Abstract
The tumor suppressor p53 provides exquisite protection from cancer by balancing cell survival and death in response to stress. Sustained stress or irreparable damage trigger p53's killer functions to permanently eliminate genetically-altered cells as a potential source of cancer. To prevent the unnecessary loss of cells that could cause premature aging as a result of stem cell attrition, the killer functions of p53 are tightly regulated and balanced against protector functions that promote damage repair and support survival in response to low stress or mild damage. In molecular terms these p53-based cell fate decisions involve protein interactions with cofactors and modifying enzymes, which modulate the activation of distinct sets of p53 target genes. In addition, we demonstrate that part of this regulation occurs at the level of DNA binding. We show that the killer function of p53 requires the four DNA binding domains within the p53 tetramer to interact with one another. These intermolecular interactions enable cooperative binding of p53 to less perfect response elements in the genome, which are present in many target genes essential for apoptosis. Modulating p53 interactions within the tetramer could therefore present a novel promising strategy to fine-tune p53-based cell fate decisions.
Throughout lifetime the cells of our body are continuously exposed to a large variety of environmental and intrinsic hazards that cause damage to the genome. In case these genetic or epigenetic aberrations are replicated and passed on during cell division danger exists that proliferation and survival promoting mutations accumulate so that sooner or later malignant progeny arises posing a threat to the organism as a whole. Early eradication of aspiring cancer cells through activation of an apoptotic cell death program is therefore an efficient means to protect the organism from a full-blown tumor disease. However, considering that moderate damage resulting from mild stress is often reparable, the decision to kill a stressed cell needs to be well-thought-out. Unreflected killing of valuable cells could eventually result in a depletion of stem cell pools and premature aging as a consequence. Every single cell is therefore continuously confronted with the choice: repair and live or die. Too much death poses the risk of aging, too little death the risk of cancer. Balancing these risks for the benefit of the organism is a central task of the tumor suppressor protein p53. Summoned under conditions of stress, p53 functions like a hub in a highly-connected intracellular signaling network to integrate a plethora of inputs from the inside and outside of the cell to trigger a well-balanced cell fate decision.Citation1
The Choice of Targets
How p53 executes this cell fate decision is therefore a question of considerable biomedical interest (). Since it is known that p53 functions as a sequence-specific DNA binding transcription factor, tremendous efforts have been made in the last decade to identify the p53-regulated targets in the genome that execute the appropriate cell fate responses.Citation2 The induction of a transient cell cycle arrest that allows for damage repair depends critically on the genes p21 (CDKN1A), 14-3-3σ (SFN) and GADD45A, with the first being crucial for cell cycle arrest in the G1 phase and the latter two for arrest in G2.Citation3 In the case of prolonged damage p53-mediated transactivation of the sestrins (SESN1 and SESN2) causes inhibition of mTOR signaling and helps to maintain the arrest reversible, while activation of mTOR under these conditions triggers a shift to cell cycle exit termed senescence.Citation4–Citation8 Another way for p53 to permanently stop cell proliferation without compromising cell viability is induction of differentiation.Citation9 For example, differentiation follows experimental reactivation of p53 in a murine model of Ras-dependent liver cancer or genotoxic stress induced-p53 activation in acute myeloid leukemia cells bearing an activated Ras oncogene.Citation10,Citation11
Only when cells have encountered sustained and irreparable damage that is incompatible with further survival, p53 shifts to the most extreme and irrevocable antiproliferative response—apoptotic cell death.Citation12,Citation13 In line with the importance of this activity numerous studies have identified many different proapoptotic p53 target genes including BAX, FAS, TP53I3 (PIG3), TNFRSF10B (KILLER/DR5), LRDD (PIDD), P53AIP1, APAF1, PERP, PMAIP1 (NOXA) and BBC3 (PUMA)—to name just the most commonly cited. Of note, accumulating evidence shows that p53-induced apoptosis does not only require activation of these proapoptotic target genes but also involves transcription-independent functions of p53 in the cytoplasm.Citation14–Citation16
Not enough, a recent review lists a total of 129 transcriptional targets of p53 with experimentally validated binding sites and global approaches using chromatin-immunoprecipitation in conjunction with microarrays (ChIP-chip) or massively parallel sequencing reveal increasingly more sites within the genome that are bound by p53.Citation2,Citation17–Citation20 The majority of these genomic sequences contain a common consensus motif to which p53 binds with high affinity and specificity. This motif is composed of two decameric half-sites RRR CWW GYY Y, where R is a purine, Y a pyrimidine and W is either adenine (A) or thymine (T), separated by a spacer, usually composed of 0–21 base pairs.Citation2,Citation21,Citation22 Considering that most of the p53-regulated genes contain response elements that more or less concur with the consensus motif, it remains a mystery how p53 can distinguish between the various genomic binding sites with their associated target genes and selectively activate a subset of them to drive cell fate into the desired direction.Citation2,Citation12,Citation13,Citation23–Citation25
The Role of Cofactor Recruitment
One way to target p53 to the promoters of specific target genes is through interaction with partner proteins. Considering the vast amount of p53 binding proteins described so far we will focus on a small fraction with a clear role in redirecting p53 towards a specific cellular outcome.
For example, the proteins of the ASPP family have turned out to be potent regulators of p53's apoptotic function.Citation26 The apoptosis promoting members, ASPP1 and ASPP2, specifically stimulate p53 binding to the promoters of the proapoptotic target genes BAX and PIG3 but not to the promoters of p21 or MDM2.Citation27 On the other hand, the inhibitory ASPP family member, iASPP, competes with the other ASPP proteins and blocks p53-mediated apoptosis.Citation28 Interestingly, iASPP discriminates between two common polymorphic variants of p53 that differ at codon 72.Citation29 iASPP preferentially binds the proline 72 (P72) variant and inhibits its activity, providing an intriguing explanation for why the arginine 72 (R72) variant is a more potent inducer of apoptosis than the P72 variant.
