Abstract
The accumulation of excess histone proteins in cells has deleterious consequences such as genomic instability in the form of excessive chromosome loss, enhanced sensitivity to DNA damaging agents and cytotoxicity. Hence, the synthesis of histone proteins is tightly regulated at multiple steps and transcriptional as well as posttranscriptional regulation of histone proteins is well established. Additionally, we have recently demonstrated that histone protein levels are regulated posttranslationally by the DNA damage checkpoint kinase Rad53 and ubiquitin-proteasome dependent proteolysis in the budding yeast. However, the underlying mechanism/s via which excess histones exert their deleterious effects in vivo are not clear. Here we have investigated the mechanistic basis for the deleterious effects of excess histones in budding yeast. We find that the presence of excess histones saturates certain histone modifying enzymes, potentially interfering with their activities. Additionally, excess histones appear to bind non-specifically to DNA as well as RNA, which can adversely affect their metabolism. Microarray analysis revealed that upon overexpression of histone gene pairs, about 240 genes were either up- or downregulated by 2-fold or more. Overall, we present evidence that excess histones are likely to mediate their cytotoxic effects via multiple mechanisms that are primarily dependent on inappropriate electrostatic interactions between the positively charged histones and diverse negatively charged molecules in the cell. Our findings help explain the basis for the existence of multiple distinct mechanisms that contribute to the tight control of histone protein levels in cells and highlight their importance in maintaining genomic stability and cell viability.
Introduction
Histones are basic proteins that help package the lengthy genomic DNA in all eukaryotes to form nucleoprotein filaments called chromatin and fit them inside the nucleus of each cell.Citation1 Two molecules each of core histones H4, H3, H2A and H2B form an octameric protein core around which 147 base pairs of DNA is wrapped to form a nucleosome core particle.Citation2 A linker or H1 histone molecule then associates with the nucleosome core particle to form a nucleosome that serves as the basic repeating unit of chromatin.Citation3 By packaging the DNA, histones also regulate access to the genetic information contained within the DNA.Citation4 Histone proteins are extensively modified posttranslationally and these modifications as well as the enzymes that carry them out have been the object of intense scrutiny over the past two decades due to their potential for modulating the expression of the genetic information, thereby affecting both normal and disease states in humans.Citation5 For most of the ∼125 years since the discovery of histones,Citation6 these proteins had been considered extremely stable and histone half-lives were experimentally determined to be in the order of several months depending on the proliferative and differentiation state of cells.Citation7–Citation9 This high metabolic stability is likely important for histones to fulfill their roles in the stable inheritance of the genetic material (DNA), as well as epigenetic states through cell division. However, these half-lives are likely to mainly reflect the contribution of chromatin bound histones as we have shown that non-chromatin bound (i.e., “free” or “excess”) histones are degraded with a half-life of ∼30 minutes in the budding yeast.Citation10,Citation11
Histone proteins are essential for viability and cells need to achieve a very delicate balance between histone and DNA synthesis during the packaging of its genome into chromatin. Histone proteins are regulated transcriptionally,Citation12,Citation13 posttranscriptionally,Citation14–Citation16 translationallyCitation17 and posttranslationally.Citation10,Citation11,Citation18 Why are the histone proteins subjected to such a high degree of regulation? This is probably because on one hand scarcity of histones results in inviability,Citation19 while on the other hand the presence of excess histones has been shown to result in excessive mitotic chromosome loss,Citation20 increased DNA damage sensitivity and cytotoxicity.Citation10,Citation11 However, the mechanisms underlying these deleterious effects of excess histone accumulation are unclear and so we were interested in investigating them. Our main hypothesis is that due to their high positive charge, histones may exhibit non-specific electrostatic interactions with many negatively charged molecules in the cells, including nucleic acids such as DNA and RNA, as well as negatively charged proteins. This postulate predicts that excess histones could potentially exert their deleterious effects via at least four main mechanisms: (1) Excess histones can compete with and prevent the appropriate assembly of variant histones. This has already been shown to be the case upon overexpression of histone H3 that interferes with the correct deposition of the centromere specific histone H3 variant CENP-A, resulting in a chromosome loss phenotype.Citation21,Citation22 (2) Excess histones can potentially swamp histone chaperones and histone modifying enzymes by binding to them and perhaps tying them up in futile catalytic cycles. We have previously obtained some evidence supporting this possibility by demonstrating that overexpression of certain histone chaperones can alleviate the toxicity due to histone overexpression.Citation10 (3) Excess histones can stick non-specifically to the DNA, thus affecting chromatin structure and thereby potentially altering gene expression. (4) Similarly, when present in excess, histones can stick to coding as well as non-coding structural and regulatory RNAs, presumably altering translation and other activities of RNA. It should be noted that the four potential mechanisms listed above are not mutually exclusive and it is very likely that one or more or all of these contribute to the deleterious effects of excess histones, although the relative contribution of each mechanism may vary. Here we have systematically explored the last three of the four mechanisms listed above and find that excess histones mediate their deleterious effects via multiple mechanisms in the budding yeast, largely by virtue of inappropriate electrostatic interactions with cellular macromolecules carrying the opposite charge.
Results
Budding yeast mutants lacking certain histone modifying enzyme subunits are sensitive to the presence of excess histones.
