Abstract
Medulloblastoma, a brain tumor arising in the cerebellum, is the most common solid childhood malignancy. The current standard of care for medulloblastoma leaves survivors with life-long side effects. Gaining insight into mechanisms regulating transformation of medulloblastoma cells-of-origin may lead to development of better treatments for these tumors. Cerebellar granule neuron precursors (CGNPs) are proposed cells-of-origin for certain classes of medulloblastoma, specifically those marked by aberrant Sonic hedgehog (Shh) signaling pathway activation. CGNPs require signaling by Shh for proliferation during brain development. In mitogen-stimulated cells, nuclear localized cyclin dependent kinase (cdk) inhibitor p27 (Kip1) functions as a checkpoint control at the G1- to S-phase transition by inhibiting cdk2. Recent studies have suggested cytoplasmically localized p27kip1 acquires oncogenic functions. Here, we show that p27Kip1 is cytoplasmically localized in CGNPs and mouse Shh-mediated medulloblastomas. Tranasgenic mice bearing an activating mutation in the Shh pathway and lacking one or both p27Kip1 alleles have accelerated tumor incidence compared to mice bearing both p27Kip1 alleles. Interestingly, mice heterozygous for p27Kip1 have decreased survival latency compared to p27Kip1-null animals. Our data indicate that this may reflect the requirement for at least one copy of p27Kip1 for recruiting cyclin D/cdk4/6 to promote cell cycle progression yet insufficient expression in the heterozygous or null state to inhibit cyclin E/cdk2. Finally, we find that mis-localized p27Kip1 may play a positive role in motility in medulloblastoma cells. Together, our data indicate that the dosage of p27Kip1 plays a role in cell cycle progression and tumor suppression in Shh-mediated medulloblastoma expansion.
Introduction
Medulloblastoma is the most common solid tumor found in children. These tumors arise in young children in the cerebellum, a part of the brain that develops post-natally in children and mice.Citation1 Some types of medulloblastoma are proposed to arise from proliferating CGNPs that fail to exit the cell cycle, migrate and/or differentiate.Citation2 CGNP proliferation requires activation of the Shh pathway. Shh is secreted from Purkinje cells in the cerebellum and binds to its receptor Patched (Ptc) on CGNPs, which prevents inhibition of Smoothened (Smo) and activates transcription of Shh targets, such as the Gli transcription factors, N-myc and the D-type cyclins, to drive CGNP proliferation.Citation3–Citation5 Mutations causing aberrant activation of the Shh signaling pathway are implicated in human and mouse medulloblastomas.Citation6,Citation7 To promote proliferation and oncogenic transformation, Shh alone is not sufficient; cooperative interactions with other signaling pathways are required.Citation4,Citation8–Citation12
CGNP proliferation is regulated by cyclin dependent kinases (Cdk4/6) in cooperation with D-type cyclins.Citation13–Citation14 However, cell cycle progression in cerebellar development is often controlled by Cdk inhibitors, p18INK4c and p27Kip1.Citation15 While p18INK4c preferentially targets Cdk4 and Cdk6, p27Kip1 has three distinct roles: (1) mediates cyclin D/Cdk4/6 assembly formation; (2) controls the late G1 phase by binding to and inhibiting Cdk2 and cyclin E; (3) regulates cell motility by binding to and inhibiting RhoA.Citation16–Citation24 In the cerebellum, nuclear p27Kip1 is found in post-mitotic and differentiated CGNPs, suggesting its ability to regulate cell cycle exit.Citation25 Indeed, losing p27Kip1 in cerebellar development accelerates CGNP proliferation.Citation15,Citation25 Low levels of p27Kip1 have been linked with high-grade tumors, including brain tumors such as astrocytoma and glioblastoma multiforme.Citation26–Citation30 Previous studies have shown that mislocalized p27Kip1 is associated with aggressive tumors, suggesting an oncogenic role in this context.Citation31–Citation34 We previously reported that p27Kip1 is mislocalized in Shh-mediated medulloblastoma, and this localization is Akt- and TSC2-dependent.Citation11,Citation35 More recently, Ayrault et al. observed that mice heterozygous for Patched and either heterozygous or nullizygous for p27Kip1 develop medulloblastoma and with high penetrance and accelerated rate.Citation8 In addition, these tumors retain the progenitor cell marker Math-1, which is expressed in proliferating CGNPs. These finding suggested that reduced levels of p27Kip1 could contribute to maintenance of a precursor cell phenotype that could be vulnerable to transforming events.
Here, using mice that express an activated mutant allele of the Shh receptor component Smoothened (SmoA1), we show that p27Kip1 is mislocalized in CGNPs and Shh-mediated medulloblastomas. We also find that SmoA1 mice lacking one or both p27Kip1 alleles have reduced survival compared to mice having two wild-type p27Kip1 alleles. Interestingly, SmoA1 mice heterozygous for p27Kip1 have decreased survival latency compared to mice lacking both copies of p27Kip1. Our results suggest that this may be due to retention of a single copy of p27Kip1 being sufficient to recruit cyclin D/Cdk4/6 to promote cell cycle progression yet insufficient to inhibit cyclin E/Cdk2. Finally, we show that p27Kip1 can interact with RhoA, perhaps contributing to tumor cell motility and invasion, and may underlie the more aggressive nature of p27Kip1 heterozygous medulloblastomas in comparison with the tumors arising in the p27Kip1-null mice.
Results and Discussion
p27Kip1 is mislocalized in Shh-mediated medulloblastoma.
