopamine-mediated Smoothened inhibition reduced levels of total and phosphorylated βArr1 in the cytoplasm and nucleus. Moreover, inhibiting Smoothened activity using the compound Smoothened Antagonist (SANT) also resulted in inhibition of Shh-mediated bArr1 induction (Sup. Fig. 1).
βArr1 modulates the CGNP proliferative response to Shh.
To determine the role of βArr1 in Shh-driven CGNP proliferation we used short hairpin (sh)RNA-expressing lentivirusmediated βArr1 knockdown () or retroviral transduction to overexpress βArr1-FLAG-tagged constructs () in CGNPs. Five Sigma-validated lentiviral constructs targeting βArr1 were purchased and tested for efficiency and specificity in mouse cell lines to generate a pool used to infect CGNPs (data not shown). CGNPs were infected with these viruses as previously described.Citation19 CGNPs were harvested for western blot analysis or immuno-stained for the proliferation marker Ki67. The percent of cells that were Ki67-positive was determined as described in the methods. Surprisingly, knockdown of βArr1 by approximately 50 percent significantly increased cyclin D2 levels in Shh treated cells (). Shh-treated CGNPs infected with βArr1 shRNA lentiviruses show a two-fold increase in Ki67 positive cells compared to Shh-treated uninfected cells or CGNPs infected with scrambled control shRNA-bearing lentiviruses ( graph, Sup. Fig. 2). These results suggest that βArr1 may negatively regulate CGNP proliferation.
In contrast to our findings with βArr1 knockdown, infecting CGNPs with viruses to ectopically express FLAG-tagged βArr1 resulted in reduced cyclin D2 levels and in a significant reduction in proliferating cells as determined by quantification of the percent of Ki67-positive cells in Shh treated CGNPs as compared to Shh treatment alone or infection with empty vector viruses ( and Fig. S2). This was not a result of βArr1 overexpression causing apoptosis, as we did not observe increased levels of cleaved caspase-3. Neither overexpression nor knockdown had a significant effect on vehicle-treated samples. To determine whether acute abrogation of βArr1 levels affected Shh-mediated CGNP proliferation in the setting of the intact external granule layer, we utilized cerebellar slice explants. As shown in , addition of Shh to the medium of slice explants infected with control scrambled lentiviruses is associated with proliferation as determined by Ki67 immunostaining. However, infection of the slice explants with βArr1-knockdown lentiviruses resulted in a dramatic increase in proliferation, supporting our conclusions from our CGNP monolayer culture studies that βArr1 plays a negative role downstream of mitogenic Shh signaling.
βArr1 phosphorylation is necessary and sufficient to mediate βArr1 nuclear translocation.
Our data show that Shh treatment increases levels of total and Ser412-phosphorylated βArr1 (). We wished to determine whether βArr1 phosphorylation plays a role in βArr1 Shh-mediated nuclear translocation. Because the kinase responsible for this post-translational modification remains unclear, we were unable to use kinase-specific inhibitors. Therefore, as an alternative to inhibiting the kinase acting at Ser412, we targeted the phosphatase PP2A ( and B), using okadaic acid (OA). PP2A has been shown to be in an immunocomplex with βArr1, and inhibition of PP2A with OA or Small-T antigen increases Ser412 phosphorylation.Citation20 To test the possibility that βArr1 Ser412 phosphorylation regulates βArr1 nuclear localization, we cultured CGNPs in the presence or absence of Shh. After 24 h, CGNPs were treated with OA, and cells were either harvested for western blot () or immuno-stained for endogenous βArr1 (). OA treatment alone was sufficient to mediate an accumulation of PS412-βArr1. The combination of OA and Shh did not increase this effect, suggesting that both lie in a common pathway (). Interestingly, treatment with OA also drove translocation of βArr1 to the nucleus even in the absence of Shh ().
To confirm our results with OA, we constructed retroviruses expressing FLAG-tagged βArr1 wild-type (WT), Ser-Ala (βArr1-S412A) or Ser-Asp (βArr1-S412D) point mutations to mimic unphosphorylated and phosphorylated βArr1, respectively. Infected CGNPs were treated with vehicle or Shh prior to a BrdU pulse. CGNPs were stained for FLAG (red), BrdU (green) and DAPI (blue) (). Shh-treated CGNPs expressing WT βArr1 show both cytoplasmic and nuclear staining, while vehicle treated samples exhibit primarily cytoplasmic βArr1 (, left parts). Furthermore, in Shh-treated samples, nuclear βArr1 is excluded from BrdU-positive cells. Conversely, infection with βArr1-S412A retrovirus maintains βArr1 in the cytoplasm even in the presence of Shh (, middle parts). However, CGNPs expressing βArr1-S412D show almost exclusively nuclear βArr1 either in the presence or absence of Shh, including in cells that are BrdU-positive (, right parts). These data suggest that the sub-cellular localization of βArr1 is regulated by post-translational phosphorylation of Ser412 and that Shh may be mediating βArr1 relocation to the nucleus through this mechanism.
We next wanted to determine whether phosphorylation of βArr1 was required for inhibiting Shh mitogenic response in CGNPs. To this end, we infected CGNPs with retroviruses overexpressing βArr1-WT, S412A or S412D as described above. After 48 h in culture, protein lysates were obtained or CGNPs were pulsed with BrdU for two hours, fixed, stained and the number of BrdU-positive cells was determined ( and E). Only CGNPs expressing either WT or S412D βArr1 showed a significant decrease in proliferation compared to Shh controls ( and E). Infection with the βArr1 S412A phospho-mutant did not drive a significant decrease in proliferation, suggesting that both phosphorylation of βArr1 and its sub-cellular localization are responsible for negatively regulating Shh signaling in CGNPs.