There are many histone modifying and chromatin remodelling enzymes that often function as multi-subunit complexes. Further, many such activities have overlapping roles and function redundantly. We reasoned that some of these non-essential histone modifying activities might be sensitive to the presence of excess histones if one or more of these redundant activities were deleted from the cells. Hence, we screened several yeast deletion strains from the genome deletion collection (Open Biosystems) lacking subunits of non-essential chromatin modifying factors for sensitivity to histone overexpression as described previously.Citation10,Citation11 We found that deletion strains corresponding to set2 (histone methyltransferase), hda2 (histone deacetylase), sas3 (catalytic subunit of the NuA4 histone acetyltransferase complex) and taf14 (a subunit of INO80 and SWI/SNF chromatin remodelling complexes, NuA4 histone acetyltransferase complex, as well as TFIID and TFIIF transcription factor complexes) were sensitive to overexpression of histone H3 (), consistent with the idea that in these strains the absence of one histone modifying factor places undue burden on the remaining histone modifying activities still available in the cell, which are quickly overwhelmed upon histone overexpression, as verified by Western blotting (). A caveat in our studies is that most of our experiments involve overexpression of just one core histone, while situations involving the generation of excess endogenous histones presumably include all four core histones in excess and may not be fully comparable to our experiments. However, in all the experiments that we have repeated using co-overexpression of two or four core histonesCitation10 (Sup. Fig. 1A and data not shown), we have obtained similar results.
Excess histones do not appear to alter the bulk nucleosomal structure but lead to subtle alterations in the fine structure of chromatin.
If excess histones could stick non-specifically to the DNA, it is possible that they can alter the gross and fine structure of chromatin. In vitro, even a slight stoichiometric excess of histones over DNA is sufficient to trigger chromatin aggregation and block transcription.Citation23 Hence, we next tested if overexpression of histone H3 affects chromatin structure in vivo. For this we analyzed the chromatin structure of cells with or without histone overexpression following cleavage with micrococcal nuclease that cleaves in between nucleosomes and provides low resolution information regarding the underlying bulk chromatin structure.Citation24 We found that overexpression of histone H3 did not result in any significant changes in the bulk chromatin structure and the nucleosomal ladder generated by micrococcal nuclease cleavage was intact (). However, we did notice a subtle but reproducible decrease in the nucleosomal repeat length by ∼10–15 bp upon histone H3 overexpression, suggesting that the nucleosomes were closer together and that the internucleosomal linker length was reduced. We also analyzed the micrococcal nuclease cleavage pattern in detail at two loci using indirect endlabelling analysis using primer extension.Citation25 As opposed to the largely unaltered bulk nucleosomal ladder, end-labelled primers specific for the mating type MATa locus and the RDN (ribosomal DNA) locus revealed significant alterations in the micrococcal nuclease cleavage patterns at both these loci (). Similar results were obtained for the actin (ACT1) locus (data not shown). Several micrococcal nuclease cleavage sites are blocked while a few new cleavage sites are generated following histone H3 overexpression, suggesting that although the bulk chromatin structure is unaffected, subtle alterations in the fine structure of chromatin may be widespread in the presence of excess histones.
We decided to confirm the alterations in the fine structure of chromatin upon histone overexpression using high resolution in vivo cleavage of the chromosomal DNA upon the galactose inducible expression of DNase ICitation26 that cleaves in the minor groove of DNA and hence exhibits a ∼10 bp periodicity of cleavage on naked DNA, although DNA bound to proteins shows considerable protection from cleavage. Indirect end-labelling analysis using primer extensionCitation25 of DNA cleaved with DNase I in vivo confirmed our results obtained with micrococcal nuclease and clearly showed that the fine structure of chromatin at all the three loci (MATa, ACT1 and RDN) analysed was significantly altered upon histone H3 overexpression (). Control experiments using microscopy and immunoprecipitation techniques clearly showed that the overexpressed histone was present in the nucleus and was largely associated with the chromatin (data not shown). Although we have not yet attempted to deduce the exact nature of the changes in chromatin structure that are detected by the altered nuclease cleavage patterns upon histone overexpression, it is possible that any such alterations in the fine structure of chromatin could potentially affect DNA transactions such as transcription.
Overexpression of histone gene pairs, but not individual histones, results in the alteration of transcript levels of numerous genes.
To directly assay if the binding of excess histones to chromatin alters the levels of yeast transcripts, we used a microarray based analysis of genome-wide gene expression.Citation27 Transcripts corresponding to 5,849 budding yeast genes were analyzed before and after histone overexpression using Nimblegen's Saccharomyces cerevisiae 4 × 72 K array as described in the Materials and Methods section (). Surprisingly, although overexpression of histone H3 alone results in significant toxicity in cell viability assays ()Citation10,Citation11 and alters the fine structure of the chromatin ( and ), it did not alter transcript levels beyond the 2-fold threshold for significant changes that we had arbitrarily chosen for our microarray experiments ( and D). Nevertheless, overexpression of the histone H3 and H4 gene pair resulted in a more than 2-fold change in the transcript levels of ∼225 genes ( and D and Sup. Table 1), while overexpression of all four core histones resulted in a significant alteration of ∼240 transcripts ( and D and Sup. Table 2). About 40% of the transcripts significantly upregulated upon the overexpression of all core histones were also upregulated upon the overexpression of the H3-H4 gene pair alone (Sup. Table 3). Several of the transcripts altered upon histone gene pair overexpression correspond to essential genes and these may have a significant negative impact on cell viability and thus contribute to the cytotoxic effects of excess histones. However, histone overexpression results in the alteration of only ∼4% of the budding yeast transcriptome, which is different compared to the results obtained from the converse microarray experiment involving depletion of histone H4, where a substantial fraction (∼25%) of the yeast transcriptome was found to be affected.Citation28
Interestingly, further analysis of our microarray data revealed that about 40% of the open reading frames (ORFs) whose transcript levels are significantly affected by histone overexpression are located in “mini clusters” of mainly 2, but occasionally up to 3–4 ORFs that lie adjacent to each other either on the Watson or the Crick strand and appear to be co-ordinately regulated (Sup. Tables 1–3). Further, the majority (∼70%) of these mini clusters are located relatively close to the telomeres and sometimes the centromeres (i.e., the distance between the mini cluster and the telomere or centromere is less than a third of the total length of the chromosome arm on which the cluster is located). One such mini cluster comprises of ORFs (YKR079C, YKR080C, YKR081C) that are upregulated 2–3 fold upon the overexpression of the H3-H4 gene pair and is located on chromosome XI relatively close to the telomere. A mini cluster that is downregulated 2–3 fold upon the overexpression of the H3-H4 gene pair is located on chromosome XIV relatively close to the telomere and comprises of genes (YNR056C, YNR057C, YNR058W) that are involved in biotin biosynthesis and whose expression maybe sensitive to iron levels. Another telomere proximal mini cluster can be found on chromosome XV and comprises of genes that appear to be involved in iron metabolism (YOR382W, YOR383C, YOR385W) and are downregulated more than 3-fold upon overexpression of the H3-H4 gene pair as well as the overexpression of all core histones. These telomere proximal mini clusters of ORFs that appear to be either co-ordinately up or downregulated upon histone overexpression may represent loci with certain inherent chromatin structural features such as specific nucleosome positioning or occupancy that may be crucial for their regulated expression under normal conditions. As such, these loci would be particularly sensitive to changes in histone levels as these chromatin structural features could be readily altered upon histone overexpression.