We have previously observed that p27Kip1 is found in the cytoplasm of medulloblastomas arising in NeuroD2-SmoA1 transgenic mice. These mice express an activated mutant allele of the Shh receptor component Smoothened in the cerebellar progenitor compartment.Citation36 Approximately 60% of these mice develop medulloblastoma by 6 months of age. To explore the contribution of nuclear and cytoplasmic p27Kip1 to Shh-driven CGNP proliferation and medulloblastoma, we analyzed cerebella in wild-type and SmoA1 mice at post-natal day 7, the peak of CGNP proliferation. In wild-type mice, p27Kip1 is present in the nucleus of CGNPs in the post-mitotic region of the external granule layer (EGLb) and in the differentiated region inner granule layer (IGL) (). In contrast, SmoA1 mice have cytoplasmic p27Kip1 throughout the EGL, and this correlates with increased proliferation as determined by staining for Proliferating Cell Nuclear Antigen (PCNA, ).
Next, to confirm whether aberrant Shh signaling leads to increased CGNP proliferation, we cultured CGNPs from wild type and SmoA1 mice in the presence or absence of exogenous Shh. We found that vehicle-treated SmoA1 CGNPs have more phospho-Histone H3 (+) cells than wild-type cells (). Addition of Shh to the growth medium increased proliferation of wildtype and SmoA1 CGNPs, with SmoA1 CGNPs proliferating at a significantly higher rate, suggesting that these mutant CGNPs are Shh-responsive and potentially benefit from cytoplasmic p27Kip1.
Tumor cells benefit by phosphorylation and/or mislocalization of tumor suppressor genes.Citation34,Citation37–Citation40 To determine whether this is the case for p27Kip1 in Shh-driven medulloblastomas, we utilized whole cell lysates from SmoA1 medulloblastoma tumors and adjacent cerebella. Using western blot analysis, we found that p27Kip1 is phosphorylated at Ser10, which is responsible for its nuclear export,Citation41–Citation44 and at Thr187, which is regulated by active cyclin E and cyclin A/Cdk2 leading to Skp2-mediated ubiquitinated degradation ().Citation45,Citation46 Using immunfluorescence analysis, as shown in , adult mouse cerebellar tissue features nuclear p27Kip1 in differentiated granule neurons occupying the internal granule layer (IGL). However, in SmoA1 medulloblastoma tissue, which is highly proliferative, p27Kip1 is found in the cytoplasm. Together, this suggests that misregulated Shh signaling alters p27Kip1 localization and is correlated with increased cell proliferation.
p27Kip1 gene dose affects medulloblastoma incidence and onset in SmoA1 mice.
The transmembrane protein Patched (Ptc) is a tumor suppressor that negatively regulates Smoothened activity and whose loss is associated with medulloblastoma formation in humans.Citation47,Citation48 Similarly, mice heterozygous for Ptc are predisposed to develop medulloblastomas.Citation49 Recently, it has been reported that p27Kip1 hetero- or nullizygousity increases medulloblastoma incidence in Ptc+/− mice.Citation8 Activating mutations in Smoothened are also found in human medulloblastomas,Citation50 and this can be phenocopied in mice using the NeuroD2-SmoA1 and SmoM1/M2 alleles.Citation36,Citation51–Citation52 To assess whether p27Kip1 gene dosage and localization affects medulloblastoma formation in this model of constitutive Shh pathway activation, we generated homozygous SmoA1 mice lacking one or both p27Kip1 alleles. SmoA1 mice were previously reported to develop medulloblastoma at a high incidence (∼90%) and a mean time of occurrence of 5 to 6 months.53 We observed a lower medulloblastoma incidence (∼53%) in our mouse colony (). However, losing one or both p27Kip1 alleles increased tumor incidence to ∼84% and ∼100%, respectively. Among the homozygous SmoA1 medulloblastoma-bearing mice, the p27Kip1 heterozygous mice have a reduced survival time (median survival of 61 days) than wild type (median survival of 147 days) and nullizygous (median survival of 71 days) ().
Surprisingly, when we generated hemizygous SmoA1 mice lacking one or both p27 alleles, we observed that hemizygous SmoA1 mice heterozygous for p27Kip1 have a higher tumor incidence (∼68%) than wild type (∼39%) or nullizygous for p27Kip1 (∼45%). Among the hemizygous SmoA1 medulloblastoma-bearing mice, the p27Kip1 heterozygous mice have a strikingly reduced survival time (median survival of 105 days) compared with wild-type (median survival of 186 days) and p27 nullizygous mice (median survival of 204 days) (). The increased tumor incidence and dramatically reduced survival of homozygous and hemizygous SmoA1 medulloblastoma-bearing mice suggests p27Kip1 is haploinsufficient in Shhmediated medulloblastomas. These results are consistent with reduced p27Kip1 function promoting enhanced CGNP transformation in the setting of activated Shh signaling.