βArr1 co-localizes with p27 and active CRE B in vitro and in vivo.
A function of nuclear βArr1 was not demonstrated until recently. In 2005 Kang et al. reported that βArr1 translocation to the nucleus of transfected fibroblasts enhances transcription of specific genes, including the cyclin-dependent kinase (cdk) inhibitor p27. They found that βArr1 was part of a complex with the transcription factor CREB and that βArr1 acts as a scaffold to recruit the histone acetyltransferase p300 to CREB, resulting in enhanced p27 transcription.Citation10 However, βARR1 is not absolutely required for p27 transcription, reflecting the complexity of regulation of this cdk inhibitor.
CGNP cell cycle progression requires the activity of cdk4 and cdk6 in complex with cyclin D1 and cyclin D2.Citation21 An important function of p27 is to inhibit cdk activity. This function of p27 prevents cell cycle progression. Importantly, it has been shown that accumulation of total p27 levels within cycling cells functions as a cell-intrinsic clock to ultimately drive growth arrest.Citation22 In culture and in vivo, p27 expression marks post-mitotic CGNPs. In vivo, p27 is expressed in the EGLb, an area where CGNPs have started to leave the cell cycle.Citation17 Miyazawa et al. showed that p27 gradually accumulates in CGNPs as they proliferate rather than after they have left the cell cycle. The authors suggested that p27 is part of an internal cellular clock whose accumulation signals the cells to begin differentiation programs. Consistent with this model, p27-/- mice have increased cerebellar size, and cultured p27-null CGNPs proliferate longer than wildtype CGNPs,Citation17 although these cells do eventually leave the cell cycle, indicating that p27 is not the sole driver of CGNP growth arrest. Moreover, p27 loss cooperates with Shh activity in driving medulloblastoma formation, suggesting a tumor suppressive role for p27 in CGNPs.Citation23,Citation24
To determine whether βArr1 acts as a nuclear scaffold in CGNPs and if this function has the potential to drive p27 transcription in Shh-treated cells, we first examined the localization of p27, βArr1, PS412-βArr1, CREB and PS133-CREB in vivo ( and Sup. Material). Immunohistochemical analysis shows that p27 localizes to the EGLb of PN7 mice as previously reported.Citation17 Although CREB can be found throughout the cells of the cerebellum during development, activated CREB (P-CREB) is only located in the EGLb of PN7 mice and the remaining EGL of PN15 mice (Sup. Fig. 3). Similarly, both βArr1 and PS412-βArr1 can only be found in EGL of PN7 and PN15 mice with a small nuclear accumulation occurring in the EGLb ( arrows).
To determine if these results could be recapitulated in vitro, CGNPs cultured with Shh were immunostained for βArr1 and p27 as indicated (). CGNPs that exhibited nuclear βArr1 ( red) were always p27-positive ( arrowheads) suggesting that nuclear βArr1 is found in the appropriate compartment to enhance p27 transcription. Nuclear βArr1 and Phospho-CREB also co-localize, as do p27 and Phospho-CREB (Sup. Fig. 3, arrowheads). However, there is a sub-population of Phospho-CREB-positive cells that are p27-negative (Sup. Fig. 3).
βArr1 levels affect p27 expression.
We next asked whether Shh mediates changes in p27 levels in vitro. Indeed, western blot analysis revealed that p27 total protein and p27 mRNA increased in response to Shh ( and B), in parallel with increased β-arrestin levels and Gli1 expression. Although Gli1 expression begins to decrease at later time points, CGNPs are known to proliferate in the presence of Shh for several days,Citation3 suggesting maintenance of the Shh signaling machinery over time, albeit at less than peak levels. However, when the number of cells expressing p27 was determined, the values were the same for both treatment groups (). These results suggest that CGNPs cultured in the presence of Shh express more p27 per cell than vehicle-treated CGNPs since the overall number of p27-expressing cells does not change. Consistent with βArr1 mediating an anti-proliferative effect in the presence of Shh, knockdown of βArr1 diminished both p27 mRNA and protein levels ( and E) while βArr1 overexpression increased p27 levels (). The modest induction of p27 in the presence of βArr1 alone may reflect interactions between βArr1 and other anti-mitogenic pathways whose activity is suppressed by Shh; however, this induction is unlikely to reflect βArr interactions with the p27 promoter, since in the absence of Shh endogenous and overexpressed βArr1 does not localize to the nucleus ().
βArr1, CRE B and p300 are found in complex on the p27 promoter.
Published reports suggest that βArr1 is part of a complex containing the transcription factor CREB and that this complex is bound to the p27 promoter.Citation10 In order to determine if CREB is bound to the p27 promoter in Shh-treated CGNPs, we performed chromatin imunnoprecipitation (ChIP) on CGNPs treated with vehicle or Shh ( and B). We found that CREB is bound to the p27 promoter preferentially in Shh treated CGNPs and is not found at other promoters, such as that of c-Jun (), whose levels are not known to be altered by Shh treatment. Furthermore, treatment with cyclopamine reduces the amount of CREB bound to the p27 promoter, indicating that binding is Smoothened-dependent ().