Excess histones can bind to RNA.
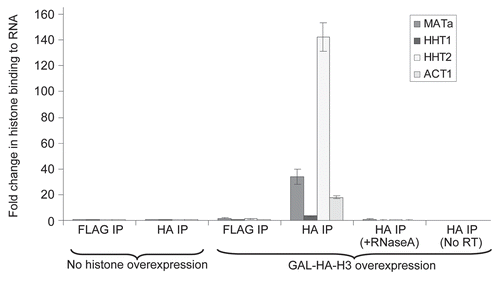
Alteration in the transcript levels of essential genes is unlikely to explain all of the cytotoxic effects of histone overexpression, since H3 overexpression results in cytotoxicity without affecting transcript levels and as such must be mediating its cytotoxic effects via other mechanisms. Due to the potentially high affinity of the positively charged histones for negatively charged molecules, it is possible that apart from sticking to DNA, excess histones can also stick to RNA. To detect if this was indeed occurring in vivo, particularly in the case of H3 overexpression alone, we performed a RNA Immunoprecipitation (RIP) assayCitation29,Citation30 similar to the popular Chromatin Immunoprecipitation (ChIP) assay in the presence and absence of galactose induced epitope tagged HA3-HHT2 (HA-H3) overexpression in a strain carrying a FLAG-tagged chromosomal HHT1 gene encoding FLAG-H3 as well. Using real time quantitative Polymerase Chain Reaction (qPCR), we clearly detected enhanced binding of the overexpressed histone HA-H3 to transcripts from three loci (ACT1, MATa, endogenous histone HHT1 as well as a mixture of endogenous and exogenous histone HHT2) that we assayed for (), suggesting that excess histones are capable of binding to RNAs in vivo and potentially altering their activities. Further, no signal was obtained in control qPCR carried out following RNaseA treatment of the immunoprecipitated material, as well as when the Reverse Transcriptase was left out of the reaction, clearly demonstrating that the signals obtained are the result of the genuine binding of histones to RNA. In the same experiment, the binding of endogenous FLAG-H3 (that was not overexpressed) to RNA was barely above the background, suggesting that under normal conditions endogenous histones are not very likely to bind to RNA and potentially alter its functions.
Discussion
Excess histone levels mediate cytotoxicity via multiple mechanisms.
We have presented evidence here to suggest that the presence of excess histones negatively impacts numerous cellular processes via multiple mechanisms based largely on potential electrostatic interactions between the highly positively charged histones and negatively charged molecules in the cell. Mutant yeast cells such as rad53Δ, tom1Δ and ubc4Δ ubc5Δ that are defective in the regulation of histone protein levels and harbor excess endogenous histones, are very sensitive to exogenous histone overexpression which is highly toxic for these cells.Citation10,Citation11 All these mutant strains are also sensitive to DNA damaging agents and exhibit genomic instability in the form of elevated chromosome loss rates. The data presented here allows us to finally explain the deleterious effects of excess histones not just in these mutants, but also in wild type cells. Excess histones can swamp the binding sites available on histone chaperonesCitation10 and then overload histone modifying enzymes. This can have serious consequences for the normal regulation of gene expression and the formation/maintenance of epigenetic marks on histones. Not surprisingly, cells lacking certain histone modifying enzymes were sensitive to histone overexpression (). Further, the stability of histone posttranslational modifications would depend not only on the enzymes that put them on and remove them, but also on the stability of the histone proteins themselves. For example, until the discovery of the first histone lysine demethylase a few years ago,Citation31 histone methylation marks were considered to be as stable as histone themselves.Citation32,Citation33 There is a constant exchange of histones between the chromatin bound and free states as a result of transcriptional eviction,Citation34,Citation35 the action of chromatin remodelling factorsCitation36 and the opposing effects of chromatin assembly and disassembly.Citation37–Citation38 We have recently obtained evidence that the tyrosine 99 residue of histone H3 may be phosphorylated only when histone H3 is not bound to the chromatin and this modification may serve as a mark to target this histone for degradation.Citation11 Hence, our findings regarding the degradation of non-chromatin bound histonesCitation10,Citation11 may have wide-ranging ramifications for the stability and maintenance of epigenetic marks on histones in chromatin by providing an additional layer of regulation.