Another family of proteins that regulates p53 is the Brn3 family of POU domain transcription factors that interact with the p53 DNA binding domain (DBD). While Brn3a stimulates p53-dependent transcription of p21 and inhibits its ability to activate the BAX and NOXA promoters, Brn3b functions in the opposite manner by assisting p53 to activate BAX but not p21 expression.Citation30–Citation32
The zinc-finger protein Hzf is a target gene of p53 and by interacting with the p53 DBD regulates its target selectivity.Citation33,Citation34 Hzf promotes p53 binding to the p21 and 14-3-3σ promoters early after DNA damage. Inactivation of Hzf—experimentally or by degradation in response to sustained DNA damage—prevents p53 binding to these promoters and allows relocalization to the response elements in the proapoptotic target genes BAX, PUMA, NOXA and PERP.Citation35 A notable exception to the regulation of target selectivity is MDM2 which appears to be unaffected by Hzf.
Similarly, Miz1 also interacts with the DNA binding domain of p53 to prevent the activation of the proapoptotic targets BAX and PUMA.Citation36 Together with Miz1 being a potent transactivator of p21 expression this results in promotion of cell survival. c-Myc via interaction with Miz1 suppresses p21 induction by p53 and thus switches the p53-response from cytostatic to apoptotic.Citation36,Citation37
The Role of Post-Translational Modifications
Discriminatory effects on target selectivity can also be exerted by interacting proteins that modulate p53's DNA binding properties via covalent post-translational modifications including phosphorylation, acetylation, methylation, ubiquitylation, neddylation, sumoylation and even addition of N-acetyl glucosamine. Here we will highlight those modifications that most prominently influence p53's promoter selectivity.
Among the phosphorylation sites, serine 46 (S46) has clear discriminatory function for p53 as a transcriptional activator. p53 is phosphorylated at this residue by homeodomain interacting protein kinase 2 (HIPK2), dual-specificity tyrosine-phosphorylation-regulated kinase 2 (DYRK2), AMPK, protein kinase C delta or p38 mitogen activated protein kinase in response to severe cellular damage.Citation38–Citation44 S46-phosphorylated p53 is recognized by the peptidyl-prolyl cis/trans isomerase Pin1 leading to dissociation of the apoptosis-inhibiting protein iASPP from p53 and induction of apoptosis via, for example, transactivation of p53AIP1, a proapoptotic factor that promotes the release of mitochondrial cytochrome c during apoptosis.Citation45,Citation46
While numerous studies have implicated acetylation of lysine residues in the C-terminus of p53 as being important for p53's transcriptional activity in general, acetylation of lysine 120 (K120) in the DNA binding domain by the MYST family histone acetyl transferases hMOF and Tip60 specifically results in increased binding to proapoptotic targets like BAX and PUMA while the nonapoptotic targets p21 and MDM2 remain unaffected.Citation47,Citation48 On the other hand, acetylation of lysine 320 (K320) by the transcriptional coactivator p300/CBP-associated factor (PCAF) predisposes p53 to activate p21 and decreases its ability to induce proapoptotic target genes. Cells ectopically expressing a mutant p53 where K320 is mutated to glutamine (K320Q) to mimic acetylation, display reduced apoptosis after some forms of DNA damage.Citation49 Vice versa K317R (corresponding to human K320R) knockin mice, where K317 acetylation is missing, consistently display increased apoptosis and higher expression of relevant target genes in several cell types.Citation50 However, K320 is not only a target for acetylation but it is also ubiquitylated by the zinc-finger protein E4F1.Citation51 This modification facilitates p53-dependent activation of p21 and Cyclin G1 expression without affecting the expression of the proapoptotic gene NOXA, overall resulting in reduced p53-mediated cell death in response to UV.
p53-mediated cell cycle arrest is also favored following methylation of at least two arginine residues (R333 and R335) by the arginine methyltransferase PRMT5.Citation52,Citation53 Consistently, depletion of PRMT5 by siRNA in cancer cell lines leads to increased apoptosis following p53 activation.
The Role of DNA Binding Cooperativity
Together these data highlight the complexity of how p53 binding proteins modulate—in a covalent or non-covalent manner—the DNA binding properties of p53 to influence the cell fate decision in favor of survival or death. Despite this substantial body of knowledge, very little is known about the molecular details. Even a structurally simple modification such as the acetylation of K120 does not directly explain why p53's specificity for certain promoter sequences changes and p53 is redirected to proapoptotic target genes. The recent progress in solving the 3D structures of p53 in contact to DNA, however, promises that it will be possible to gain a clearer view of how p53's sequence specificity is regulated by either modifications or through association with interaction partners.
One striking result of the recent structural studies was that the p53 molecules within the tetramer, which assembles as a dimer of dimers on two cognate half sites in the DNA, do not only interact through their oligomerization domains but also tightly and specifically via their DBDs. Nuclear magnetic resonance (NMR) spectroscopy, X-ray crystallography and computational studies indicate that the oppositely charged glutamate (E180) and arginine (R181) residues in the short helix H1 of the DBDs engage in intermolecular interactions to form a so-called double salt bridge as part of the DBD dimer interface.Citation54–Citation58 This dimer interface was further confirmed when Fersht and colleagues succeeded in obtaining a structure of full-length p53 bound to DNA by using a combination of small angle X-ray scattering, NMR and electron microscopy.Citation59 In vitro studies with recombinant p53 DBDs carrying targeted mutations in the critical residues highlighted that the dimer interface is crucial for a long-known property of p53, called DNA binding cooperativity, which simply means that four interacting p53 subunits cooperate to bind DNA better than four non-interacting subunits.Citation54
To better understand the relevance of this dimer interface and the resulting DNA binding cooperativity for the biology of p53, we analyzed the consequences of expressing dimer interface mutants of full-length p53 in cells.Citation9 For this, we generated a panel of p53 expression constructs with mutations in the H1 helix residues E180 and R181 that reduced or increased interactions between neighboring p53 subunits (). This mutant panel covers the whole cooperativity range from barely detectable to super-physiological DNA binding cooperativity.