It has been shown in a neuroblastoma cell line expressing opioid receptors that βArr1 acts as a nuclear scaffold that recruits the histone acetyltransferase (HAT) p300 to activated CREB, thereby enhancing p27 transcription.Citation10 To test whether this interaction plays a role in p27 expression in proliferating CGNPs responding to mitogenic Shh signaling, we cultured CGNPs in vehicle or Shh in the presence of scrambled control lentiviruses or βArr1-targeting lentiviruses. Like CREB, p300 is also recruited to the p27 promoter in the presence of Shh, and this interaction is disrupted by βArr1 shRNA infection ().
To verify that βArr1 is in a complex with CREB and determine whether (a) βArr1 in this complex is phosphorylated and (b) the complex is regulated by Shh in CGNPs, we cultured CGNPs in the presence or absence of Shh. Protein lysates were collected and immuno-precipitated with either PS412-βArr1 or CREB antibodies (). PS412-βArr1 antibodies immuno-precipitate CREB and vice versa only when CGNPs are cultured with Shh (). We conclude, therefore, that Shh may be indirectly upregulating p27 by promoting βArr1 nuclear localization. In the nucleus, βArr1 binds to CREB and p300 and together they enhance p27 transcription. Ultimately, p27 accumulation drives cell cycle exit as a result of its inhibitory effects on cdk activity.
Discussion
Our results suggest a working model to define a role for βArr1 in Shh-mediated CGNP proliferation (). Shh binds to Ptch, resulting in Smoothened activation. Shh promotes cell cycle progression by upregulating and/or activating proliferation-promoting factors such as IRS1 and N-Myc as described previously.Citation19,Citation25,Citation26 However, in addition to promoting the well-characterized negative feedback loop of Ptc upregulation by Gli,Citation27 Shh also regulates sub-cellular localization of βArr1 (). Phosphorylation of βArr1 at Ser412 is necessary and sufficient to mediate βArr1 localization to the nucleus (). Inhibiting PP2A with OA in CGNPs increases βArr1 phosphorylation, as previously reported,Citation20 and nuclear accumulation, even in the absence of Shh (). Altering the expression of βArr1 impacts CGNPs' mitogenic response to Shh, as βArr1 overexpression reduces proliferation while βArr1 knockdown increases proliferation in Shh-treated cells (). The effect of βArr1 on proliferation is dependent upon its phosphorylation since mutating Ser 412 alanine reverses the effect of βArr1 overexpression ().
The kinase acting at βArr1 Ser412 remains to be determined. In cell lines, where βArr1 Ser412 phosphorylation has been best characterized as a mediator of G protein-coupled receptor endocytosis, the MEK inhibitor PD98059 had a partial effect on βArr1 Ser412 phosphorylation.Citation20 However, it is unlikely that the MEK target ERK is playing a role in CGNPs as ERK activation does not change with Shh treatment nor does MEK inhibition alter CGNP proliferation in response to Shh.Citation19,Citation28 However, PP2A has been shown to play a role in dephosphorylation of βArr1 Ser412 in cell lines,Citation20 and we obtained similar results in CGNPs. Negative regulation of PP2A by Shh to affect βArr1 phosphorylation is unlikely since several published reports to date indicate that hedgehog and PP2A work in concert to propagate signaling.Citation19,Citation29–Citation31 One possibility, therefore, is that Shh regulates a yet-to-be-identified kinase that phosphorylates βArr1 at Ser412.
A question that has remained unanswered regarding cerebellar development is how CGNPs leave the cell cycle despite the continued presence of Shh. In fact, as CGNPs migrate they move through a gradient of Shh emanating from Purkinje neurons. Their growth arrest may be attributed to Shh-mediated upregulation of Patched, a classic Gli transcriptional target. However, our findings reveal an additional negative feedback loop in the induction and nuclear localization of βArr1, promoting enhanced transcription of p27. We found that levels of the CGNP differentiation marker p27 are upregulated in response to Shh, but the number of CGNPs expressing p27 remains the same () as that in vehicle-treated cultures. However, individual cells in Shh-treated groups express higher levels of p27 per cell than the vehicle group. This is consistent with previous reports showing that p27 accumulates in CGNPs as they proliferate rather than after they have left the cell cycle, thus cells that proliferate longer will accumulate more p27.Citation17 These results are also consistent with p27 levels functioning as an intracellular timer that signals to CGNPs to stop proliferating and begin differentiating when it has accumulated to high levels. Therefore, one exciting possibility is that increased levels of Shh, which may occur as CGNPs migrate toward the Shh gradient, trigger βArr1 nuclear translocation and more p27 accumulation.
It was recently reported that a nuclear function of βArr1 is to act as a scaffolding platform recruiting the p300 histone acetyltransferase to CREB and thereby enhancing the transcription of p27.Citation10 We found that nuclear βArr1 co-localizes with p27 and activated CREB in vitro and in vivo (). Furthermore, we found that the phosphorylated form of βArr1 preferentially binds to CREB only after Shh treatment and that CREB is bound to the p27 promoter in Shh-treated CGNPs (). Our hypothesis, therefore, is that Shh promotes both CGNP proliferation and eventual growth-arrest, the latter of which includes regulating βArr1 nuclear translocation. In the nucleus βArr1 increases p27 expression, which accumulates to ultimately trigger cell cycle exit. Loss of this function, via βArr1 knockdown, causes prolonged CGNP proliferation in response to Shh due to a concomitant loss of p27. In the absence of Shh, lower levels of p27 are required to trigger cell cycle exit due to the lack of mitogenic stimulation, therefore accumulation of nuclear βArr1 is not required to increase levels of p27. Our studies reveal a mechanism through which Shh mediates the cell-intrinsic timing of cerebellar granule neuron precursor proliferation competency during cerebellar development. Future studies will address whether diminished expression or activity of βArr1 may contribute to Shh-induced medulloblastoma occurrence or growth.