We also found that excess histones can alter the fine structure of chromatin. This could potentially impact all metabolic activities that require access to the DNA. Hence, we have now systematically analyzed the genome wide effects of excess histones on gene expression using microarrays. We found that although overexpression of histone H3 alone is toxic to the cells ()Citation10,Citation11 and alters chromatin fine structure, it did not result in an appreciable alteration of gene expression (). However, overexpression of the H3-H4 gene pair or simultaneous overexpression of all four core histones lead to a significant alteration in the expression levels of 4% of the budding yeast genome, including several genes that are essential for viability. This difference between overexpression of H3 alone or in combination with H4 may be due to the fact that histones are deposited on to the DNA as pairs of H3-H4 and H2A-H2B dimers.Citation39 Hence, although overexpression of H3 alone may lead to its non-specific association with the DNA, this association may not be very strong in vivo and as such it may not prove to be a significant obstacle for the transcriptional machinery. On the other hand, (H3-H4)2 tetramers and the core histone octamer are known to associate tightly with the DNA and so upon the overexpression of the H3-H4 gene pair or all the core histones, it is likely that these excess histones will form chromatin structures that may present a significant challenge to the transcriptional machineryCitation40 and our microarray data appears to support this notion. Overall, our microarray data suggests that although excess histones are capable of altering the expression of a small fraction of yeast genes that probably contributes to their deleterious effects in vivo, the fact that H3 overexpression does not alter gene expression and is still highly toxic strongly argues that the other mechanisms discussed here are likely to be mediating most of the cytotoxic effects of excess histones.
Numerous studies in recent years have revealed the dynamic nature of chromatin structure, particularly in the context of transcription.Citation36–Citation38 Our microarray based analysis of yeast genes with altered expression patterns upon histone overexpression revealed that a significant number of them lie in small clusters of 2–4 genes that are co-ordinately regulated (Sup. Tables 1–3). These clusters could have unique chromatin features that may facilitate the normal regulated expression of genes within them. Such special chromatin features may include promoter regions with highly positioned nucleosomes or promoters and/or coding sequences that are largely devoid of nucleosomes. A cursory survey of published data on nucleosome occupancy in yeastCitation35 suggests that this may be the case indeed with at least a few of the identified mini clusters. For example, the YNR056C, YNR057C, YNR058W cluster which is downregulated upon histone overexpression (Sup. Table 1) appears to have poor nucleosome occupancy in yeast based on published data.Citation35 It is possible that histone overexpression results in a higher nucleosome occupancy in this region, thereby resulting in lower levels of transcription consistent with our microarray data. On the other hand, the YOR382W, YOR383C, YOR385W cluster that is also downregulated upon histone overexpression appears to have well-positioned nucleosomes based on published genome wide nucleosome occupancy data.Citation35 How exactly histone overexpression brings about a reduction in the expression levels of ORFs within mini clusters that may have either poorly positioned or highly positioned nucleosomes is unclear. Future studies involving chromatin fine structure mapping will confirm if there are indeed common chromatin structural features within the identified mini clusters and reveal the exact nature of any chromatin structure changes at these loci upon histone overexpression.
Another intriguing feature of the identified mini clusters is their telomere (and occasionally centromere) proximal localization. This is reminiscent of the effect of histone H4 depletion where ∼50% of the genes within 20Kb of the telomeres were preferentially derepressed, compared to derepression of just 15% of the genes genome-wide.Citation28 Any special chromatin features in these clusters may be related to their relative proximity to the telomeres (or the centromeres) that are known to have specialized chromatin structures, although these are not normally known to spread over ∼20 Kb from the telomeres in the budding yeast and the closest mini clusters are at least that far away from the telomere.Citation41 Another possibility is that histone overexpression may result in their excessive association with certain loci where they may not be present normally and this in turn may lead to their silencing by the recruitment of Sir proteins, perhaps via subsequent localization of these loci to the nuclear periphery.Citation42 Future studies will reveal whether the relative telomere proximal location of the mini clusters affected by histone overexpression has any functional significance.
As opposed to the potential alterations in transcription caused by excess histones, the process of transcription itself may generate excess histones if the reassembly of the transcriptionally evicted histones onto the chromatin is blocked, as in conditional spt16 mutants, where the evicted histones are presumably subjected to Rad53 mediated histone proteolysis.Citation43 In fact, during the G1 phase of the cell cycle, the presence of excess transcriptionally evicted histones in the spt16 mutants or exogenously expressed histones in wild type cells triggers the downregulation of the G1 cyclin CLN3, leading to a delay in S-phase entry. Contrary to a situation of histone excess, it has also been suggested that a lack of adequate histones due to sub-optimal histone pre-mRNA processing may also trigger a similar G1 arrest.Citation15 Hence, eukaryotic cells may have evolved surveillance mechanisms similar to known cell cycle checkpoints to protect them from the harmful effects of both excess histone accumulation as well as a scarcity of histones. These mechanisms delay S-phase entry by prolonging the G1 phase of the cell cycle, presumably to provide cells with additional time to either degrade excess histones or increase histone mRNA production as needed prior to S-phase entry and start of replication, when excess or inadequate amounts of histones could potentially be highly detrimental for the cells.
Additional problems due to the presence of excess histones may arise from the translational defects occurring as a result of these histones binding to coding RNAs () and interfering with their translation. Further, since the majority of the RNAs in the cell are structural ribosomal RNAs (rRNAs) that are crucial for ribosome function, their non-specific interaction with excess histones would serve as a double setback for ongoing protein synthesis in the cell. Hence, it is not surprising that overexpression of histones is highly toxic and confers lethality in mutants such as rad53Δ that cannot efficiently degrade excess histones.Citation10 This lethality probably represents the cumulative effects of excess histones by the various mechanisms discussed above. In fact, even wild type yeast cells experience ∼20% lethality upon histone overexpression (data not shown) and exhibit genomic instability in the form of enhanced rates of chromosome loss.Citation10,Citation11,Citation20 Genomic instability is characterized by the increased rate of acquisition of alterations in the genome and is associated with most human cancers.Citation44,Citation45 Taken together, these data suggest that improper histone stoichiometry and aberrant chromatin structure may contribute to genomic instability and carcinogenesis. As such, our studies highlight the potentially crucial role of proper histone stoichiometry and chromatin structure as well as multiple histone regulatory mechanisms in maintaining viability and genomic stability.