The expression of these mutants in p53-null cell lines resulted in distinct biological outcomes. Low cooperativity mutants induced p21 and MDM2 expression leading to a selective cell cycle arrest while high cooperativity mutants activated BAX, NOXA and other proapoptotic target genes causing cell death. Likewise, when p53 function in p53-/- HCT116 cells was restored with the panel of cooperativity mutants at physiological expression levels the extent of apoptosis induced by genotoxic stress correlated directly with DNA binding cooperativity, indicating that p53's killing function strongly depends on its ability to bind DNA in a cooperative manner.
DNA Binding Cooperativity Enables Binding to Imperfect Binding Elements
One hypothesis was that the binding of p53 to apoptotic target genes requires higher levels of cooperativity than binding to survival genes. To test this we compared the genomic binding profiles of a low (EL) and high (RE) cooperativity mutant by chromatin immunoprecipitation coupled to the unbiased detection of binding sites (BS) with genome-wide promoter tiling microarrays (ChIP-chip). Bioinformatic analysis combining ChIP-chip results with experimental validation rates determined by ChIP-qPCR revealed approximately 1,250 BS for the high cooperativity mutant RE in the promoter regions of the human genome (). Interestingly, the low cooperativity mutant EL showed only approximately 100 BS, which represent a subset of the RE BS. This led us to the conclusion that the DNA binding cooperativity serves to increase the number of BS in the genome.
To understand the differences between BS that are strongly dependent on cooperativity (RE-only BS) and those, which are bound independently of cooperativity (common BS of EL and RE), we performed motif analysis on experimentally validated “common EL/RE” and “RE-only” BS. De novo motif discovery as well as screening the bound sequences for p53 binding motifs of the TRANSFAC database revealed that common EL/RE but not RE-only BS were strongly enriched for the 20-meric p53 full-site motif (V$P53_01) ( and D). In contrast, the decameric p53 half-site motif (V$P53_02) was identified with equal frequency in both sets of BS. Nevertheless, in both cases, the average motif score as a measure of similarity to the consensus was significantly lower among the validated RE-only sites (), suggesting that RE tolerates mismatches to the consensus binding site better than EL. Another explanation for the absence of 20-meric full sites in RE-only sequences—despite the presence of decameric half-sites—are spacer elements that separate two half-sites. Applying a spacer-tolerant algorithm, we indeed identified spacer-containing full sites much more frequently in RE-only than in common EL/RE sequences (). Together, these results indicate that the sequence requirements for recruitment of RE are less stringent than for EL and that DNA binding cooperativity increases the number of binding sites in the genome by enabling binding to imperfect, i.e., mismatch- and spacer-containing, response elements.
A Role for Cooperativity in Binding and Activating Imperfect Binding Sites
To experimentally confirm that the extent of DNA binding cooperativity determines binding to imperfect response elements, we performed electrophoretic mobility shift assays. It has been previously shown that even subtle changes in the core CWWG sequence of a RRR CWWG YYY half-site can dramatically reduce DNA binding affinity, which is known to be maximal for CATG.Citation2 Mutation of the invariable C or G nucleotides typically results in a complete loss of binding activity, whereas changing of the central AT to AA, TT or TA reduces binding only.Citation2 Consistently, binding of wild-type p53 and even more pronounced of low cooperativity mutants (RR, LR and EL) was reduced when the core CATG sequence was mutated to CAAG, CTTG or CTAG (). In contrast, the high cooperativity mutants RE and EE + RR bound these non-CATG sequences even better than wild-type p53. Similarly, spacer elements in between the two half-sites completely abolished the binding of low cooperativity mutants whereas high cooperativity mutants were still bound (). H1 helix interactions therefore strongly influence the sequence specificity of the p53 tetramer in the way that high cooperativity renders p53 tolerant to deviations from the consensus sequence.
To investigate whether cooperativity also affects transactivation of target genes in a way predicted by the DNA binding experiments, we analyzed luciferase reporter plasmids containing the consensus-like 5′ p53 binding site of the p21 promoter in comparison to derivative constructs containing central CTAG sequences and/or variable spacers (). Activation of these reporters by our panel of cooperativity mutants was measured following transfection into p53-null H1299 cells. The parental promoter construct— with central CATG sequence and without any spacer—yielded high levels of reporter activity and was preferentially activated by low cooperativity mutants (). Mutation of the central CATG to CTAG in both half-sites as well as the insertion of a 5 or 14 bp spacer reduced the maximal activity of the reporter (). However, this decrease primarily affected the transactivation by low cooperativity mutants so that the difference between low and high cooperativity mutants became less apparent (). In fact, insertion of a 14 bp spacer rendered the promoter with a CATG core independent of cooperativity so that all p53 H1 helix mutants induced equal reporter activity levels (). By combining a central CTAG sequence with a spacer insertion we even obtained reporters that were preferentially induced by high cooperativity mutants ( and D). Together these experiments illustrate that the level of DNA binding cooperativity determines which promoter sequences are activated by p53.