Experimental Procedures
Animal studies.
Harvest of neural precursors from neonates and preparation of cerebella from wild-type and mutant mice for histological analysis were carried out in compliance with the Memorial Sloan-Kettering Institutional animal care and use committee guidelines.
β-Arrestin plasmids and lentiviruses.
Constructs for FLAG-βArr1 were obtained from the Lefkowitz groupCitation32 and cloned into a modified retrovirus vector containing a GFP tag (pIG). Retrovirus constructs for both βArr1 phosphomutants were constructed using the QuikChange® II XL Site-Directed Mutagenesis Kit (Stratagene).
Cerebellar granule neuron precursor culture.
CGNP cultures were generated as previously described.Citation28 Briefly, cerebella from postnatal day (PN) 5 Swiss-Webster 129 (SW129) mice were dissected into HBSS supplemented with glucose. The meninges were removed, cerebella were pooled and treated with Trypsin-EDTA for dissociation by trituration into HBSS. Triturated cells were centrifuged and resuspended in Dulbecco's modified Eagle's medium-F-12 (DMEM/F12) (Gibco) supplemented with 25 mM KCl, N2 supplement (Gibco), antibiotic and 10% fetal calf serum (Sigma). Cells were plated in individual poly-DL-ornithine (Sigma) pre-coated wells of a 6-well plate, and for treated samples 3 µg/mL Shh (R&D Systems) was added to the media. After 6–12 hours the media was changed to DMEM/F12/N2/KCl minus serum, with or without Shh as indicated. Unless otherwise stated, cells were left undisturbed for 24 hours prior to further treatment or analysis.
Protein preparation and immunobloting.
For immunoblot analysis cells were scraped cells into their medium then pelleted by low speed centrifugation. Cells were washed once in PBS and protein extracts were prepared as previously described.Citation28 Protein content was determined by using the Bio-Rad protein assay. Assays were performed in duplicate for each sample.
50 µg of each sample was separated by sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) on 8% polyacrylamide gels and then transferred in 20% methanol buffer at 4°C to Immobilon polyvinylidene difluoride (Millipore) membranes.
Standard western blot proceduresCitation28 were used to determine protein levels. Antibodies used for western blotting were: Cyclin D2 (Santa Cruz), C-Jun/AP1 (Calbiochem), GAPDH (Cell Signaling), beta Arrestin 1 and 2 (Abcam), phospho-beta Arrestin 1 (Cell Signaling) and β-tubulin (Sigma). Peroxidase activity was detected using Amershams's ECL reagents and exposing membranes to Kodak Biomax film. Multiple exposures were taken to avoid saturating film. The film was scanned and the digitalized images were quantified by densitometry using Adobe Photoshop 9.0 software.
Sub-cellular fractionation.
Sub-cellular fractionation was performed using the NE-PER Nuclear and Cytoplasmic Extraction Kit (Thermo Scientific) as per manufacturer's protocols. 1 × 106 CGNPs were treated with either vehicle or Shh for 48 hours prior to harvesting. Western blot analysis was performed on the extracts as described above.
Immunofluorescence.
The cells were fixed with 4% paraformaldehyde for 20 minutes followed by two 10-minute washes with PBS. Cells were blocked for 30 minutes then exposed to primary and secondary antibodies according to standard methods. All antibodies, beta Arrestin 1 (Abcam), Ki67 (Vector Labs), p27 (BD Pharmagen), P-CREB (Cell Signaling) and BrdU (Becton Dickinson) were used at a 1:100 dilution in blocking solution.
Immunohistochemistry.
Paraffin-embedded sections from post-natal day 0, 7, 15 or 21 cerebella were stained with indicated antibodies using the Ventana Medical Systems Discovery XT Staining Module. Antibodies were used at a 1:100 dilution.
shRNA virus production and infection.
Lentiviral constructs expressing short hairpin RNAs targeting βArr1 (Sigma, Mission shRNA) were individually transfected into NIH3T3 cells, which were then analyzed by western blotting to determine efficiency of βArr1 knockdown. 293T (Invitrogen) packaging cells were co-transfected with constructs that successfully knocked down βArr1 or a constructs carrying a nonsense shRNA sequence, delta 8.9 and vesicular stomatitis virus G glycoprotein plasmids, using Fugene 6 transfection reagent (Roche). The media was changed 12 hours after transfection and supernatants (10 mL) were harvested every 24 hours for 72 hours and kept at 4°C until they were pooled, filtered through 0.45 µm syringe filters, aliquoted and stored at −80°C until use. After a 1 hour incubation with Shh, CGNPs were infected with 3 mLs of virus supernatant for 4 hours. Viral supernatant was then removed and replaced with ±Shh as indicated.
Image capturing.
Staining of cultured primary cells and tissue sections was visualized with a Leica DM5000B microscope and images were taken using Leica FW400 software. For quantification of BrdU uptake into newly synthesized DNA, TIFF images of four random fields were taken for each experimental group using the 10× objective. The percentage of cells staining positive for BrdU over the total number of cells was determined using Image Pro Plus software (MediaCybernetics). Con-focal images were visualized with Leica TCS AOBS SP2 (Inverted Stand) and images captured with Leica LCS Lite software.
Immunoprecipitation.