Materials and Methods
Yeast strains, plasmids and western blotting.
Yeast strains are listed in Supplemental Table 4 in the Supplemental Material section provided with the electronic version of the manuscript. Plasmid pYES2-HTH-HHT2 has been described previouslyCitation11 and was used for the galactose induced overexpression of epitope-tagged histone H3 to assay the sensitivity of yeast mutants to histone overexpression in , as well as for histone H3 overexpression to monitor the effects on chromatin structure in . Plasmid pSUN1 has been described elsewhere26 and was used for galactose mediated DNaseI expression in the in vivo footprinting experiment described in . Plasmids pYES6/CT-HA3-HHT2 and pYES6/CT-HA3-HHF2 carry galactose inducible H3 and H4 genes respectively sub-cloned between the BamHI and XbaI sites in the multiple cloning site of the high copy 2 µ based plasmid with a Blasticidin selectable marker (pYES6/CT from Clontech). Plasmid pHM90 was a gift from Dr. Hiroshi Masumoto and carries a construct for the galactose inducible overexpression of the H3-H4 gene pair (GAL1-10-FLAG-HHT1-HHF1) upon integration at the TRP1 locus, as in the microarray experiments in . Dr. Mary Ann Osley generously provided p67 (CEN-HIS3-GAL1-10-FLAG-HTA2-HTB2), a low copy plasmid for the galactose inducible overexpression of the H2A-H2B gene pair, which was used in our microarray experiments shown in . Our antibodies and western blotting procedure have been described in detail elsewhere.Citation10,Citation11
Chromatin structure analysis by nuclease digestion and primer extension.
Wild type W303-1A cells carrying the empty vector pYES2-HTH or the pYES2-HTH-HHT2 plasmid were grown overnight in minimal media lacking uracil and with raffinose as the carbon source. Equal amounts of cells were then treated with 2% galactose for 4 hours to induce histone H3 overexpression (GAL-HTH-H3) following which they were fixed with 1% formaldehyde for 15 minutes. For each sample, nuclei isolated from 250 million cells were digested with 1 unit of micrococcal nuclease (MNase) for 10 minutes at 37°C in the presence of 1 mM CaCl2 as described previously.Citation24 Following MNase cleavage, the formaldehyde crosslinks were reversed and the cleaved DNA was purified by phenol/chloroform extraction followed by ethanol precipitation. The purified DNA was used as such for analysis by agarose gel electrophoresis () and primer extension (). Primer extension of the MNaseI digested DNA was carried out essentially as described previouslyCitation25 using linear polymerase chain reaction (PCR).
For the analysis of chromatin fine structure upon histone overexpression using in vivo footprinting,Citation26 wild type W303-1A cells carrying either the pSUN1 plasmid alone or in combination with the pYES6/CT-HA3-HHT2 plasmid were used. The cells were grown in minimal media lacking the appropriate selection markers and raffinose as the carbon source, prior to overnight treatment with 2% galactose. 250 million cells were then harvested for each sample and the DNaseI cleaved DNA was purified by phenol/chloroform extraction followed by ethanol precipitation. The DNA was then digested to completion with the restriction endonuclease StyI and re-purified as before prior to use in primer extension reactions as described previously.Citation25
Microarray analysis.
Wild-type W303-1a yeast cells harbouring an empty vector (pYES2-HTH);Citation11 a high copy plasmid (pYES2-HTH-HHT2)Citation11 for galactose inducible histone H3 overexpression (GAL-H3); an integrated construct (pHM90) for galactose inducible H3-H4 gene pair (GAL-H3-H4); or GALH3-H4 plus a low copy plasmid (p67) for the galactose inducible expression of H2A-H2B gene pair (GAL-H2A-H2B-H3-H4) were grown to a density of 10 million cells per ml in minimal media lacking appropriate selection markers. Galactose was then added to a final concentration of 2% for 4 hours to induce histone overexpression prior to harvesting 50 million cells for extraction of total RNA using the RNeasy kit from Qiagen following the manufacturer's instructions. After appropriate quality control analysis of the extracted RNA (on BioRad's Experian Automated Electrophoresis system), the RNA was reverse transcribed using random hexamers to generate cDNA. The cDNA was then labelled and hybridized in duplicate to the Nimblegen Saccharomyces cerevisiae 4 × 72 K array following instructions provided in the Nimblegen Arrays User's Guide. The array was scanned and hybridization signals from the histone overexpression samples were normalized to the signals obtained from the empty vector sample. The raw data was analyzed using ArrayStar, before exporting it to Microsoft Excel for presentation.
RNA immunoprecipitation (RIP).
The wild-type yeast strain (YAG1021) carrying a FLAG-tagged endogenous H3 gene (FLAG-HHT2) at its normal chromosomal location along with the pYES6/CT-HA3-HHT2 plasmid for galactose inducible HA3-H3 overexpression was used for the RIP experiments. This strain allows us to detect the binding of endogenous levels of epitope tagged H3 (FLAG-H3) to RNA in the absence of galactose, while in the presence of galactose it permits the detection of the binding of overexpressed HA3-H3 to RNA. Cells were grown overnight in 100 ml of rich media with raffinose as the carbon source and Blasticidin as the selection antibiotic until they reached a density of 10 million cells/ml. Equal amounts of the culture was then treated with or without 2% galactose for 4 hours prior to addition of 1% formaldehyde to the culture media to allow crosslinking of proteins to nucleic acids for 15 minutes. Crosslinked cells were harvested to prepare whole cell extracts that were sonicated and treated with RNase-free DNase I to digest all DNA. Then RNA immunoprecipitation (RIP) was carried out using equal amounts of whole cell extracts essentially as described previouslyCitation30 with a minor modification. To absolutely ensure complete removal of any contaminating DNA in our RIP experiments, we initially treated the whole cell extracts with RNase-free DNaseI and followed this up with a second round of DNaseI treatment of the material immunoprecipitated by the antibody beads as well. FLAG antibody beads were used to immunoprecipitate (IP) endogenous FLAG-H3 (FLAG IP), while HA antibody beads were used to IP the exogenously overexpressed HA3-H3 (HA IP). The RNAs immunoprecipitated with the antibody beads were recovered after reversing the formaldehyde crosslinks and reverse transcribed to give cDNAs that were quantified by real time quantitative PCR (qPCR) using primers and probes specific for transcripts arising from the ACT1, MATa, HHT1 and HHT2 loci.