The Consensus Sequence Binding-Transactivation Paradox
Curiously, high cooperativity mutants often bound perfect consensus-like response elements at least equally well if not even stronger than low cooperativity mutants, but failed to efficiently transactivate reporter constructs made up of these binding sites. Although not fully understood at present, we can envision two possible mechanisms. First, because high cooperativity mutants bind to many more sites in the genome than low cooperativity mutants, essential cofactors that might be present in limiting amounts could be sequestered, so that the local availability of these factors on a given promoter might be insufficient to support high expression levels of the target gene. This idea is experimentally supported by our data showing that coexpression of a high cooperativity mutant also limits transactivation in a heterologous reporter system, which depends on the transactivation domain but not the DNA binding domain of p53.Citation9 Second, it still remains unclear how p53 and other transcription factors (TF) efficiently drive a promoter to maturation.Citation60–Citation62 Many different chromatin-modifying enzymes and chromatin remodellers have been identified as essential players involved in this transactivation process.Citation63,Citation64 In one scenario, TFs stably associate with a binding site in the promoter and serve as a docking site for the various cofactors that one after the other are recruited to the promoter for its activation. In a contrasting model, various different preformed TF-cofactor complexes exist in the nucleoplasm and the TF functions as a shuttling factor to transport these factors to the target gene promoters.Citation65–Citation67 In the latter model, stable association of p53 with the promoter DNA (as in the case of high cooperativity mutants on consensus binding sites) could compromise the hypothetical shuttling function and be detrimental to the transactivation process. Therefore, an efficient shuttling of highly cooperative p53 would only be possible on imperfect, low-affinity binding sites. No matter which model applies, excessively high levels of cooperativity prevent efficient transactivation of genes with perfect p53 binding elements so that high cooperativity contributes to shifting the expression profile to target genes with imperfect binding sites.
Imperfect binding sites are enriched in proapoptotic target genes.
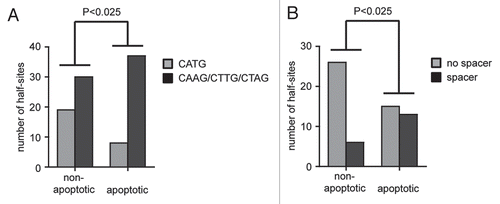
Importantly, there is evidence that low-affinity and spacer-containing sequences are more common in proapoptotic than in nonapoptotic genes, which could explain that the apoptotic potential of p53 correlates with the level of DNA binding cooperativity. It has been known for a while that the cellular level of p53 can dictate the response of the cell such that lower levels of p53 result in arrest whereas higher levels result in apoptosis.Citation68 It has therefore been hypothesized that only high levels of p53 protein, for example following stabilization in response to massive DNA damage, allow for sufficient binding to proapoptotic target genes, which in many cases contain p53 binding elements that only poorly resemble the consensus binding sequence and which—compared to response elements in cell cycle arrest targets—show very little evolutionary conservation.Citation69 To investigate whether the imperfect response elements in proapoptotic target genes resemble the binding sequences that we found to be preferentially bound by high cooperativity mutants, we analyzed 60 p53 binding sites found in 39 experimentally validated bona fide p53 target genes (Sup. Table 1).Citation70 The p53 response elements in non-apoptotic genes were indeed significantly enriched for the half-site RRR CATG YYY, whereas central CAAG, CTTG or CTAG sequences as well as spacers between the two half-sites were significantly more common in the proapoptotic genes (). Our study therefore provides the first direct experimental evidence that the activation of the apoptosis program indeed requires p53 binding to imperfect binding sites, which are overrepresented in the promoters of many known proapoptotic target genes, and that this depends on the cooperative nature of DNA binding by the p53 tetramer.
Open Questions
Considering the relevance of DNA binding cooperativity for binding and activation of proapoptotic target genes, it can be hypothesized that known p53 binding proteins or post-translational modifications that affect p53-based cell fate decisions act via modulating this cooperativity. For example, chromatin-associated factors only present on proapoptotic promoters could be envisioned to attach to p53 and stimulate DBD interactions to allow a more stable binding to the imperfect binding sequences in these promoters. So far, direct evidence for this is missing and will be difficult to obtain, because the cooperativity status of p53, i.e., the interaction strength of neighboring p53 subunits in a p53 tetramer, cannot be easily measured in living cells. However, there is some indirect evidence that at least a few of the known apoptosis-promoting factors might function via modulation of cooperativity.Citation9 First of all, known apoptosis-promoting conditions such as ectopic expression of ASPP2 or the apoptosis-enhancing mutation of serine 46 to phenylalanine appear to be less effective when cooperativity is impaired. Second, ASPP2 was able to increase apoptosis induced by low cooperativity mutants but could not further increase the apoptotic function of the engineered high cooperativity p53 “EE+RR”, suggesting that ASPP2 binding to the p53 DBD—possibly in a hit-and-run mechanism— enhances cooperativity to enable p53 to bind to proapoptotic target genes. While it is clear that cooperativity is essential for high-level induction of apoptosis by p53, it remains to be elucidated, whether an increase in cooperativity mediates the proapoptotic activity of p53-stimulating cofactors or modifying enzymes.
Importantly, mutations that reduce cooperativity by interfering with H1 helix interactions are found in human tumors. Apart from many somatic mutations affecting residues E180 and R181, even families with Li Fraumeni-like syndrome carrying germline mutations in the H1 helix have been described. Interestingly, the R181H mutation (EH) has been identified very early in a family with familial breast carcinoma, but was excluded as a cancer-promoting mutation in part because the mutant protein retained the ability to suppress proliferation of p53-null Saos-2 cells in culture.Citation71 Nevertheless, similar germline mutations (R181C and R181L) have been found in other families. We confirmed that these p53 mutant proteins were indeed able to induce cell arrest, but showed strongly impaired apoptotic activity.Citation9 This implies that H1 helix mutations cause a loss of DNA binding cooperativity resulting in an increased cancer risk. However, formal confirmation, which could be provided by the analysis of cooperativity mutant mice, remains to be obtained. In summary, cooperativity appears to be essential for both p53's apoptotic activity and its tumor suppressor function.
Figures and Tables
Figure 1 DNA binding cooperativity—a new variable in the p53-based cell fate decision. Posttranslational modifications of p53: P, phosphorylation; Ac, acetylation; me, methylation; Ubi, ubiquitylation; Nedd, neddylation.