CGNP protein lysates were prepared as described above. 1 mg of protein was precleared with 50% protein A or protein G sepharose (Sigma) for one hour at 4°C with rotation. Next, lysates were incubated with PSer412-βArr1 (Cell Signaling), CREB (Cell Signaling), mouse IgG or rabbit IgG (Upstate) overnight at 4°C with rotation. The beads were washed four times with pull down buffer (20 mM Hepes pH 7.5, 0.5 mM EDTA, 10% glycerol, 0.1% TritionX-100, 1 mM DTT) with 10-minute rotations at 4°C in between. Immuno-complexes were eluted using 0.1 M glycine pH 2.5 and normalized with 1 M Tris pH 7.4. Western blots were performed as described.
Chromatin Immunoprecipitation (ChIP).
The assay was performed according to the protocol for the ChIP assay kit (Upstate Biotechnologies). Briefly, 2 × 106 CGNPs were fixed and washed in PBS containing protease inhibitor cocktail (Sigma). After sonication, DNA-protein immunocompexes were immunoprecipitated with anti-CREB (Cell Signaling), p300 (Millipore) or IgG (Upstate) controls overnight at 4°C with rotation. The presence of the target p27 and C-Jun promoter sequence in both the input DNA and the recovered DNA-protein immunocomplexes was detected by qPCR. The data was normalized to the corresponding DNA input control. Sequences for the qPCR were as follows: negative control 5′-TTG TTT GCT TCC TGC CTT CT-3′; 5′-GGT TTC CAC CCT GTG ACA TC-3′, p27 5′-GGA AAA CAC CCC CAA AAG CAC GAG-3′; 5′-TCC CGG CGC CGA GAC CAA T-3′, C-Jun 5′-GGG CGG GCC GGG AAA AAC-3′; 5′-CAT TGG CTC GCG TCG CTC TCA GG-3′.
Quantitative PCR.
RNA from CGNPs was collected using TRIZOL® invitrogen reagent as per manufacturer's instructions. RNA samples were resuspended in 87.5 µL DEPC-treated water. In order to fully remove any residual DNA from the samples, RNA was further purified using the RNeasy Mini Kit (Qiagen) according to manufacturer's instructions. DNase (Qiagen) digestion, as per Qiagen instructions, was performed in solution prior to further RNA purification via the RNeasy column.
cDNA was generated with SuperScript First-Strand Synthesis System for RT-PCR (Invitrogen) per manufacturer's instructions. Syber Green Expression Assays (Applied Biosystems) using probes for GAPDH, βArr1 and the promoters of p27 and c-Jun were performed in quadruplicate according to manufacturer's protocol on an ABI 7000 Sequence Detection System. Data was analyzed with ABI GeneAmp SDS software (Applied Biosystems). The primers used were as follows: actin 5′-ACG ACC AGA GGC ATA CAG G-3′; 5′-CCC CAT TGA ACA TGG CAT TG-3′, βArr1 5′-GAA GGC AGA CGG AGC AGT TA-3′; 5′-TCT CAA GAG CTT CTG ATC TTC CA-3′. The average threshold cycle (CT) was determined to quantify initial transcript levels and results reported as fold changes.
Statistics.
Statistical analysis was performed using one-way ANOVA followed by a Bonferroni-Dunn test for multiple comparisons within a group or a two-tailed t-test for comparisons between groups, as indicated by the figure legends; p < 0.05 was considered significant and is marked by an asterisk (*). All results are represented as average, with error bars indicating the standard error of the mean. Experiments in vitro were performed at least 3–4 times, with separate litters of mice, to confirm reproducibility and consistency.
Abbreviations
| Shh | = | sonic hedgehog |
| GPCR | = | G-protein coupled receptor |
| βArr1 | = | β-arrestin1 |
| CGNP | = | cerebellar granule cell precursor |
| EGL | = | external granule layer |
Figures and Tables
Figure 1 Shh upregulates βArr1 protein, phosphorylation and nuclear localization. (A) CGNPs were treated with or without Shh as indicated for 48 hours, and lysates were blotted for βArr1, PSer412-βArr1, βArr2 and cyclin D2. Tubulin demonstrates equal protein loading. (B) Quantification of (A). Average fold change for each protein was calculated by normalization to Tubulin levels. Each Shh-mediated fold change was further normalized to the corresponding vehicle sample. βArr1, PSer412-βArr1 and cyclin D2 are significantly increased in Shh treated CGNPs. (C) RNA isolated from vehicle or Shh treated CGNPs was analyzed by qRT-PCR to evaluate βArr1 expression. Shh treatment does not induce βArr1 mRNA as indicated by fold change over β-actin control. (D) CGNPs treated with or without Shh for 24 h prior to treatment with 1 µg/mL cyclopamine. Cells were fixed and stained for βArr1 (red) and nuclei were visualized by DAPI (blue). βArr1 is primarily cytoplasmic in vehicle-treated (left part and inset) and Shh + cyclopamine-treated CGNPs (right part and inset) compared to Shh-treated cells (center part and inset). (E) Percentage of cells expressing βArr1 and percentage of those with nuclear βArr1 from staining in (D) (n > 300 cells in 3 or more fields). There is a significant increase in the number of cells with nuclear βArr1 in the Shh-treated group. (F) Sub-cellular fractionation followed by western blot analysis of vehicle, Shh or Shh plus cyclopamine-treated CGNPs to confirm increased nuclear localization of βArr1 with Shh. In the presence of cyclopamine, total and phospho-βArr1 levels in the nucleus decrease. cJun/Ap1 was used as nuclear protein control and GAPDH as cytoplasmic control. See also Supplemental Figure 1 for verification of βArr1 antibody specificity and analysis of Smoothened antagonist on βArr1.