To determine how p27Kip1 loss contributes to SmoA1 medulloblastoma formation, we investigated how G1- and S-phase cell cycle regulators are affected by p27Kip1 reduction or loss in adult cerebella and medulloblastoma of SmoA1 mice. We observed high protein levels of D-type cyclins in tumors compared to non-tumors (). These tumors also maintained high levels of N-myc, a Shh transcriptional target;Citation4,Citation54 Bmi-1, a progenitor cell marker implicated in Shh-driven medulloblastoma;Citation11,Citation55–Citation56 cyclin A and cyclin E. Consistent with our previous report, active mTOR signaling, which is vital for cell growth, was found in these tumors, based on ribosomal protein-S6 phosphorylation.Citation11 Finally, previous reports have shown that tyrosine phosphorylation of p27Kip1 by c-Abl and Src family kinases initiates the transition of p27Kip1 from inhibitor of cyclin E/Cdk2 in G0 phase to substrate of cyclin/Cdk2 in G1 phase, therefore making p27Kip1 a poor Cdk2 inhibitor and potentially a promoter of cyclin D/ Cdk4 assembly.Citation20,Citation57–Citation58 While we find Src to be present in both non-tumor tissue and tumors, we observed high levels of c-Abl in Shhmedulloblastoma. Thus, all of the tumors are highly proliferative, regardless of p27 gene status.
Cyclin D/Cdk4/6 assembly is dependent on p27Kip1.
Progression from G1 to S phase of the cell cycle requires assembly of D-type cyclin:Cdk complexes.Citation19 It has been shown that p27Kip1 has an essential role in assembly and stability of cyclin D/Cdk4/6 complexes.Citation20–Citation21 We speculated that the increased aggressiveness of p27Kip1 heterozygous medulloblastomas in the hemizygous mice might be due to differential assembly of D-type cyclin:Cdk complexes in comparison to the p27Kip1-null mice. Using coimmunprecipitation and western blotting from cerebella and medulloblastomas from SmoA1 mice, we found that p27Kip1 is bound with cyclin D1, Cdk4 and Cdk6 in tumors but not in the normal cerebella tissue (). Next, to see if this assembly is affected by p27Kip1 gene dosage, we performed immunoprecipitation using tumors and cerebellar tissue from hemizygous SmoA1 mice that are either wild type, heterozygous or nullizygous for p27Kip1. We chose hemizygous SmoA1 mice because of the significant effect p27 heterozygosity has on tumor latency in that genetic background. We observed decreased interaction between cyclin D1 and Cdk6 as tumors lose p27Kip1 (), which would be consistent with impaired cyclin D1:Cdk6 complex assembly, which may impede G1 cell cycle progression. In its role as a tumor suppressor and negative cell cycle regulator, p27Kip1 binds to and inhibits cyclin E/Cdk2 to prevent late G1-to-S progression, leading to cell cycle exit.Citation19 Medulloblastomas that are heterozygous or null for p27Kip1 show less cyclin E bound to p27Kip1, suggesting ongoing activity of the cyclin E:Cdk2 complex, which promotes pRb inactivation. Together, this may reflect the requirement for p27Kip1 in recruiting cyclin D/Cdk4/6 to promote cell cycle progression, yet insufficient to inhibit cyclin E/ Cdk2 in these tumors.
Cytosolic p27Kip1 plays a role in cell motility in SmoA1 medulloblastoma.
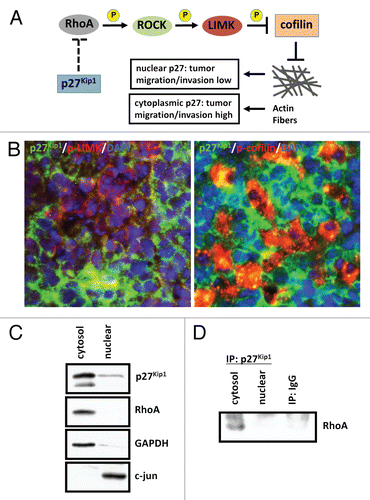
The observation that p27Kip1 is predominantly mislocalized rather than undergoing degradation in SmoA1 medulloblastomas suggests it may have a functional role in the cytoplasm of these tumor cells. Recent studies have suggested that cytoplasmic p27Kip1 plays an oncogenic role in mediating cell motility by preventing activation of RhoA, a regulator of actin cytoskeleton in the formation of stress fibers ().Citation17,Citation22,Citation37,Citation59–Citation61 Cell migration is controlled by RhoA signaling and is dependent on RhoA to convert from the GDPinactive state to GTP-active state.Citation62 RhoA-GTP stabilizes actin stress fibers through the activation of its substrate Rhoassociated, coiled-coil containing protein kinase (ROCK), which then phosphorylates and activates LIM domain-containing protein kinases (LIMK). LIMK in turn phosphorylates and inhibits the actin depolymerization-promoting protein cofilin, resulting in stabilization of stress fibers. See et al. showed that p27Kip1 deficiency in PDGF-expressing glial cells correlated with elevated levels of Rho-GTP and reduced cell migration.Citation63 We therefore asked whether p27Kip1 might play a role in cell motility in Shh-mediated medulloblastomas. We first co-stained p27Kip1 with phospho-LIMK and phospho-cofilin in these tumors and found an inverse relationship between p27Kip1 and these markers of motility inhibition, therefore suggesting that these tumor cells carrying cytoplasmic p27Kip1 have increased cell motility ().