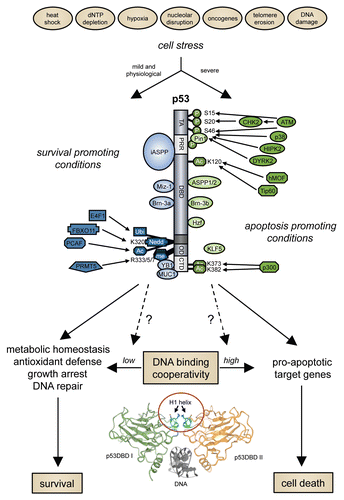
Figure 2 Role of cooperativity for DNA binding of p53 in the human genome. (A) Schematic representation of the dimerization patterns of wild-type p53 and the H1 helix mutants used in this study. The small insert shows the 3D structure of the double salt bridge in the wild-type molecule. To disrupt the intradimer interface we introduced modest charge-neutralizing (E180→L “LR” and R181→L “EL”) and more severe charge-inverting (E180→R “RR” and R181→E “EE”) mutations into the H1 helix of the full-length p53 molecule. The short names denote the amino acid sequence at positions 180 and 181 in the mutant proteins, e.g., “ER” for E180, R181 in the wild-type. To assure that functional defects are truly due to defective core domain interactions and are not caused by structural misfolding of the core domain or disturbed interaction with other cellular proteins, we also introduced the two most severe mutations E180R and R181E together into a single p53 molecule (double mutant E180R, R181E “RE”) and used the two complementing mutants “EE” and “RR” in functional rescue studies. (B) p53 DNA binding cooperativity determines the number of binding sites in the genome. The number of binding sites was estimated by bioinformatic analysis combining ChIP-chip results with experimental validation rates determined by ChIP-qPCR.Citation9 (C) De novo motif discovery in validated common EL/RE and RE-only binding sequences. Twenty-meric and decameric consensus motifs are shown for comparison. (D and E) Frequency and average motif scores of the TRANSFAC motifs V$P53_01 (full site), V$P53_02 (half-site) and V$E2F_01 (E2F site as a control) in validated common EL/RE and RE-only binding sequences. Results are presented as the mean ± SD. (F) Distribution of spacer lengths in validated common EL/RE and RE-only binding sequences as determined by the spacer-tolerant p53MH algorithm.
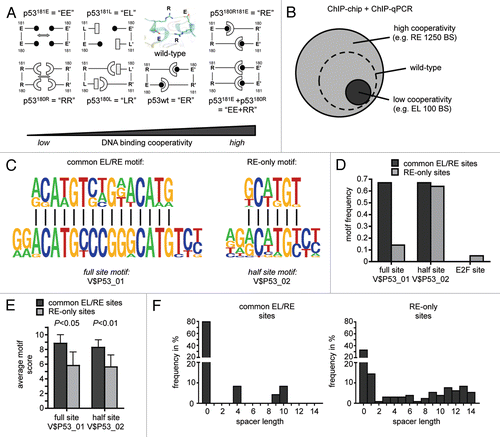
Figure 3 Impact of DNA binding cooperativity on sequence selectivity of p53. (A) Shown are electrophoretic mobility shift assays (EMSA) for DNA binding of in vitro translated wild-type p53 and the indicated H1 helix mutants to dsDNA oligonucleotides (5′-GGG AGC TTA GGC WWG TCT AGG CWW GTC TA-3′) with WW denoting AT, AA, TT or TA sequences in the center of each half site. EMSAs were performed as previously described.Citation9,Citation72 Compared to H1 helix mutants with reduced DNA binding cooperativity (EE, RR, LR, EL), mutants with increased DNA binding cooperativity (RE and EE+RR) revealed an increased ability to bind the lower affinity non-CATG sequences. (B) Same as in (A) using dsDNA oligonucleotides containing the 5′ p53 binding site in the p21 promoter (5′-TCT GGC CGT CAG GAA CATG TCC (N)1–14 CAA CATG TTG AAG CTC TGG CAT A-3′) with increasing central spacer sequences (N)1–14. The high cooperativity mutant (RE) showed an increased ability to bind the spacer-containing motifs, while the low cooperativity mutant (EL) was largely unable to bind these spacer-containing elements.
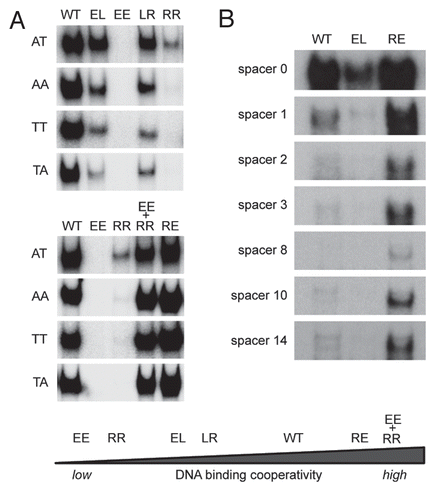
Figure 4 Impact of DNA binding cooperativity on sequence selectivity of transactivation. (A) Shown are p53 binding sequences that differ from the consensus sequence with respect to the CATG in the core of a half-site (bold) and with respect to spacer length (underlined). (B) Luciferase reporter assays. Single copies of the sequences in (A) were cloned into pGL4.23[luc2/minP] and tested for transactivation by the indicated p53 cooperativity mutants. Firefly luciferase activity was measured 48 hours following co-transfection of 100 ng reporter plasmid and 5 ng p53 expression plasmid (pCMVneo-BamHI) into p53-null H1299 cells. The p53 mutants are shown in the order of increasing DNA binding cooperativity. Mean ± SD. (C) Shown is the maximum p53-induced reporter activation for the different promoter sequences. (D) Shown is the ratio of the reporter activities induced by “EE + RR” (high cooperativity) and “RR” (low cooperativity) for the different promoter sequences.