Abbreviations
| ChIP | = | chromatin immunoprecipitation |
| DNase I | = | deoxyribonuclease I |
| IP | = | immunoprecipitation |
| MNase | = | micrococcal nuclease |
| ORFs | = | open reading frames |
| PCR | = | polymerase chain reaction |
| qPCR | = | quantitative PCR |
| RIP | = | RNA immunoprecipitation |
| RNase A | = | ribonuclease A |
Figures and Tables
Figure 1 Yeast cells lacking histone modifying enzyme subunits are sensitive to histone overexpression. (A) Sensitivity of mutant yeast cells lacking histone modifying enzyme subunits to histone overexpression. Wild type (WT) or the indicated mutant strains were transformed with a plasmid carrying galactose inducible HA-epitope tagged H3 (pYES2-HTH-HHT2) or the empty vector (pYES2-HTH).Citation11 The rad53Δ strain carries the crt1Δ as well to suppress the loss of viability due to the essential nature of Rad53.Citation46 10-fold serial dilutions of the indicated strains were plated on glucose or galactose media and incubated for 3 days at 30°C prior to being photographed. (B) Histone H3 overexpression in yeast cells lacking histone modifying enzyme subunits. Four independently isolated transformants for each of the indicated mutant strains carrying the plasmid pYES2-HTH-HHT2 were grown overnight in minimal media lacking uracil and with raffinose as the carbon source. Cells were then treated with galactose for 90 minutes prior to harvesting 10 million cells per sample. Whole cell lysates were prepared by the alkaline lysis methodCitation47 and processed for Western blotting to detect both the endogenous and overexpressed epitope-tagged histone H3 using the H3-C polyclonal antibody as described previously.Citation10,Citation11 Asterisks indicate the transformants used in the experiment shown above in (A).
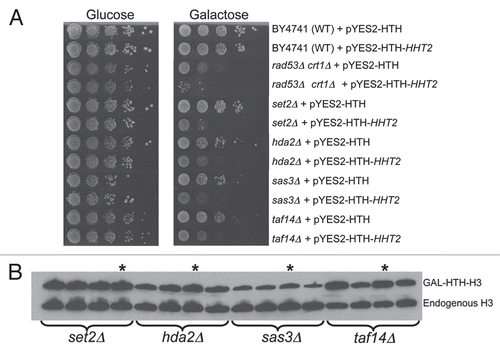
Figure 2 Histone overexpression does not affect the bulk chromatin structure but alters the fine structure of chromatin. (A) Micrococcal nuclease digestion of bulk chromatin. MNase cleaved DNA isolated from cells in the presence or absence of histone overexpression was purified as described in the Materials and Methods section. This DNA was then resolved on a 2% agaorse gel and stained with ethidium bromide to visualize the nucleosomal ladder. The positions of mono-, di-, tri- and tetra-nucleosomes are indicated. (B) Analysis of chromatin structure at specific loci using MNase cleavage. The MNase digested genomic DNA purified above in (A) was used as a template for analysis by indirect end-labeling with multiple cycle primer extensionCitation25 using 32P-end-labeled primers specific to the MATa and RDN loci to perform linear PCR. The reaction products were resolved on a 5% denaturing polyacrylamide gel which was processed for autoradiography. On the right hand side of the parts, a “-” sign indicates loss of a MNase cleavage site while a “+” sign indicates the gain of a site upon GAL-HTH-H3 overexpression.
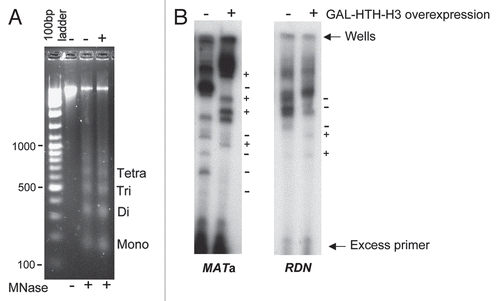
Figure 3 Indirect end-labeling analysis of chromatin following in vivo DNase I cleavage. Galactose inducible DNase I was expressed in wild type cells either expressing or not expressing GAL-HA-H3 as described previously.Citation26 The DNase I cleaved genomic DNA was purified and analyzed by indirect end-labeling with multiple cycle primer extensionCitation25 using a 32P-end-labeled primer specific to the ACT1 locus in addition to the primers for MATa and RDN loci described above in . The reaction products were resolved on a 5% denaturing polyacrylamide gel which was processed for autoradiography. On the right hand side of the parts, a “−” sign indicates loss of a DNase I hypersensitive site while a “+” sign indicates the gain of a site upon GAL-HTH-H3 overexpression.