p27Kip1 can regulate cell migration by binding to RhoA, potentially interfering its GDP state-to-GTP state conversion and/or its relationship to activate ROCK.Citation17,Citation64–Citation65 To determine a potential relationship between p27Kip1 and RhoA, we then carried out subcellular fractionation and immunprecipitation/western blotting of these tumor cells and found that these proteins interact in the cytoplasm (, ). Though p27Kip1 serves as a tumor suppressor to regulate cell cycle progression in the nucleus, it may also be stabilized in the cytoplasm to contribute to oncogenesis independent of its cell cycle functions to promote cell motility and invasion. Other members of the Cip/Kip class possess similar characteristics. Cytoplasmic p21Cip1 can inhibit ROCK activity, whereas p57Kip2 inhibits the function of LIMK by sequestering it in the cytoplasm, away from its substrate cofilin.Citation23,Citation38–Citation39,Citation66–Citation67 We have previously shown the expression analysis of CDKN1B (p27Kip1) in human Shh-subgroup medulloblastomas and found a moderate decrease compared with adult cerebellum.Citation11 p27Kip1 is not a classic tumor suppressor, as it is rarely mutated or deleted in cancer, but rather often deregulated in cancer by post-translational modifications.Citation68–Citation69 While our data suggest that p27Kip1 may be a useful biomarker, it is unknown whether cytoplasmic p27 is seen in human medulloblastoma. Therefore, more work is needed in human medulloblastoma studies to define its regulation, as it serves an important role in controlling tumor cell growth and could potentially serve as a therapeutic target in clinical trials.
Materials and Methods
Mice.
Harvest of neural precursors from neonatal mice and preparation of cerebella and tumor tissue from wild-type and mutant mice for histological analysis were carried out in compliance with the Memorial Sloan-Kettering Institutional animal care and use committee guidelines. C57-BL6 wild-type mice (Jackson Laboratories), heterozygous and nullizygous p27 mice (kindly given by Andrew Koff of Memorial Sloan-Kettering Cancer Center)Citation70 and NeuroD2-SmoA1 mice (kindly provided by Jim Olson of Fred Hutchinson Cancer Research Center)Citation36 were used.
Genotyping.
p27Kip1-null male mice were bred to SmoA1 female mice to generate SmoA1/+; p27+/− F1 mice. Intercrossing these mice led to other genotypes used for analysis. SmoA1 mice were genotyped by PCR as previously described.Citation36 To distinguish hemizygous from homozygous SmoA1 mice, QPCR was performed as described in www.jax.org.
CGNP culture.
CGNP cultures were generated as previously described.Citation4 Cells were plated on individual poly-DL-ornithine (Sigma) pre-coated plates or pre-coated glass coverslips for immunostaining. Where indicated, Shh (R&D Systems) was used at a concentration of 3 µg/mL for 48 hrs.
Immunoblotting, immunoprecipitation and subcellular fractionation.
Protein extracts were prepared as previously described.Citation5 A total of 50 µg murine cerebella and medulloblastoma protein, were run on 8–12% SDS-polyacrylamide gels and transferred to a PVDF membrane (Millipore). The blots were blocked in 5% milk for one hour at room temperature and incubated with primary antibodies in 3% BSA (in TBS-T) or 5% milk overnight at 4°C. Blots were washed three times and incubated with secondary antibodies in 5% milk in TBS-T for two hours at room temperature. After washing, the signals were developed using the enhanced chemiluminescence method (Amersham), and the membranes were exposed to Kodak Biomax film. Primary antibodies were: total p27 (BD Transduction Labs), phospho-p27 S10 and T187 (Santa Cruz), Cdk4 (C-22; Santa Cruz), Cdk6 (C-21; Santa Cruz), cyclin D1 (H-295; Santa Cruz), cyclin D2 (M-20; Santa Cruz), cyclin E (M-20; Santa Cruz), cyclin A (H-432; Santa Cruz), N-myc (C-19; Santa Cruz), Bmi-1 (Upstate), phospho- and total rp-S6 (Cell Signaling), Src (Cell Signaling), c-Abl (K-12; Santa Cruz) and β-tubulin (Sigma). HRP conjugated secondary antibodies were: goat anti-rabbit IgG (H+L) (Thermo Scientific) and donkey anti-mouse IgG (H+L) (Jackson Immuno Research).
For immunoprecipitation studies, 1 mg of protein extract was used in each case. Ten µg of antibody were incubated with protein A-sepharose for 2 h. Protein extracts were precleared with protein A-sepharose for 2 h and then incubated with the antibody plus protein A-sepharose overnight. The precipitate was washed four times and proteins were eluted with 0.2 M glycine and neutralized with 1M Tris pH 7.4. Antibodies used for immunoprecipitation were same as above. Rabbit IgG was used as control (Upstate Biotechnologies).
Subcellular fractionation was performed using NE-PER Nuclear and Cytoplasmic Extraction Reagents (Pierce) following the manufacturer's instructions. Protein content was determined by using the Bio-Rad protein assay. Antibodies used for subcellular fractionation were: p27 (BD Transduction Labs), RhoA (119; Santa Cruz), GADPH (Cell Signaling), and c-jun (Calbiochem).
Immunofluorescence.
CGNPs were fixed in 4% PFA for 10 minutes. Cells were then washed with 1xPBS and permeabilized in 1% TritonX-100 for 5 minutes. Cells were blocked in 5% goat serum in PBS-T (1xPBS and 0.1% TritonX-100) for one hour atroom temperature, washed once with 1xPBS and then incubated with primary antibody in 2.5% goat serum (in PBS-T) overnight at 4°C. They were washed three times with 1xPBS and incubated with secondary antibody for two hours at room temperature, then washed and mounted in DAPI-containing mounting medium (Vector Labs).
Paraffin-embedded tissue slides were processed as previously described before incubation with primary antibodies.Citation11 Primary antibodies used were: p27 (BD Transduction Labs), PCNA (Calbiochem), phospho-Histone H3 S10 (Cell Signaling) phospho-LIMK1/2 T508/505 (Santa Cruz) and phospho-cofilin S3 (Cell Signaling). Secondary fluorescent-tagged antibodies were: Alexa Fluor goat anti-rabbit 488/594 (Invitrogen) and Alexa Fluor goat anti-mouse 488/594 (Invitrogen).