![Figure 4 Impact of DNA binding cooperativity on sequence selectivity of transactivation. (A) Shown are p53 binding sequences that differ from the consensus sequence with respect to the CATG in the core of a half-site (bold) and with respect to spacer length (underlined). (B) Luciferase reporter assays. Single copies of the sequences in (A) were cloned into pGL4.23[luc2/minP] and tested for transactivation by the indicated p53 cooperativity mutants. Firefly luciferase activity was measured 48 hours following co-transfection of 100 ng reporter plasmid and 5 ng p53 expression plasmid (pCMVneo-BamHI) into p53-null H1299 cells. The p53 mutants are shown in the order of increasing DNA binding cooperativity. Mean ± SD. (C) Shown is the maximum p53-induced reporter activation for the different promoter sequences. (D) Shown is the ratio of the reporter activities induced by “EE + RR” (high cooperativity) and “RR” (low cooperativity) for the different promoter sequences.](/cms/asset/05218fd9-d50b-49ab-b998-8fa03a7e678e/kccy_a_10913595_f0004.gif)
Figure 5 Proapoptotic target genes in a list of experimentally validated p53 target genes (Sup. Table 1) are enriched for p53 response elements with spacers and non-CATG core sequences. Statistical significance was calculated by Pearson's Chi-square test. (A) Number of half-sites with a central CATG versus CAAG, CTTG or CTAG sequence in non-versus proapoptotic target genes. (B) Presence of spacer containing response elements in non-versus proapoptotic target genes.
Additional material
Download Zip (34.2 KB)Acknowledgements
We thank Andreas Rosenwald, Caroline Kisker and Martin Eilers for their cooperation and all members of the laboratory—in particular Rasa Beinoraviciute-Kellner, Markus Sauer and Marie Zeitlinger—for their contribution to this study. This work was funded by grants to T.S. from the Deutsche Forschungsgemeinschaft (Transregio TR17 Teilprojekt B2, Klinische Forschergruppe KFO210 STI 182/3-1), Deutsche Krebshilfe (107904), LOEWE research program, “Tumor & Inflammation” and von Behring-Röntgen-Stiftung (57-0012).
References
- Vogelstein B, Lane D, Levine AJ. Surfing the p53 network. Nature 2000; 408:38 - 44
- Riley T, Sontag E, Chen P, Levine A. Transcriptional control of human p53-regulated genes. Nat Rev Mol Cell Biol 2008; 9:402 - 412
- Levine AJ, Oren M. The first 30 years of p53: growing ever more complex. Nat Rev Cancer 2009; 9:749 - 758
- Budanov AV, Karin M. p53 target genes sestrin1 and sestrin2 connect genotoxic stress and mTOR signaling. Cell 2008; 134:451 - 460
- Korotchkina LG, Demidenko ZN, Gudkov AV, Blagosklonny MV. Cellular quiescence caused by the Mdm2 inhibitor nutlin-3A. Cell Cycle 2009; 8:3777 - 3781
- Steelman LS, McCubrey JA. Intriguing novel abilities of Nutlin-3A: induction of cellular quiescence as opposed to cellular senescence—implications for chemotherapy. Cell Cycle 2009; 8:3634 - 3635
- Demidenko ZN, Korotchkina LG, Gudkov AV, Blagosklonny MV. Paradoxical suppression of cellular senescence by p53. Proc Natl Acad Sci USA 2010; 107:9660 - 9664
- Korotchkina LG, Leontieva OV, Bukreeva EI, Demidenko ZN, Gudkov AV, Blagosklonny MV. The choice between p53-induced senescence and quiescence is determined in part by the mTOR pathway. Aging (Albany NY) 2010; 2:344 - 352
- Schlereth K, Beinoraviciute-Kellner R, Zeitlinger MK, Bretz AC, Sauer M, Charles JP, et al. DNA binding cooperativity of p53 modulates the decision between cell cycle arrest and apoptosis. Mol Cell 2010; 38:356 - 368
- Xue W, Zender L, Miething C, Dickins RA, Hernando E, Krizhanovsky V, et al. Senescence and tumour clearance is triggered by p53 restoration in murine liver carcinomas. Nature 2007; 445:656 - 660
- Meyer M, Rubsamen D, Slany R, Illmer T, Stabla K, Roth P, et al. Oncogenic RAS enables DNA damage- and p53-dependent differentiation of acute myeloid leukemia cells in response to chemotherapy. PLoS One 2009; 4:7768
- Aylon Y, Oren M. Living with p53, dying of p53. Cell 2007; 130:597 - 600
- Murray-Zmijewski F, Slee EA, Lu X. A complex barcode underlies the heterogeneous response of p53 to stress. Nat Rev Mol Cell Biol 2008; 9:702 - 712
- Green DR, Kroemer G. Cytoplasmic functions of the tumour suppressor p53. Nature 2009; 458:1127 - 1130
- Morselli E, Galluzzi L, Kepp O, Kroemer G. Nutlin kills cancer cells via mitochondrial p53. Cell Cycle 2009; 8:1647 - 1648
- Vaseva AV, Marchenko ND, Moll UM. The transcription-independent mitochondrial p53 program is a major contributor to nutlin-induced apoptosis in tumor cells. Cell Cycle 2009; 8:1711 - 1719
- Cawley S, Bekiranov S, Ng HH, Kapranov P, Sekinger EA, Kampa D, et al. Unbiased mapping of transcription factor binding sites along human chromosomes 21 and 22 points to widespread regulation of noncoding RNAs. Cell 2004; 116:499 - 509