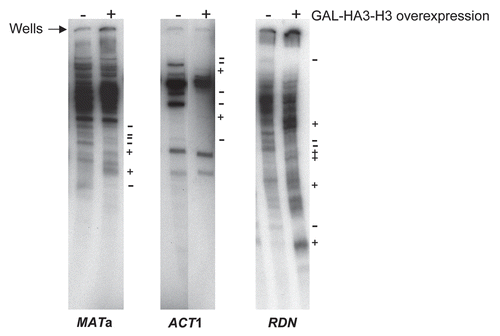
Figure 4 Genome wide comparison of gene expression in wild type yeast cells upon overexpression of individual or pairs of histone genes. (A) Overexpression of histone H3 alone does not result in appreciable changes in gene expression. Microarray analysis was carried out on total RNA isolated from the wild type W303-1A strains carrying either the empty vector or a galactose inducible histone H3 gene as described in the Materials and Methods section. A scatter plot is shown and illustrates the expression level of genes along the x- and y-axes representing transcript levels reflecting gene expression values on a log2 scale for the strains indicated. The three solid green lines represent fold-changes in transcript levels from the baseline, which is represented by the green line in the middle and indicates identical transcript levels in the two strains being compared. The upper solid green line represents a two-fold increase in transcript levels, while the lower green line represents a two-fold reduction in transcript levels. The dashed purple line is the linear regression with the coefficient of correlation R2 indicated on the bottom right corner. Each transcript is represented by a color coded data point to reflect where it is in comparison to the middle green line, with red color for upregulated transcripts and blue color for downregulated transcripts (the darker the color, the greater the fold-change in the transcript levels). Only the transcript levels of the overexpressed histone H3 (HHT2) was significantly elevated in this experiment and the corresponding data point is indicated by the arrow. (B) Changes in gene expression upon co-overexpression of the histone H3-H4 gene pair. Scatter plot for the indicated strains as in (A). (C) Changes in gene expression upon simultaneous overexpression of all four core histones. Scatter plot for the indicated strains as in (A). (D) Summary of the gene expression data derived from microarray analyses. The number of transcripts altered two-fold or more upon overexpression of different histones is summarized in a tabular form.
Figure 5 Excess histones can bind to RNA. An exponentially growing yeast strain with a FLAG epitope on the chromosomal HHT2 gene (FLAG-H3) and carrying the plasmid pYES6/CT-HA3-HHT2 for galactose inducible HA3-H3 overexpression was treated with or without galactose prior to performing RNA immunoprecipitation (RIP) using FLAG or HA antibodies as described in Materials and Methods. The amount of ACT1, MATa, HHT1 and HHT2 transcripts co-immunoprecipitated using the FLAG and HA antibody beads in the absence of galactose mediated histone overexpression (i.e., the background signal) was arbitrarily set to “1” and the relative binding of HA-H3 to these RNAs upon the addition of galactose is shown here. No signal above background was obtained in control qPCR carried out following RNaseA treatment of the immunoprecipitated material, as well as when the reverse transcriptase enzyme was left out of the reverse transcription reaction used to generate the cDNA for qPCR analysis. Error bars represent standard error of the mean from three experiments.
Additional material
Download Zip (763.6 KB)Acknowledgements
The authors wish to thank Drs. Hiroshi Masumoto, Mary Ann Osley, Rodney Rothstein, Alain Verreault and Yanchang Wang for providing certain plasmids and yeast strains used in this study. We thank Dr. Munah Abdul-Rauf for construction of the pYES6/CT-HA3-HHT2 and pYES6/CT-HA3-HHF2 plasmids. We also thank Steve Miller at the Florida State University Nimblegen Microarray Facility for help with the processing of the microarray samples and Dr. SuTan Wu for data analysis. Research in AG's laboratory is supported by a Bankhead-Coley Cancer Research Program grant (07BN-02) from the Florida Department of Health and an NIH grant (R21 MH081046).
References
- van Holde KE. Chromatin 1988; New York Springer-Verlag
- Richmond TJ, Davey CA. The structure of DNA in the nucleosome core. Nature 2003; 423:145 - 150
- Brown DT. Histone H1 and the dynamic regulation of chromatin function. Biochem Cell Biol 2003; 81:221 - 227
- Wolffe A. Chromatin structure and function 1995; Second Edition San Diego Academic Press
- Singh RK, Gunjan A. Epigenetic Therapy: Targeting Histones and their modifications in human diseases. Future Med Chem 2010; 2:543 - 548
- Kossel A. Ueber einen peptonartigen Bestandtheil des Zellkerns. Hoppe Seylers Z Physiol Chem 1884; 8:511 - 515
- Commerford SL, Carsten AL, Cronkite EP. Histone turnover within nonproliferating cells. Proc Natl Acad Sci USA 1982; 79:1163 - 1165
- Tsvetkov S, Ivanova E, Djondjurov L. The pool of histones in the nucleosol and cytosol of proliferating Friend cells is small, uneven and chasable. Biochem J 1989; 264:785 - 791
- Wunsch AM, Lough J. Histones synthesized at different stages of myogenesis are differentially degraded in myotube cells. J Cell Physiol 1989; 141:97 - 102
- Gunjan A, Verreault A. A Rad53 kinase-dependent surveillance mechanism that regulates histone protein levels in S. cerevisiae. Cell 2003; 115:537 - 549
- Singh RK, Kabbaj MH, Paik J, Gunjan A. Histone levels are regulated by phosphorylation and ubiquitylation-dependent proteolysis. Nat Cell Biol 2009; 11:925 - 933
- Stein GS, Stein JL, Marzluff WF. Histone genes: Structure organization and regulation 1984; New York John Wiley & Sons