Immunostaining performed on cultured cells or tissue sections was visualized using a Leica DM5000B microscope, and images were captured with Leica FW400 software. For quantification of phospho-Histone H3 immunostaining, TIFF images of four random fields were taken for each experimental group using the 10X objective. The percentage of P-Histone-H3-positive cells over the total number of cells, as determined by DAPI staining, was calculated using Image Pro Plus software (MediaCybernetics).
Figures and Tables
Figure 1 p27Kip1 is mislocalized in Shh-mediated medulloblastoma. (A) In cerebellar development of post-natal day 7, the peak for CGNP proliferation, p27Kip1 (green) is expressed in post-mitotic region, external granule layer b (EGLb), molecular layer (ML) and differentiated region inner granule layer (IGL), whereas aberrant Shh signaling leads to p27Kip1 mislocalization and increased CGNP proliferation throughout the EGL. Proliferation marker PCNA (red) stains CGNPs in proliferating region EGLa. (B) Wild-type and SmoA1 CGNPs were plated, treated with Shh and measured for proliferation by quantification of phospho-Histone H3. (C) While non-tumor cells undergo cell cycle exit and/or differentiation via p27Kip1 nuclear localization in the IGL, the Shh-induced medulloblastoma maintains misregulated phosphorylation of p27Kip1 at Ser10 and Thr187, which leads to mislocalization and inhibition of cell cycle regulation. (D) Non-tumors in NeuroD2-SmoA1 mice have nuclear p27Kip1 (first column), whereas SmoA1 medulloblastoma have cytoplasmic p27Kip1 (second column) and high proliferation (third and fourth column).
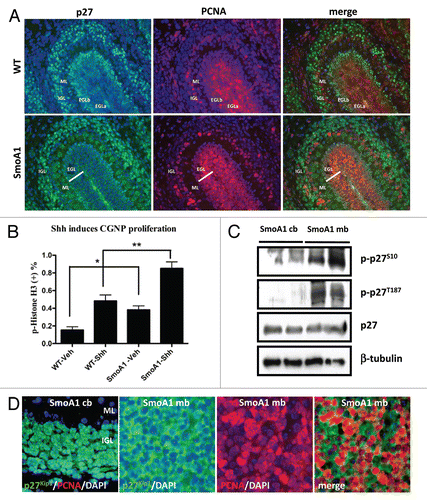
Figure 2 (A–C) p27Kip1 loss accelerates medulloblastoma incidence in SmoA1 mice. (A) p27Kip1 loss accelerates medulloblastoma incidence in homozygous and hemizygous mice for SmoA1 transgene. (B and C) SmoA1 mice heterozygous for p27Kip1 have a dramatic decreased survival latency compared to homozygous and nullizygous for p27Kip1.
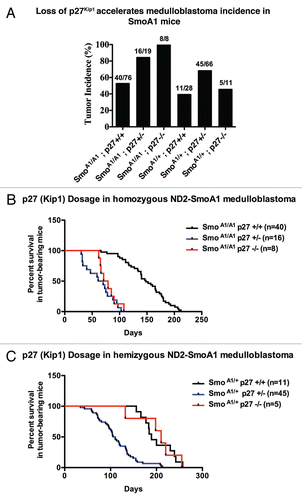
Figure 2D p27Kip1 loss accelerates medulloblastoma incidence in SmoA1 mice. (D) Western blot analysis of cell cycle indicators in NeuroD2-SmoA1 medulloblastomas arising in wild-type, p27+/− and p27−/− mice.
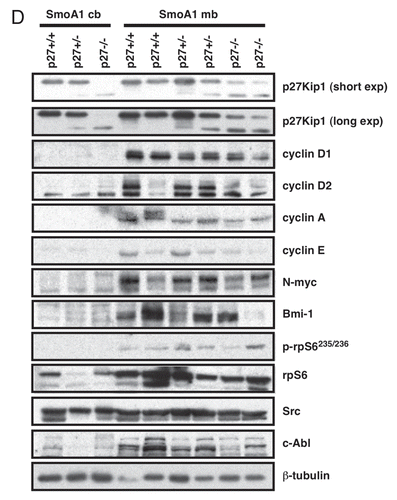
Figure 3 Cyclin D/Cdk4/6 assembly is dependent on p27Kip1. (A) Immunoprecipitation shows p27Kip1 interacts with cyclin D1, Cdk4/6 in SmoA1 medulloblastoma. (B) Immunoprecipitation shows decreased cyclin D1/Cdk6 interaction as a result of p27Kip1 loss. Tumor proliferation benefits as cyclin E/p27Kip1 is decreased, suggesting p27Kip1 is haploinsufficient as a tumor suppressor.
Figure 4 p27Kip1 plays a role in cell motility in SmoA1 medulloblastoma. (A) Schematic diagram of p27Kip1 regulating cell motility in tumor cells. (B) Immunofluorescence shows p27Kip1 is inversely correlated with stabilized cell motility, as evidenced by p27Kip1 (green), active phosphorylated-LIMK to prevent cell motility (red, left panel) and inactive phosphorylation of LIMK substrate and actin-depolymerization protein cofilin (red, right panel) in SmoA1 medulloblastoma. (C) Subcellular fractionation of SmoA1 medulloblastoma shows p27Kip1 and cell motility regulator RhoA are localized mainly in the cytoplasm. GAPDH serves as cytosolic control and c-jun as nuclear control. (D) Immunoprecipitation of p27Kip1 reveals interaction with RhoA. This data suggests p27Kip1 is needed to positively regulate cell motility in SmoA1 medulloblastoma.