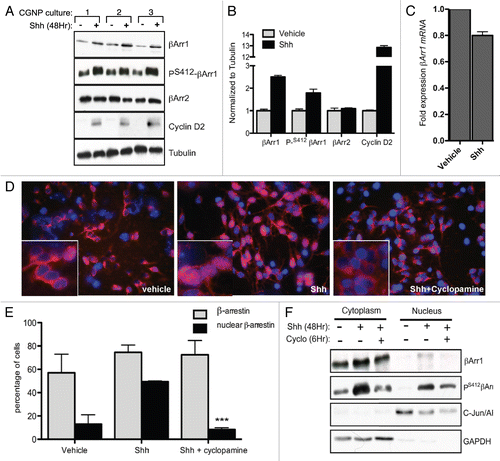
Figure 2 βArr1 knockdown increases Shh-mediated CGNP proliferation while overexpression reduces Ki67 and cyclin D2 levels. (A) CGNPs were infected with a pool of lentiviruses containing sequences targeting βArr1. Cells were fixed, stained for Ki67 and DAPI, and the number of Ki67-positive cells was counted (graph). Lysates were analyzed by western blot to demonstrate βArr1 knockdown and increased cyclin D2 levels. (B) CGNPs were infected with retroviruses containing FLAG-tagged βArr1 and treated with Shh for 48 hours. Cells were fixed, stained for Ki67 and DAPI, and the number of Ki67-positive cells was determined (graph). Lysates were analyzed by western blot to show βArr1 overexpression, as determined by FLAG levels and reduced cyclin D2 levels. Cleaved caspase-3 levels indicate that reduced proliferation is not a result of increased apoptosis. (C) Infection of cerebellar slices with lentiviruses targeting βArr1 causes increased Ki67 staining (red) in the EGL (designated by white line). **/++ or *** indicates statistically significant difference compared to vehicle treated CGNPs (p < 0.005 and p < 0.001 respectively, n = 5). See also Supplemental Figure 2 for analysis of control infections.
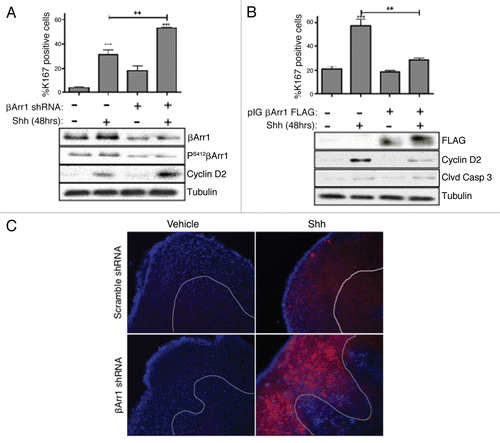
Figure 3 βArr1 phosphorylation at Ser412 is necessary and sufficient to mediate βArr1 accumulation in the nucleus. (A) CGNPs were treated with Shh for 48 h prior to treatment with okadaic acid (OA, 100 nM) for 3 hrs. Western blot analysis shows that OA treatment increases phosphorylation of βArr1 at Ser412, even in the absence of Shh. (B) Treatment of CGNPs with OA for 3 h mediates the translocation of endogenous βArr1 (red) from the cytoplasm to the nucleus in both vehicle and Shh treated cells. Nuclei were visualized with DAPI (blue). (C) CGNPs were infected with βArr1 FLAG retroviruses WT or Ser412 phospho-mutants (S412A or S412D) and were treated with Shh as indicated for 48 h. Cells were then immuno-stained with antibodies against FLAG (red), BrdU (green) and DAPI (blue). Ser-Ala (S412A) mutation prevents βArr1 nuclear localization in Shh treated CGNPs, as determined by FLAG-positive cells (red, middle parts). However, Ser-Asp (S412D) mutants show exclusively nuclear βArr1 in both vehicle and Shh treated CGNPs (arrowheads, right parts). (D) CGNPs infected with wild-type and mutant βArr1 retroviruses were collected for western blot analysis to evaluate FLAG, cyclin D2 and β-tubulin levels. Cyclin D2 is reduced in FLAG βArr1 wild-type- and S412D-infected cells. (E) CGNPs were pulsed with BrdU for 8 h prior to fixation and staining. Cells infected with either βArr1 WT or βArr1 Ser-Asp mutation (βArr1S412D) show reduced proliferation in response to Shh. ** or ***indicates statistically significant difference to vehicle treated CGNPs (p < 0.005 and p < 0.001 respectively, n = 3).
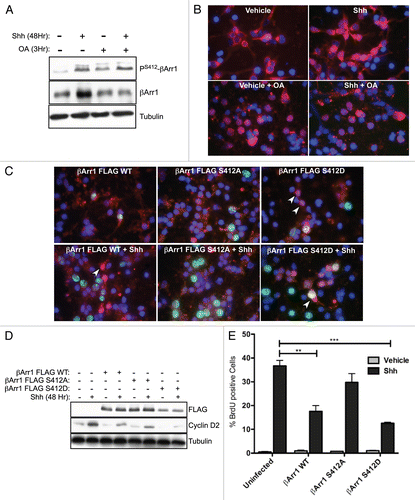
Figure 4 βArr1, p27 and PS412-βArr1 co-localize in vivo and in vitro. (A) Serial sections from PN day 0, 7 and 15 WT mice were stained for either p27, βArr1 or PS412-βArr1, the indicated proteins as described in the methods. Expression of p27 can be seen in the post-mitotic CGNP layer (EGLb) from PN0 and most clearly at PN7. Lower levels of p27 are seen in the proliferative layer of the EGL (EGLa), higher magnification images. βArr1 and PS412-βArr1 are most highly expressed at PN7, with peak nuclear localization occurring in the EGLb (black arrows, higher magnification images). They are also expressed in the remaining EGL of PN15 mice but not in the IGL. (B) CGNPs were cultured in the presence of Shh for 48 h before fixation and stained with βArr1 (red) and p27 (green). Nuclei were visualized by DAPI (blue). Arrowheads indicate cells with nuclear co-localization of βArr1 with p27. See also Supplemental Figure 3 for in vivo analysis of CREB and P-CREB.