- Hearnes JM, Mays DJ, Schavolt KL, Tang L, Jiang X, Pietenpol JA. Chromatin immunoprecipitation-based screen to identify functional genomic binding sites for sequence-specific transactivators. Mol Cell Biol 2005; 25:10148 - 10158
- Smeenk L, van Heeringen SJ, Koeppel M, van Driel MA, Bartels SJ, Akkers RC, et al. Characterization of genome-wide p53-binding sites upon stress response. Nucleic Acids Res 2008; 36:3639 - 3654
- Wei CL, Wu Q, Vega VB, Chiu KP, Ng P, Zhang T, et al. A global map of p53 transcription-factor binding sites in the human genome. Cell 2006; 124:207 - 219
- Funk WD, Pak DT, Karas RH, Wright WE, Shay JW. A transcriptionally active DNA-binding site for human p53 protein complexes. Mol Cell Biol 1992; 12:2866 - 2871
- el-Deiry WS, Kern SE, Pietenpol JA, Kinzler KW, Vogelstein B. Definition of a consensus binding site for p53. Nat Genet 1992; 1:45 - 49
- Das S, Boswell SA, Aaronson SA, Lee SW. p53 promoter selection: choosing between life and death. Cell Cycle 2008; 7:154 - 157
- Blattner C. Regulation of p53: the next generation. Cell Cycle 2008; 7:3149 - 3153
- Georges SA, Chau BN, Braun CJ, Zhang X, Dobbelstein M. Cell cycle arrest or apoptosis by p53: are microRNAs-192/215 and -34 making the decision?. Cell Cycle 2009; 8:680 - 681
- Trigiante G, Lu X. ASPP [corrected] and cancer. Nat Rev Cancer 2006; 6:217 - 226
- Samuels-Lev Y, O'Connor DJ, Bergamaschi D, Trigiante G, Hsieh JK, Zhong S, et al. ASPP proteins specifically stimulate the apoptotic function of p53. Mol Cell 2001; 8:781 - 794
- Bergamaschi D, Samuels Y, O'Neil NJ, Trigiante G, Crook T, Hsieh JK, et al. iASPP oncoprotein is a key inhibitor of p53 conserved from worm to human. Nat Genet 2003; 33:162 - 167
- Bergamaschi D, Samuels Y, Sullivan A, Zvelebil M, Breyssens H, Bisso A, et al. iASPP preferentially binds p53 proline-rich region and modulates apoptotic function of codon 72-polymorphic p53. Nat Genet 2006; 38:1133 - 1141
- Perez-Sanchez C, Budhram-Mahadeo VS, Latchman DS. Distinct promoter elements mediate the co-operative effect of Brn-3a and p53 on the p21 promoter and their antagonism on the Bax promoter. Nucleic Acids Res 2002; 30:4872 - 4880
- Budhram-Mahadeo VS, Bowen S, Lee S, Perez-Sanchez C, Ensor E, Morris PJ, et al. Brn-3b enhances the proapoptotic effects of p53 but not its induction of cell cycle arrest by cooperating in trans-activation of bax expression. Nucleic Acids Res 2006; 34:6640 - 6652
- Hudson CD, Morris PJ, Latchman DS, Budhram-Mahadeo VS. Brn-3a transcription factor blocks p53-mediated activation of proapoptotic target genes Noxa and Bax in vitro and in vivo to determine cell fate. J Biol Chem 2005; 280:11851 - 11858
- Sugimoto M, Gromley A, Sherr CJ. Hzf, a p53-responsive gene, regulates maintenance of the G2 phase checkpoint induced by DNA damage. Mol Cell Biol 2006; 26:502 - 512
- Sharma S, Dimasi D, Higginson K, Della NG. RZF, a zinc-finger protein in the photoreceptors of human retina. Gene 2004; 342:219 - 229
- Das S, Raj L, Zhao B, Kimura Y, Bernstein A, Aaronson SA, et al. Hzf Determines cell survival upon genotoxic stress by modulating p53 transactivation. Cell 2007; 130:624 - 637
- Miao L, Song Z, Jin L, Zhu YM, Wen LP, Wu M. ARF antagonizes the ability of Miz-1 to inhibit p53-mediated transactivation. Oncogene 2010; 29:711 - 722
- Herold S, Wanzel M, Beuger V, Frohme C, Beul D, Hillukkala T, et al. Negative regulation of the mammalian UV response by Myc through association with Miz-1. Mol Cell 2002; 10:509 - 521
- D'Orazi G, Cecchinelli B, Bruno T, Manni I, Higashimoto Y, Saito S, et al. Homeodomain-interacting protein kinase-2 phosphorylates p53 at Ser 46 and mediates apoptosis. Nat Cell Biol 2002; 4:11 - 19
- Rinaldo C, Prodosmo A, Mancini F, Iacovelli S, Sacchi A, Moretti F, et al. MDM2-regulated degradation of HIPK2 prevents p53Ser46 phosphorylation and DNA damage-induced apoptosis. Mol Cell 2007; 25:739 - 750
- Taira N, Nihira K, Yamaguchi T, Miki Y, Yoshida K. DYRK2 is targeted to the nucleus and controls p53 via Ser46 phosphorylation in the apoptotic response to DNA damage. Mol Cell 2007; 25:725 - 738
- Hofmann TG, Moller A, Sirma H, Zentgraf H, Taya Y, Droge W, et al. Regulation of p53 activity by its interaction with homeodomain-interacting protein kinase-2. Nat Cell Biol 2002; 4:1 - 10
- Perfettini JL, Castedo M, Nardacci R, Ciccosanti F, Boya P, Roumier T, et al. Essential role of p53 phosphorylation by p38 MAPK in apoptosis induction by the HIV-1 envelope. J Exp Med 2005; 201:279 - 289
- Yoshida K, Liu H, Miki Y. Protein kinase C delta regulates Ser46 phosphorylation of p53 tumor suppressor in the apoptotic response to DNA damage. J Biol Chem 2006; 281:5734 - 5740
- Okoshi R, Ozaki T, Yamamoto H, Ando K, Koida N, Ono S, et al. Activation of AMP-activated protein kinase induces p53-dependent apoptotic cell death in response to energetic stress. J Biol Chem 2008; 283:3979 - 3987