Acknowledgements
We especially thank Dr. Andrew Koff and Dr. Zaher A. Nahle for assistance and advice. These studies were funded by the NIH (NINDS R01NS061070) (A.M.K.) and the Memorial Sloan-Kettering Brain Tumor Center (B.B.).
References
- Marino S. Medulloblastoma: Developmental mechanisms out of control. Trends Mol Med 2005; 11:17 - 22
- Rubin JB, Rowitch DH. Medulloblastoma: A problem of developmental biology. Cancer Cell 2002; 2:7 - 8
- Knoepfler PS, Kenney AM. Neural precursor cycling at sonic speed: N-Myc pedals, GSK-3 brakes. Cell Cycle 2006; 5:47 - 52
- Kenney AM, Cole MD, Rowitch DH. Nmyc upregulation by sonic hedgehog signaling promotes proliferation in developing cerebellar granule neuron precursors. Development 2003; 130:15 - 28
- Kenney AM, Rowitch DH. Sonic hedgehog promotes G(1) cyclin expression and sustained cell cycle progression in mammalian neuronal precursors. Mol Cell Biol 2000; 20:9055 - 9067
- Pietsch T, Waha A, Koch A, Kraus J, Albrecht S, Tonn J, et al. Medulloblastomas of the desmoplastic variant carry mutations of the human homologue of Drosophila patched. Cancer Res 1997; 57:2085 - 2088
- Raffel C, Jenkins RB, Frederick L, Hebrink D, Alderete B, Fults DW, et al. Sporadic medulloblastomas contain PTCH mutations. Cancer Res 1997; 57:842 - 845
- Ayrault O, Zindy F, Rehg J, Sherr CJ, Roussel MF. Two tumor suppressors, p27Kip1 and patched-1, collaborate to prevent medulloblastoma. Mol Cancer Res 2009; 7:33 - 40
- Uziel T, Karginov FV, Xie S, Parker JS, Wang YD, Gajjar A, et al. The miR-17∼92 cluster collaborates with the Sonic Hedgehog pathway in medulloblastoma. Proc Natl Acad Sci USA 2009; 106:2812 - 2817
- Uziel T, Zindy F, Xie S, Lee Y, Forget A, Magdaleno S, et al. The tumor suppressors Ink4c and p53 collaborate independently with Patched to suppress medulloblastoma formation. Genes Dev 2005; 19:2656 - 2667
- Bhatia B, Northcott PA, Hambardzumyan D, Govindarajan B, Brat DJ, Arbiser JL, et al. Tuberous sclerosis complex suppression in cerebellar development and medulloblastoma: separate regulation of mammalian target of rapamycin activity and p27Kip1 localization. Cancer Res 2009; 69:7224 - 7234
- Fernandez LA, Northcott PA, Dalton J, Fraga C, Ellison D, Angers S, et al. YAP1 is amplified and upregulated in hedgehog-associated medulloblastomas and mediates Sonic hedgehog-driven neural precursor proliferation. Genes Dev 2009; 23:2729 - 2741
- Ciemerych MA, Kenney AM, Sicinska E, Kalaszczynska I, Bronson RT, Rowitch DH, et al. Development of mice expressing a single D-type cyclin. Genes Dev 2002; 16:3277 - 3289
- Pogoriler J, Millen K, Utset M, Du W. Loss of cyclin D1 impairs cerebellar development and suppresses medulloblastoma formation. Development 2006; 133:3929 - 3937
- Zindy F, Knoepfler PS, Xie S, Sherr CJ, Eisenman RN, Roussel MF. N-Myc and the cyclin-dependent kinase inhibitors p18Ink4c and p27Kip1 coordinately regulate cerebellar development. Proc Natl Acad Sci USA 2006; 103:11579 - 11583
- Hengst L, Dulic V, Slingerland JM, Lees E, Reed SI. A cell cycle-regulated inhibitor of cyclin-dependent kinases. Proc Natl Acad Sci USA 1994; 91:5291 - 5295
- Larrea MD, Wander SA, Slingerland JM. p27 as Jekyll and Hyde: Regulation of cell cycle and cell motility. Cell Cycle 2009; 8:3455 - 3461
- Koff A, Ohtsuki M, Polyak K, Roberts JM, Massague J. Negative regulation of G1 in mammalian cells: Inhibition of cyclin E-dependent kinase by TGF-beta. Science 1993; 260:536 - 539
- Sherr CJ, Roberts JM. CDK inhibitors: Positive and negative regulators of G1-phase progression. Genes Dev 1999; 13:1501 - 1512
- James MK, Ray A, Leznova D, Blain SW. Differential modification of p27Kip1 controls its cyclin D-Cdk4 inhibitory activity. Mol Cell Biol 2008; 28:498 - 510
- Larrea MD, Liang J, Da Silva T, Hong F, Shao SH, Han K, et al. Phosphorylation of p27Kip1 regulates assembly and activation of cyclin D1-Cdk4. Mol Cell Biol 2008; 28:6462 - 6472
- Besson A, Gurian-West M, Schmidt A, Hall A, Roberts JM. p27Kip1 modulates cell migration through the regulation of RhoA activation. Genes Dev 2004; 18:862 - 876
- Besson A, Assoian RK, Roberts JM. Regulation of the cytoskeleton: An oncogenic function for CDK inhibitors?. Nat Rev Cancer 2004; 4:948 - 955