- Osley MA. The regulation of histone synthesis in the cell cycle. Annu Rev Biochem 1991; 60:827 - 861
- Kaygun H, Marzluff WF. Regulated degradation of replication-dependent histone mRNAs requires both ATR and Upf1. Nat Struct Mol Biol 2005; 12:794 - 800
- Marzluff WF, Wagner EJ, Duronio RJ. Metabolism and regulation of canonical histone mRNAs: life without a poly(A) tail. Nat Rev Genet 2008; 9:843 - 854
- Reis CC, Campbell JL. Contribution of Trf4/5 and the nuclear exosome to genome stability through regulation of histone mRNA levels in Saccharomyces cerevisiae. Genetics 2007; 175:993 - 1010
- Borun TW, Gabrielli F, Ajiro K, Zweidler A, Baglioni C. Further evidence of transcriptional and translational control of histone messenger RNA during the HeLa S3 cycle. Cell 1975; 4:59 - 67
- Gunjan A, Paik J, Verreault A. The emergence of regulated histone proteolysis. Curr Opin Genet Dev 2006; 16:112 - 118
- Han M, Chang M, Kim UJ, Grunstein M. Histone H2B repression causes cell cycle-specific arrest in yeast: effects on chromosomal segregation, replication and transcription. Cell 1987; 48:589 - 597
- Meeks-Wagner D, Hartwell LH. Normal stoichiometry of histone dimer sets is necessary for high fidelity of mitotic chromosome transmission. Cell 1986; 44:43 - 52
- Glowczewski L, Yang P, Kalashnikova T, Santisteban MS, Smith MM. Histone-histone interactions and centromere function. Mol Cell Biol 2000; 20:5700 - 5711
- Au WC, Crisp MJ, DeLuca SZ, Rando OJ, Basrai MA. Altered dosage and mislocalization of histone H3 and Cse4p lead to chromosome loss in Saccharomyces cerevisiae. Genetics 2008; 179:263 - 275
- Steger DJ, Workman JL. Transcriptional analysis of purified histone acetyltransferase complexes. Methods 1999; 19:410 - 416
- Martens JA, Winston F. Evidence that Swi/Snf directly represses transcription in S. cerevisiae. Genes Dev 2002; 16:2231 - 2236
- Axelrod JD, Majors J. An improved method for photofootprinting yeast genes in vivo using Taq polymerase. Nucleic Acids Res 1989; 17:171 - 183
- Wang X, Simpson RT. Chromatin structure mapping in Saccharomyces cerevisiae in vivo with DNase I. Nucleic Acids Res 2001; 29:1943 - 1950
- Horak CE, Snyder M. Global analysis of gene expression in yeast. Funct Integr Genomics 2002; 2:171 - 180
- Wyrick JJ, Holstege FC, Jennings EG, Causton HC, Shore D, Grunstein M, et al. Chromosomal landscape of nucleosome-dependent gene expression and silencing in yeast. Nature 1999; 402:418 - 421
- Gilbert C, Kristjuhan A, Winkler GS, Svejstrup JQ. Elongator interactions with nascent mRNA revealed by RNA immunoprecipitation. Mol Cell 2004; 14:457 - 464
- Gilbert C, Svejstrup JQ. RNA immunoprecipitation for determining RNA-protein associations in vivo. Curr Protoc Mol Biol 2006; 27:4
- Shi Y, Lan F, Matson C, Mulligan P, Whetstine JR, Cole PA, et al. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004; 119:941 - 953
- Byvoet P, Shepherd GR, Hardin JM, Noland BJ. The distribution and turnover of labeled methyl groups in histone fractions of cultured mammalian cells. Arch Biochem Biophys 1972; 148:558 - 567
- Thomas G, Lange HW, Hempel K. Relative stability of lysine-bound methyl groups in arginie-rich histones and their subfrations in Ehrlich ascites tumor cells in vitro. Hoppe Seylers Z Physiol Chem 1972; 353:1423 - 1428
- Lee CK, Shibata Y, Rao B, Strahl BD, Lieb JD. Evidence for nucleosome depletion at active regulatory regions genome-wide. Nat Genet 2004; 36:900 - 905
- Dion MF, Kaplan T, Kim M, Buratowski S, Friedman N, Rando OJ. Dynamics of replication-independent histone turnover in budding yeast. Science 2007; 315:1405 - 1408
- Shivaswamy S, Bhinge A, Zhao Y, Jones S, Hirst M, Iyer VR. Dynamic remodeling of individual nucleosomes across a eukaryotic genome in response to transcriptional perturbation. PLoS Biol 2008; 6:65
- Kim HJ, Seol JH, Han JW, Youn HD, Cho EJ. Histone chaperones regulate histone exchange during transcription. EMBO J 2007; 26:4467 - 4474
- Takahata S, Yu Y, Stillman DJ. FACT and Asf1 regulate nucleosome dynamics and coactivator binding at the HO promoter. Mol Cell 2009; 34:405 - 415
- English CM, Adkins MW, Carson JJ, Churchill ME, Tyler JK. Structural basis for the histone chaperone activity of Asf1. Cell 2006; 127:495 - 508
- Chang CH, Luse DS. The H3/H4 tetramer blocks transcript elongation by RNA polymerase II in vitro. J Biol Chem 1997; 272:23427 - 23434
- Bühler M, Gasser SM. Silent chromatin at the middle and ends: lessons from yeasts. EMBO J 2009; 28:2149 - 2161
- Taddei A, Van Houwe G, Nagai S, Erb I, van Nimwegen E, Gasser SM. The functional importance of telomere clustering: global changes in gene expression result from SIR factor dispersion. Genome Res 2009; 19:611 - 625
- Morillo-Huesca M, Muñoz-Centeno MC, Singh RK, Reddy GU, Oreal V, et al. Accumulation of transcription-evicted histones induces a CLN3-dependent cell cycle delay in G1. PLoS Genetics 2010; (In Press)
- Lengauer C, Kinzler KW, Vogelstein B. Genetic instabilities in human cancers. Nature 1998; 396:643 - 649
- Hoeijmakers JH. Genome maintenance mechanisms for preventing cancer. Nature 2001; 411:366 - 374
- Huang M, Zhou Z, Elledge SJ. The DNA replication and damage checkpoint pathways induce transcription by inhibition of the Crt1 repressor. Cell 1998; 94:595 - 605
- Kushnirov VV. Rapid and reliable protein extraction from yeast. Yeast 2000; 16:857 - 860