Figure 5 Nuclear βArr1 regulates p27 levels. (A) Time course of CGNPs treated with Shh for increasing periods of time shows increased p27 and βArr1 protein levels by western blotting. PS412-βArr1 levels also increase over time, correlating to the Shh- and time-dependent increase in Gli1 transcription (graph). Tubulin is shown as a loading control. (B) RNA was isolated from CGNPs and treated with Shh for the indicated time points and qPCR performed to evaluate p27 expression. Expression level was normalized to that in vehicle-treated CGNPs. p27 mRNA levels increase over time in the presence of Shh, leveling off after 72 h. (C) CGNPs treated with Shh for 48 h were immunostained for p27, and the ratio of p27-positive cells to DAPI-only (i.e., p27-negative) cells was determined; there is no change in the overall number of cells expressing p27. (D and E) CGNPs were infected with lentiviruses expressing shRNAs targeting βArr1 and the effects on p27 were determined. βArr1 knockdown is associated with reduced p27 protein (western blot, D) and mRNA (graph, E). (F) Western blot analysis of CGNPs transduced with βArr1 FLAG retroviruses demonstrates increased p27 levels. * or ***indicates statistically significant difference compared to vehicle treated CGNPs (p < 0.05 and p < 0.001 respectively n = 3).
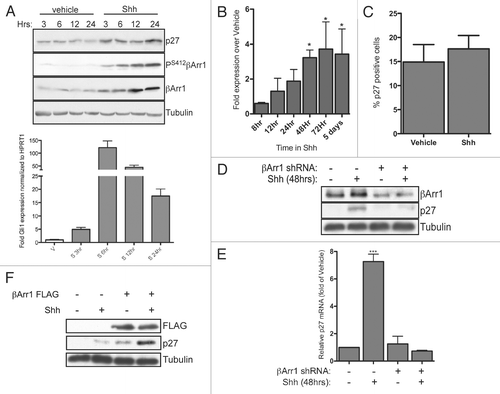
Figure 6 βArr1 is part of an immuno-complex with CREB, and both Shh and βArr1 regulate CREB and p300 binding to the p27 promoter. (A) CGNPs treated with vehicle or Shh for 48 h were collected for ChIP analysis with CREB antibody as described in the methods. Cells treated with cyclopamine were treated for 12 h prior to lysis. Analysis was performed by qPCR and results quantified as fold-over negative control primers. CREB is bound to the p27 but not the C-Jun promoter in response to Shh in CGNPs. (B) CGNPs were treated as above or infected with βArr1-targeting lentiviruses. Cells were collected for ChIP analysis using a p300 antibody and analysis performed by qPCR. p300 is recruited specifically to the p27 promoter in response to Shh, and this is reversed by βArr1 knockdown. (C) CGNP extracts were immunoprecipitated with IgG, CREB or PS412βArr1 antibody and the immunocomplexes were analyzed by western blots for IgG, CREB and PS412βArr1. 10% of the total lysate was loaded as a control. See also Supplemental Figure 4 for ChIP analysis with control antibodies.
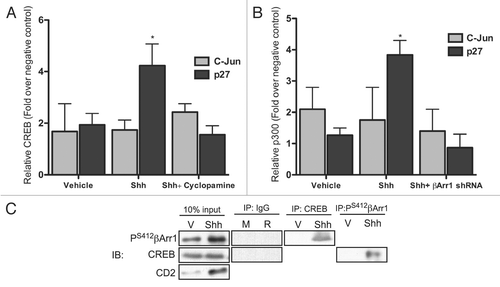
Figure 7 Proposed model for how Shh signaling pathway negatively regulates its mitogenic potential through β-arrestin 1. Activation of Shh signaling leads to cell cycle progression via the upregulation/activation of Gli, N-Myc and IRS1. Shh also activates a negative feedback loop through its induction and phosphorylation of βArr1, mediating its nuclear translocation, where it enhances p27 transcription in complex with CREB and p300. This sets the stage for accumulation of p27 to ultimately drive CGNP cell cycle exit.
Additional material
Download Zip (4.1 MB)Acknowledgements
We thank Betsy Ross and her lab members for helpful discussion in preparation of this manuscript. Marc Caron kindly provided βArr1 plasmids. These studies were funded by NRSA F32 AG030888 from the National Institute on Aging (Susana Parathath) and NIH R01 NS061070 (Anna Marie Kenney).