- Besson A, Hwang HC, Cicero S, Donovan SL, Gurian-West M, Johnson D, et al. Discovery of an oncogenic activity in p27Kip1 that causes stem cell expansion and a multiple tumor phenotype. Genes Dev 2007; 21:1731 - 1746
- Miyazawa K, Himi T, Garcia V, Yamagishi H, Sato S, Ishizaki Y. A role for p27/Kip1 in the control of cerebellar granule cell precursor proliferation. J Neurosci 2000; 20:5756 - 5763
- Razis E, Selviaridis P, Labropoulos S, Norris JL, Zhu MJ, Song DD, et al. Phase II study of neoadjuvant imatinib in glioblastoma: evaluation of clinical and molecular effects of the treatment. Clin Cancer Res 2009; 15:6258 - 6266
- Tatard VM, Xiang C, Biegel JA, Dahmane N. ZNF238 is expressed in postmitotic brain cells and inhibits brain tumor growth. Cancer Res 2010; 70:1236 - 1246
- Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2009; 8:2685
- Kirla RM, Haapasalo HK, Kalimo H, Salminen EK. Low expression of p27 indicates a poor prognosis in patients with high-grade astrocytomas. Cancer 2003; 97:644 - 648
- Gillies JK, Lorimer IA. Regulation of p27Kip1 by miRNA 221/222 in glioblastoma. Cell Cycle 2007; 6:2005 - 2009
- Liang J, Zubovitz J, Petrocelli T, Kotchetkov R, Connor MK, Han K, et al. PKB/Akt phosphorylates p27, impairs nuclear import of p27 and opposes p27-mediated G1 arrest. Nat Med 2002; 8:1153 - 1160
- Shin I, Yakes FM, Rojo F, Shin NY, Bakin AV, Baselga J, et al. PKB/Akt mediates cell-cycle progression by phosphorylation of p27(Kip1) at threonine 157 and modulation of its cellular localization. Nat Med 2002; 8:1145 - 1152
- Viglietto G, Motti ML, Bruni P, Melillo RM, D'Alessio A, Califano D, et al. Cytoplasmic relocalization and inhibition of the cyclin-dependent kinase inhibitor p27(Kip1) by PKB/Akt-mediated phosphorylation in breast cancer. Nat Med 2002; 8:1136 - 1144
- Viglietto G, Motti ML, Fusco A. Understanding p27(kip1) deregulation in cancer: Down-regulation or mislocalization. Cell Cycle 2002; 1:394 - 400
- Bhatia B, Nahle Z, Kenney AM. Double trouble: When Sonic hedgehog signaling meets TSC inactivation. Cell Cycle 2010; 9:456 - 459
- Hallahan AR, Pritchard JI, Hansen S, Benson M, Stoeck J, Hatton BA, et al. The SmoA1 mouse model reveals that notch signaling is critical for the growth and survival of sonic hedgehog-induced medulloblastomas. Cancer Res 2004; 64:7794 - 7800
- Denicourt C, Saenz CC, Datnow B, Cui XS, Dowdy SF. Relocalized p27Kip1 tumor suppressor functions as a cytoplasmic metastatic oncogene in melanoma. Cancer Res 2007; 67:9238 - 9243
- Lee S, Helfman DM. Cytoplasmic p21Cip1 is involved in Ras-induced inhibition of the ROCK/LIMK/cofilin pathway. J Biol Chem 2004; 279:1885 - 1891
- Yokoo T, Toyoshima H, Miura M, Wang Y, Iida KT, Suzuki H, et al. p57Kip2 regulates actin dynamics by binding and translocating LIM-kinase 1 to the nucleus. J Biol Chem 2003; 278:52919 - 52923
- Jiao W, Lin HM, Datta J, Braunschweig T, Chung JY, Hewitt SM, et al. Aberrant nucleocytoplasmic localization of the retinoblastoma tumor suppressor protein in human cancer correlates with moderate/poor tumor differentiation. Oncogene 2008; 27:3156 - 3164
- Rodier G, Montagnoli A, Di Marcotullio L, Coulombe P, Draetta GF, Pagano M, et al. p27 cytoplasmic localization is regulated by phosphorylation on Ser10 and is not a prerequisite for its proteolysis. EMBO J 2001; 20:6672 - 6682
- Ishida N, Kitagawa M, Hatakeyama S, Nakayama K. Phosphorylation at serine 10, a major phosphorylation site of p27(Kip1), increases its protein stability. J Biol Chem 2000; 275:25146 - 25154
- Ishida N, Hara T, Kamura T, Yoshida M, Nakayama K, Nakayama KI. Phosphorylation of p27Kip1 on serine 10 is required for its binding to CRM1 and nuclear export. J Biol Chem 2002; 277:14355 - 14358
- Connor MK, Kotchetkov R, Cariou S, Resch A, Lupetti R, Beniston RG, et al. CRM1/Ran-mediated nuclear export of p27(Kip1) involves a nuclear export signal and links p27 export and proteolysis. Mol Biol Cell 2003; 14:201 - 213
- Sheaff RJ, Groudine M, Gordon M, Roberts JM, Clurman BE. Cyclin E-CDK2 is a regulator of p27Kip1. Genes Dev 1997; 11:1464 - 1478