- Oda K, Arakawa H, Tanaka T, Matsuda K, Tanikawa C, Mori T, et al. p53AIP1, a potential mediator of p53-dependent apoptosis and its regulation by Ser46-phosphorylated p53. Cell 2000; 102:849 - 862
- Mantovani F, Tocco F, Girardini J, Smith P, Gasco M, Lu X, et al. The prolyl isomerase Pin1 orchestrates p53 acetylation and dissociation from the apoptosis inhibitor iASPP. Nat Struct Mol Biol 2007; 14:912 - 920
- Sykes SM, Mellert HS, Holbert MA, Li K, Marmorstein R, Lane WS, et al. Acetylation of the p53 DNA-binding domain regulates apoptosis induction. Mol Cell 2006; 24:841 - 851
- Tang Y, Luo J, Zhang W, Gu W. Tip60-dependent acetylation of p53 modulates the decision between cell cycle arrest and apoptosis. Mol Cell 2006; 24:827 - 839
- Knights CD, Catania J, Di Giovanni S, Muratoglu S, Perez R, Swartzbeck A, et al. Distinct p53 acetylation cassettes differentially influence gene-expression patterns and cell fate. J Cell Biol 2006; 173:533 - 544
- Chao C, Wu Z, Mazur SJ, Borges H, Rossi M, Lin T, et al. Acetylation of mouse p53 at lysine 317 negatively regulates p53 apoptotic activities after DNA damage. Mol Cell Biol 2006; 26:6859 - 6869
- Le Cam L, Linares LK, Paul C, Julien E, Lacroix M, Hatchi E, et al. E4F1 is an atypical ubiquitin ligase that modulates p53 effector functions independently of degradation. Cell 2006; 127:775 - 788
- Jansson M, Durant ST, Cho EC, Sheahan S, Edelmann M, Kessler B, et al. Arginine methylation regulates the p53 response. Nat Cell Biol 2008; 10:1431 - 1439
- Durant ST, Cho EC, La Thangue NB. p53 methylation—the Argument is clear. Cell Cycle 2009; 8:801 - 802
- Dehner A, Klein C, Hansen S, Muller L, Buchner J, Schwaiger M, et al. Cooperative binding of p53 to DNA: regulation by protein-protein interactions through a double salt bridge. Angew Chem Int Ed Engl 2005; 44:5247 - 5251
- Madhumalar A, Jun LH, Lane DP, Verma CS. Dimerization of the core domain of the p53 family: a computational study. Cell Cycle 2009; 8:137 - 148
- Klein C, Planker E, Diercks T, Kessler H, Kunkele KP, Lang K, et al. NMR spectroscopy reveals the solution dimerization interface of p53 core domains bound to their consensus DNA. J Biol Chem 2001; 276:49020 - 49027
- Kitayner M, Rozenberg H, Kessler N, Rabinovich D, Shaulov L, Haran TE, et al. Structural basis of DNA recognition by p53 tetramers. Mol Cell 2006; 22:741 - 753
- Ho WC, Fitzgerald MX, Marmorstein R. Structure of the p53 core domain dimer bound to DNA. J Biol Chem 2006; 281:20494 - 20502
- Tidow H, Melero R, Mylonas E, Freund SM, Grossmann JG, Carazo JM, et al. Quaternary structures of tumor suppressor p53 and a specific p53 DNA complex. Proc Natl Acad Sci USA 2007; 104:12324 - 12329
- Hager GL, McNally JG, Misteli T. Transcription dynamics. Mol Cell 2009; 35:741 - 753
- Perissi V, Rosenfeld MG. Controlling nuclear receptors: the circular logic of cofactor cycles. Nat Rev Mol Cell Biol 2005; 6:542 - 554
- Misteli T. Beyond the sequence: cellular organization of genome function. Cell 2007; 128:787 - 800
- Beckerman R, Prives C. Transcriptional regulation by p53. Cold Spring Harb Perspect Biol 2010; 2:935
- An W, Kim J, Roeder RG. Ordered cooperative functions of PRMT1, p300 and CARM1 in transcriptional activation by p53. Cell 2004; 117:735 - 748
- Karpova TS, Kim MJ, Spriet C, Nalley K, Stasevich TJ, Kherrouche Z, et al. Concurrent fast and slow cycling of a transcriptional activator at an endogenous promoter. Science 2008; 319:466 - 469
- McNally JG, Muller WG, Walker D, Wolford R, Hager GL. The glucocorticoid receptor: rapid exchange with regulatory sites in living cells. Science 2000; 287:1262 - 1265
- Metivier R, Penot G, Hubner MR, Reid G, Brand H, Kos M, et al. Estrogen receptor-alpha directs ordered, cyclical and combinatorial recruitment of cofactors on a natural target promoter. Cell 2003; 115:751 - 763
- Chen X, Ko LJ, Jayaraman L, Prives C. p53 levels, functional domains and DNA damage determine the extent of the apoptotic response of tumor cells. Genes Dev 1996; 10:2438 - 2451
- Horvath MM, Wang X, Resnick MA, Bell DA. Divergent evolution of human p53 binding sites: cell cycle versus apoptosis. PLoS Genet 2007; 3:127
- Ma B, Pan Y, Zheng J, Levine AJ, Nussinov R. Sequence analysis of p53 response-elements suggests multiple binding modes of the p53 tetramer to DNA targets. Nucleic Acids Res 2007; 35:2986 - 3001
- Frebourg T, Kassel J, Lam KT, Gryka MA, Barbier N, Andersen TI, et al. Germ-line mutations of the p53 tumor suppressor gene in patients with high risk for cancer inactivate the p53 protein. Proc Natl Acad Sci USA 1992; 89:6413 - 6417
- Sauer M, Bretz AC, Beinoraviciute-Kellner R, Beitzinger M, Burek C, Rosenwald A, et al. C-terminal diversity within the p53 family accounts for differences in DNA binding and transcriptional activity. Nucleic Acids Res 2008; 36:1900 - 1912