References
- Eberhart CG. Even cancers want commitment: Lineage identity and medulloblastoma formation. Cancer Cell 2008; 14:105 - 107
- Hatten M, Heintz N. Mechanisms of neural patterning and specification in the developing cerebellum. Annu Rev Neurosci 1995; 18:385 - 408
- Wechsler-Reya RJ, Scott MP. Control of neuronal precursor proliferation in the cerebellum by Sonic Hedgehog. Neuron 1999; 22:103 - 114 [see comments]
- Dahmane N, Ruiz-i-Altaba A. Sonic hedgehog regulates the growth and patterning of the cerebellum. Development 1999; 126:3089 - 3100
- Wallace VA. Purkinje-cell-derived Sonic hedgehog regulates granule neuron precursor cell proliferation in the developing mouse cerebellum. Curr Biol 1999; 9:445 - 448
- Alcedo J, Ayzenzon M, Von Ohlen T, Noll M, Hooper JE. The Drosophila smoothened gene encodes a seven-pass membrane protein, a putative receptor for the Hedgehog signal 1996; 86:221 - 232
- DeCamp DL, Thompson TM, de Sauvage FJ, Lerner MR. Smoothened activates Galphai-mediated signaling in frog melanophores. J Biol Chem 2000; 275:26322 - 26327
- Ho KS, Scott MP. Sonic hedgehog in the nervous system: Functions, modifications and mechanisms. Curr Opin Neurobiol 2002; 12:57 - 63
- Lefkowitz RJ, Rajagopal K, Whalen EJ. New roles for β-arrestins in cell signaling: Not just for deven-transmembrane receptors 2006; 24:643 - 652
- Kang J, Shi Y, Xiang B, Qu B, Su W, Zhu M, et al. A nuclear function of β-arrestin1 in GPCR signaling: Regulation of histone acetylation and gene transcription 2005; 123:833 - 847
- Mo W, Zhang L, Yang G, Zhai J, Hu Z, Chen Y, et al. Nuclear β-arrestin1 functions as a scaffold for the dephosphorylation of STAT1 and moderates the antiviral activity of IFNβ 2008; 31:695 - 707
- DeWire S, Ahn S, Lefkowitz R, Shenoy S. beta-arrestins and cell signaling. Annu Rev Physiol 2007; 69:483 - 510
- Chen W, Ren XR, Nelson CD, Barak LS, Chen JK, Beachy PA, et al. Activity-dependent internalization of Smoothened mediated by {beta}-arrestin 2 and GRK2. Science 2004; 306:2257 - 2260
- Meloni AR, Fralish GB, Kelly P, Salahpour A, Chen JK, Wechsler-Reya RJ, et al. Smoothened signal transduction is promoted by G protein-coupled receptor kinase 2. Mol Cell Biol 2006; 26:7550 - 7560
- Kovacs JJ, Whalen EJ, Liu R, Xiao K, Kim J, Chen M, et al. {beta}-Arrestin-mediated localization of Smoothened to the primary cilium. Science 2008; 320:1777 - 1781
- Wang P, Wu Y, Ge X, Ma L, Pei G. Subcellular localization of beta-Arrestins is determined by their intact N domain and the nuclear export signal at the C terminus. J Biol Chem 2003; 278:11648 - 11653
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 2000; 20:5756 - 5763
- Lin FT, Krueger KM, Kendall HE, Daaka Y, Fredericks ZL, Pitcher JA, et al. Clathrin-mediated endocytosis of the beta-Adrenergic receptor is regulated by phosphorylation/dephosphorylation of beta-Arrestin1. J Biol Chem 1997; 272:31051 - 31057
- Parathath SR, Mainwaring LA, Fernandez LA, Campbell DO, Kenney AM. Insulin receptor substrate 1 is an effector of sonic hedgehog mitogenic signaling in cerebellar neural precursors. Development 2008; 135:3291 - 3300
- Hupfeld CJ, Resnik JL, Ugi S, Olefsky JM. Insulin-induced {beta}-Arrestin1 Ser-412 phosphorylation is a mechanism for desensitization of ERK activation by G{alpha}i-coupled receptors. J Biol Chem 2005; 280:1016 - 1023
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, et al. Development of mice expressing a single D-type cyclin. Genes Dev 2002; 16:3277 - 3289
- Durand B, Raff M. A cell-intrinsic timer that operates during oligodendrocyte development. Bioessays 2000; 22:64 - 71
- Ayrault O, Zindy F, Rehg J, Sherr C, Roussel M. Two tumor suppressors p27Kip1 and Patched-1 collaborate to prevent medulloblastoma. Mol Cancer Res 2009; 7:33 - 40
- Bhatia B, Malik A, Fernandez LA, Kenney A. P27kip1, a double-edged sword in Shh-mediated medulloblastoma: Tumor accelerator and suppressor. Cell Cycle 2010; In press
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003; 130:15 - 28
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA 2003; 100:7331 - 7336
- Hepker J, Wang QT, Motzny CK, Holmgren R, Orenic TV. Drosophila cubitus interruptus forms a negative feedback loop with patched and regulates expression of Hedgehog target genes. Development 1997; 124:549 - 558
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 2000; 20:9055 - 9067
- Jia H, Liu Y, Yan W, Jia J. PP4 and PP2A regulate Hedgehog signaling by controlling Smo and Ci phosphorylation. Development 2009; 136:307 - 316
- Krauss S, Foerster J, Schneider R, Schweiger S. Protein phosphatase 2A and rapamycin regulate the nuclear localization and activity of the transcription factor GLI3. Cancer Res 2008; 68:4658 - 4665
- Sjostrom SK, Finn G, Hahn WC, Rowitch DH, Kenney AM. The cdk1 complex plays a prime role in regulating N-myc phosphorylation and turnover in neural precursors. Dev Cell 2005; 9:327 - 338
- Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. b-Arrestin-dependent formation of 2 adrenergic receptor-Src protein kinase complexes. Science 1999; 283:655 - 661