- Vlach J, Hennecke S, Amati B. Phosphorylation-dependent degradation of the cyclin-dependent kinase inhibitor p27. EMBO J 1997; 16:5334 - 5344
- Johnson RL, Rothman AL, Xie J, Goodrich LV, Bare JW, Bonifas JM, et al. Human homolog of patched, a candidate gene for the basal cell nevus syndrome. Science 1996; 272:1668 - 1671
- Hahn H, Wicking C, Zaphiropoulous PG, Gailani MR, Shanley S, Chidambaram A, et al. Mutations of the human homolog of Drosophila patched in the nevoid basal cell carcinoma syndrome. Cell 1996; 85:841 - 851
- Goodrich LV, Milenkovic L, Higgins KM, Scott MP. Altered neural cell fates and medulloblastoma in mouse patched mutants. Science 1997; 277:1109 - 1113
- Lam CW, Xie J, To KF, Ng HK, Lee KC, Yuen NW, et al. A frequent activated smoothened mutation in sporadic basal cell carcinomas. Oncogene 1999; 18:833 - 836
- Taipale J, Chen JK, Cooper MK, Wang B, Mann RK, Milenkovic L, et al. Effects of oncogenic mutations in smoothened and patched can be reversed by cyclopamine. Nature 2000; 406:1005 - 1009
- Mao J, Ligon KL, Rakhlin EY, Thayer SP, Bronson RT, Rowitch D, et al. A novel somatic mouse model to survey tumorigenic potential applied to the Hedgehog pathway. Cancer Res 2006; 66:10171 - 10178
- Hatton BA, Villavicencio EH, Tsuchiya KD, Pritchard JI, Ditzler S, Pullar B, et al. The Smo/Smo model: hedgehog-induced medulloblastoma with 90% incidence and leptomeningeal spread. Cancer Res 2008; 68:1768 - 1776
- Oliver TG, Grasfeder LL, Carroll AL, Kaiser C, Gillingham CL, Lin SM, et al. Transcriptional profiling of the Sonic hedgehog response: A critical role for N-myc in proliferation of neuronal precursors. Proc Natl Acad Sci USA 2003; 100:7331 - 7336
- Leung C, Lingbeek M, Shakhova O, Liu J, Tanger E, Saremaslani P, et al. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004; 428:337 - 341
- Michael LE, Westerman BA, Ermilov AN, Wang A, Ferris J, Liu J, et al. Bmi1 is required for Hedgehog pathway-driven medulloblastoma expansion. Neoplasia 2008; 10:1343 - 1349
- Grimmler M, Wang Y, Mund T, Cilensek Z, Keidel EM, Waddell MB, et al. Cdk-inhibitory activity and stability of p27Kip1 are directly regulated by oncogenic tyrosine kinases. Cell 2007; 128:269 - 280
- Chu I, Sun J, Arnaout A, Kahn H, Hanna W, Narod S, et al. p27 phosphorylation by Src regulates inhibition of cyclin E-Cdk2. Cell 2007; 128:281 - 294
- Larrea MD, Hong F, Wander SA, da Silva TG, Helfman D, Lannigan D, et al. RSK1 drives p27Kip1 phosphorylation at T198 to promote RhoA inhibition and increase cell motility. Proc Natl Acad Sci USA 2009; 106:9268 - 9273
- Besson A, Gurian-West M, Chen X, Kelly-Spratt KS, Kemp CJ, Roberts JM. A pathway in quiescent cells that controls p27Kip1 stability, subcellular localization, and tumor suppression. Genes Dev 2006; 20:47 - 64
- Nguyen L, Besson A, Heng JI, Schuurmans C, Teboul L, Parras C, et al. p27kip1 independently promotes neuronal differentiation and migration in the cerebral cortex. Genes Dev 2006; 20:1511 - 1524
- Bar-Sagi D, Hall A. Ras and Rho GTPases: a family reunion. Cell 2000; 103:227 - 238
- See WL, Heinberg AR, Holland EC, Resh MD. p27 deficiency is associated with migration defects in PDGF-expressing gliomas in vivo. Cell Cycle 2010; 9:1562 - 1567
- Papakonstanti EA, Ridley AJ, Vanhaesebroeck B. The p110delta isoform of PI 3-kinase negatively controls RhoA and PTEN. EMBO J 2007; 26:3050 - 3061
- Charette ST, McCance DJ. The E7 protein from human papillomavirus type 16 enhances keratinocyte migration in an Akt-dependent manner. Oncogene 2007; 26:7386 - 7390
- Denicourt C, Dowdy SF. Cip/Kip proteins: More than just CDKs inhibitors. Genes Dev 2004; 18:851 - 855
- Besson A, Dowdy SF, Roberts JM. CDK inhibitors: Cell cycle regulators and beyond. Dev Cell 2008; 14:159 - 169
- Chu IM, Hengst L, Slingerland JM. The Cdk inhibitor p27 in human cancer: prognostic potential and relevance to anticancer therapy. Nat Rev Cancer 2008; 8:253 - 267
- Slingerland J, Pagano M. Regulation of the Cdk inhibitor p27 and its deregulation in cancer. J Cell Physiol 2000; 183:10 - 17
- Kiyokawa H, Kineman RD, Manova-Todorova KO, Soares VC, Hoffman ES, Ono M, et al. Enhanced growth of mice lacking the cyclin-dependent kinase inhibitor function of p27(Kip1). Cell 1996; 85:721 - 732